
Abstract
Three standalone tetrahedral copper hydride clusters, [Cu4(μ4-H)(μ2-X)2(PPh2Py)4]+ (X = Cl, Br; Py = pyridyl), containing a tetrahedral [Cu4(μ4-H)] unit have been synthesized and structurally characterized. The six Cu–Cu distances of the [Cu4(μ4-H)] unit can be divided into three groups (2.65, 2.85, and 2.95 Å), lowering the idealized point group of the Cu4 core to D2 symmetry, thereby resulting in intrinsically chiral metal clusters which exist as racemic pairs of enantiomers in the centrosymmetric crystal structures. Strong photoluminescence (attributable to the existence of the two short Cu–Cu distances of 2.65 Å) was observed in solution and in the solid state upon near-UV irradiation. According to the Jellium model, the title clusters can be considered as two-shell Jelliumatic systems with superatomic electron counts of 2e@0e corresponding to the two shells of H−@[Cu4X2(PPh2Py)4]2+. The four-coordinated hydride in the tetrahedral Cu4 cavity adopts the superatomic electronic configuration of 1S2.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, copper hydride clusters have raised widespread attention owing to their potential applications in catalysis [1,2,3,4,5,6,7], hydrogen storage [8,9,10,11], etc. In order to provide better understanding of their chemical and physical properties, many copper hydride clusters and nanoclusters have been synthesized and structurally characterized [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26]. To date copper hydride clusters of ever increasing nuclearity and complexity in structure, often accompanied by intriguing properties, are known; notable examples are: Cu2H [12, 13], Cu3H, Cu3H3 [14, 15], Cu4H3 [16], Cu6H6 [1, 17, 18], Cu7H [19, 20], Cu8H [21, 22], Cu14H12 [23], Cu18H7 [24], Cu20H11 [25], Cu28H15 [26], Cu32H20 [27], etc. (the ligands were omitted for clarity). Among these hydride copper clusters, Cu7H, Cu8H, Cu20H11, Cu28H15 and Cu32H20 contain μ4-H− in their tetrahedral cavities. In particular, the Cu7H [19], Cu20H11 [25], Cu28H15 [26] and Cu32H20 [27] clusters were characterized by neutron diffraction.
Although [Cu4(μ4-H)]3+ unit is an essential component in the above mentioned polynuclear copper hydride clusters, to date, there is as yet no report of standalone tetrahedral Cu4(μ4-H) copper hydride cluster in the literature. In view of the fact that tetrahedron is the simplest form of 3-D polyhedra, which often acts as a building block, or a capping component, in cluster formation, it is desirable to assess the structural and electronic characteristics of a standalone tetrahedral copper hydride cluster. We should mention here that among the tetranuclear copper clusters reported in the literature, the most common motif is the so-called cubane-like L4Cu4X4 (L = monodentate ligands, X = Cl, Br, I, O, S, Se, C ≡ CR) [28,29,30,31,32,33,34,35,36].
Herein we describe the synthesis and characterization of three copper hydride clusters, [Cu4(μ4-H)(μ2-X)2(PPh2Py)4]+ clusters (X = Cl, Br; Py = pyridyl), which represent first examples of a standalone tetranuclear copper hydride cluster having a μ4-H in the tetrahedral Cu4 cavity. (Scheme 1) Interestingly, clusters 1 and 2 were found to emit strong photoluminescence upon near UV irradiation.

Synthesis of 1–3′
Experimental
Reagents
Copper chloride (CuCl2·2H2O, 98+%), sodium borohydride (NaBH4, 96%), methanol (CH3OH, A.R.), dichloromethane (CH2Cl2, A.R.) and n-hexane (n-C6H14, A.R.) were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China), 2-(diphenylphosphino) Pyridine (PPh2Py, 99%), copper bromide (CuBr2, 99+%), potassium hexafluorophosphate (KPF6, 98%) and dibromomethane (CH2Br2, 99%) were purchased from Adamas Reagent Co. Ltd. (Shanghai, China). All reagents were used as received without further purification. The water used in all experiments was ultrapure.
Synthesis of [Cu4HCl2(PPh2Py)4]·[CuCl2]·CH2Cl2 (1)
In the preparation, 17 mg of CuCl2·2H2O was dissolved in 20 mL CH3OH, followed by the addition of 50 mg PPh2Py in 30 mL CH2Cl2. The mixture was cooled to 0 °C in an ice bath, after 20 min, a freshly prepared 10 mL NaBH4 aqueous solution (7.6 mg/mL) was added dropwise under vigorous stirring, the color of the mixture turned from pale green into orange. The mixture was aged for 5 days. The color of the mixture turned from orange into brilliant yellow gradually during this period. The aqueous phase was then removed. The organic phase was washed several times with water. Pale-yellow single crystals suitable for X-ray diffraction study were grown by a double-layer of n-hexane/CH2Cl2 after 2 weeks.
Synthesis of [Cu4HCl2(PPh2Py)4]·PF6·1.5CH2Cl2·0.5H2O (2)
In a typical preparation, 34 mg of CuCl2·2H2O was dissolved in 20 mL CH3OH, followed by the addition of 50 mg PPh2Py in 30 mL CH2Cl2, the resulting mixture was then cooled to 0 °C in an ice bath. After 20 min, a freshly prepared 10 mL aqueous solution of NaBH4 (7.6 mg/mL) was added dropwise under vigorous stirring, followed by the addition of 10 mL of an aqueous KPF6 solution (3.6 mg/mL). The mixture was aged for 5 days. The color change was the same as that in synthesis of cluster 1. The aqueous phase was then removed, the organic phase was washed several times with water. Pale-yellow single crystals were obtained by a double-layer of n-hexane/CH2Cl2 after 2 weeks.
Synthesis of [Cu4HBr2(PPh2Py)4]·PF6·3CH2Br2 (3)
44 mg of CuBr2 was dissolved in 20 mL of CH3OH, followed by the addition of 50 mg PPh2Py in 30 mL of CH2Br2, the mixture was cooled to 0 °C in an ice bath. After 20 min, a freshly prepared 10 mL aqueous solution of NaBH4 (7.6 mg/mL) was added dropwise under vigorous stirring, followed by the addition of 10 mL aqueous KPF6 solution (3.6 mg/mL). The mixture was aged for 5 days. The color change was the same as that in synthesis of cluster 1. The aqueous phase was then removed, the organic phase was washed several times with water. Pale-yellow single crystals were obtained by a double-layer of n-hexane/CH2Br2 after several weeks.
Synthesis of [Cu4HBr2(PPh2Py)4]·PF6·2.5CH2Cl2 (3′)
The procedure for 3′ is the same as with 3, the only difference being the use of CH2Cl2 as solvent. The color change was the same as that in synthesis of cluster 1.
Single-Crystal X-ray Diffraction
The diffraction data were collected on a Bruker APEX CCD diffractometer equipped with a graphite-monochromated Mo Kα radiation source (λ = 0.71073 Å). All crystallographic data were collected at low temperature in a nitrogen flow cooled with liquid nitrogen. The absorption correction was performed with SADABS.
Single Crystal X-ray Structure Determination
The structures were solved by direct method using SHELXS-97 and refined by full-matrix least-squares techniques using SHELXL-2014. All non-hydrogen atoms were refined anisotropically. The H atoms on ligands were positioned geometrically and were allowed to ride on the C atoms in the subsequent refinement [aromatic C–H = 0.94 Å, U(H) = 1.2 Ueq(C)]. In each case, the encapsulated hydride was detected from the difference electron density map, and its position was allowed to refine freely for 1 and 3. In the refinement for 2 and 3′, the Cu–H distances of the encapsulated hydride were restrained to be equal (via SADI).
Part of the aromatic rings, and PF6– or solvent molecules, were disordered in 2; their position parameters were refined individually with the same set of anisotropic thermal parameters in the refinements (SIMU). The occupancy factors of the two disordered components were least-square refined so that their sum is constrained to be unity. The crystallographic details are listed in Table S1 (SI). CCDC Nos. for 1–3′ are 1819960, 1819961, 1819962 and 1819963.
ESI–MS
Chemical compositions were identified by electrospray ionization time-of-flight mass spectrometry (ESI–TOF–MS) in positive mode.
PXRD
X-ray powder diffractions were measured on a PANalytical X’pert pro diffractometer with Cu Kα radiation. The powder X-ray diffraction patterns of clusters 1 and 2 are shown in Fig. S1 (SI).
UV/Vis
Pure crystals of cluster 1 and 2 were dissolved in dichloromethane (CH2Cl2) for spectral measurements. UV/Vis absorption spectra were recorded on a Shimadzu UV2550.
FT-IR
FT-IR spectra were recorded on a Nicolet AVATAR FT-IR 330 spectrometer as KBr pellets in the frequency range 4000–400 cm−1. FT-IR spectra of 1 and 2 are shown in Fig. S2 (SI).
Photoluminescence
Photoluminescence spectra were measured on a Hitachi F-7000 spectrometer and shown in Figs. S3–S5 (SI).
Results and Discussion
Structure Descriptions
The title clusters contain the common tetrahedral unit, [Cu4(μ4-H−)]3+, in addition to extra coordination of the doubly-bridging halides, X− = Cl−, Br−, at opposite edges of the tetrahedron. The P and N atoms of four chelating ligands, PPh2Py, bridge the remaining four edges of the tetrahedron.
In cluster 1, the monocationic cluster, [Cu4(μ4-H)(μ2-Cl)2(PPh2Py)4]+, is located on a crystallographic twofold axis in the centrosymmetric monoclinic unit cell with C2/c space group (Z = 4). In the crystal structure of 1 (Fig. 1), the [Cu4(μ4-H−)]3+ core is surrounded by two Cl− and two PPh2Py ligands, results in the formation of a twofold symmetric cation [Cu4(μ4-H)(μ2-Cl)2(PPh2Py)4]+. Each Cu is coordinated to one P atom and one N atom from two different PPh2Py ligands. The asymmetric unit also contains one [CuCl2]− as the counteranion, and two solvent CH2Cl2 molecules.

Structure of [Cu4(μ4-H)(μ2-Cl)2(PPh2Py)4]+ (1), as [CuCl2]− salt (50% probability, C, H at 30% probability, C–H hydrogen atoms are omitted): a general view. b View along crystallographic twofold axis. Symmetry code: a = − x+1, y, − z+1/2
In the crystal structure of 2 (Fig. 2), the [Cu4(μ4-H)(μ2-Cl)2(PPh2Py)4]+ is the same as that in 1, with the exception that the counterion is the PF6− anion and the solvent molecules are CH2Cl2 and H2O.

Structure of [Cu4(μ4-H)(μ2-Cl)2(PPh2Py)4]+ (2), as [PF6]− salt (50% probability, C, H at 30% probability, C–H hydrogen atoms are omitted)
In the crystal structure of 3 (Fig. 3), the cation [Cu4(μ4-H)(μ2-Br)2(PPh2Py)4]+ is different from that in 1, the arrangements of the PPh2Py ligands are different. One of the Cu (Cu3) is coordinated to two P atoms, and another Cu (Cu2) to two N atoms. The charge compensation anion is PF6− and the solvent molecules are CH2Br2.

Structure of [Cu4(μ4-H)(μ2-Br)2(PPh2Py)4]+ (3), as [PF6]− salt (50% probability, C, H at 30% probability, C–H hydrogen atoms are omitted)
The crystal structure of 3′ is the same as that of 3. The charge compensation anion is also PF6−, the solvent molecules are CH2Cl2, instead of CH2Br2.
The chelating effect of the PPh2Py plays an important factor in the assembly of these structures. Each Cu atom is coordinated by P and N atoms from two adjacent chelating PPh2Py ligands. For cluster 2, four bidentate Cu(n)–P(n)–C–N(n)–Cu(n + 1) linkages, where n = 1–4, wrap around the Cu4 tetrahedron, connecting Cu1 to Cu2 to Cu3 to Cu4. The last N4 atom binds to Cu1 to complete the cycle. The same is true for cluster 1, with the exception that the crystallographically imposed twofold symmetry reduces Cu3 and Cu4 into the symmetry-related Cu1a and Cu2a, respectively.
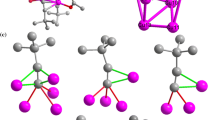
To our surprise, the chelating arrangement of cluster 3 is different from that of clusters 1 and 2. Here one of the four chelate linkages, namely, Cu2–P2–C–N2–Cu3, “flips” to become Cu2–N2–C–P2–Cu3 instead. We believe this unexpected “flip” can be traced to steric hindrance caused by the substantially larger van der Waals radius of the bromide vs chloride ligands. Figure 4 depicts the space filling models of 1, 2 and 3, showing the van der Waals contacts and intramolecular C–H···X (X = Cl, Br) interactions between the bridging Cl atom and the aromatic rings. The cavity surrounded by four PPh2Py is barely large enough to accommodate the halide atoms. One hint of this steric hindrance, albeit a significantly weaker one, is evident in the crystallographical disorder of the two phenyl groups in cluster 2 (as shown by dotted rings in Fig. 2) and the “flip” of one of the four chelate linkages of 3 in the same region. Another curious consequence of this steric effect is that the Br-bridged Cu4 tetrahedron of 3 is smaller than the Cl-bridged analog (cf. Table 1, average Cu–Cu distances of 2.77 versus 2.81 Å, respectively). This can be attributed that the longer Cu–Br (2.51(av) Å) versus Cu–Cl (2.41(av) Å) bonds and the weak Cu–Cu interactions at 2.77–2.81 Å. In other words, the steric effect(s) of the phenyl and pyridyl groups amongst themselves as well as with the halides and the requirement of normal Cu-halide bond lengths cause the Cu4 tetrahedron of 3 to shrink from average Cu–Cu distance of 2.81 Å to 2.77 Å. At these Cu–Cu distances the potential surface is pretty flat, representing at best weak Cu–Cu bonding interactions.

vdW models of clusters 1, 2, and 3 as viewed from X1 (top) and X2 (bottom) where X = Cl (clusters 1 and 2), Br (cluster 3). Disordered phenyl groups in 2 are drawn as dark red and dark green; the flipped PPh2Py ligand in 3 is represented in dark yellow (Color figure online)
The two shortest Cu–Cu distances of 2.64–2.68 Å can be considered as significant bonding, though weak in comparison to the 2.4–2.5 Å range. The other two pairs of Cu–Cu distances are basically nonbonding.
Table 1 provides a comparison of Cu–Cu distances in a number of known copper hydride clusters. The average Cu–Cu distances of clusters 1–3′, in the range of 2.817–2.760 Å, is shorter than those in [Cu7(H){S2C(aza-15-crown-5)}6] [19, 20] and [Cu8(H){S2CNR2}6]+ [21, 22] (average 3.055–2.843 Å), approximately equal to those in [Cu20(H)11{S2P(OiPr)2}9] [25] and [Cu32(H)20{S2P(OiPr)2}12] [27] (average 2.777–2.811 Å), but longer than that in [Cu28(H)15(S2CNP)12]+ [26] (average 2.644 Å).
Table 1 also provides a comparison of Cu–H distances in a number of copper hydride clusters. The average Cu–H distance of cluster 1–3′ is 1.694–1.727 Å, which is shorter than those in [Cu7(H){S2C(aza-15-crown-5)}6] [19, 20] (average 1.896 Å) and [Cu8(H){S2CNR2}6]+ [21, 22] (average 1.778 Å), and [Cu32(H)20{S2P(OiPr)2}12] [27] (average 1.739 Å), approximately equal to that in [Cu20(H)11{S2P(OiPr)2}9] [25] (average 1.708 Å), and is longer than that in [Cu28(H)15(S2CNnPr2)12]+ [26] (average 1.565 Å). The ligands used in these latter clusters and the structures are different from those used in this paper.
The van der Waals (vdW) models of these clusters are shown in Fig. 4, as viewed from X1 (top) and X2 (bottom) where X = Cl, Br. The vdW ring orientations of cluster 1 seem to be more favorable (less repulsions) than that of cluster 2. That may be why cluster 2 has the disorder of two rings. In cluster 3, the disorders were avoided by flipping the last chelate.
Cu Coordination
Each Cu atom is coordinated with P, N, X (X = Cl or Br) and three Cu atoms in a highly distorted trigonal prismatic arrangement instead of the usual distorted octahedral fashion. We believe this configuration is most likely dictated by the doubly bridging halide ligands.
Cu–H Distances in Tetrahedral Cavities
The average Cu–H distances in 1–3′ are 1.727, 1.719, 1.711 and 1.694 and the average Cu–Cu distances of 2.817, 2.801, 2.783, and 2.760 Å (cf. Table 1). These values are substantially shorter than the corresponding values in the Cu4H tetrahedral cages of Cu7H clusters recently reported by Liu and coworkers. The latter structure was characterized by neutron diffraction [19]. The average Cu–Cu distance of the latter cluster was measured to be 3.055 Å, and the Cu–H distance was 1.896 Å. The implication is that the hydride is more tightly held in the tetrahedral cavities of 1–3′ with somewhat stronger Cu–H and Cu–Cu interactions. Given these comparisons, it should be emphasized that the Cu–Cu interactions in these copper hydride clusters are weak interactions at best.
ESI–MS
We now turn to the hydride formulations of the title clusters. As described earlier, X-ray crystallography provided information with regards to the hydride position in clusters 1–3′. ESI–MS is a powerful complementing technique. As shown in Fig. 5a, c, two prominent peaks were observed centered at m/z = 1379.00 and 1466.89, corresponding to the molecular ions [Cu4HCl2(C17H14NP)4]+ and [Cu4HBr2(C17H14NP)4]+, respectively, based on the analysis of the high-resolution mass data. The observed isotopic patterns of the monovalent clusters are in good agreement with the simulation (Fig. 5b, d). It is revealed that the molecular compositions of the clusters indeed contain one hydrogen atom. The most probable location of the single hydride is at the center of the Cu4 tetrahedron, as indicated by the successful least-squares refinements of the hydride position determined from the difference electron density map of the structures.

ESI–MS spectra a, c of the single-crystals of 2 and 3′ in CH2Cl2. b, d Show both observed (black) and calculated (red) high-resolution MS spectra of the major MS peaks in (a, c) (Color figure online)
Symmetry and Chirality
A detailed analysis of the Cu–Cu distances in 1–3 indicates that the Cu4 cages deviate substantially from Td point group symmetry. The six Cu–Cu distances can be divided into three groups, with average values listed in Table 2. The numbering system corresponds to those in Figs. 2 and 3 (in Fig. 1, Cu3 is Cu1a and Cu4 is Cu2a, as dictated by the crystallographically imposed twofold symmetry). As such, 1–3 conform to an idealized D2 point group symmetry and are intrinsically chiral metal clusters, existing as racemic pairs in the crystal structures [two pairs in the C2/c unit cell of 1 (Fig. 6) and one pair each of the P − 1 unit cells of 2 (Fig. 7) and 3 (Fig. 8)]. The chelating ligand PPh2Py not only plays an important role in the assembly of these clusters, but also causes different senses or modes of distortions of the tetrahedral metal core. Chirality can be inferred from these distortions, as evidenced by the observed racemic pairs of enantiomers in the crystal structures.

Racemic pair of Cluster 1

Racemic pair of Cluster 2

Racemic pair of Cluster 3
Optical Properties
As depicted in Fig. 9a, UV–Vis spectra of clusters 1 and 2 show four absorption bands at 228, 257, 311 (shoulder) and 382 nm (shoulder). For comparison, the UV–Vis spectrum of the PPh2Py ligand is shown in Fig. 9b.

UV/Vis absorption spectra of clusters 1 and 2 in dichloromethane solution
Photoluminescence spectra (excited at λex = 365 nm at room temperature) show cluster 1 to exhibit a maximum emission at 543 nm in CH2Cl2 solution (Fig. 10a). Similarly, cluster 2 shows a maximum emission at 545 nm in CH2Cl2 solution (Fig. 10b). The excitation spectra of 1 and 2 were measured at the respective emission maxima (λem = 543 nm for 1 and λem = 545 nm for 2). At room temperature, the excitation spectra of 1 and 2 each displayed a broad band centered at ~ 365 nm.

Photoluminescence spectra of clusters 1 (a) and 2 (b) in dichloromethane solution: ex excitation, em emission
In a number of reviews, Ford et al. [37,38,39,40,41] and others [42] presented an extensive list of data regarding the emission maxima and lifetimes of a large number of multinuclear cuprous luminophors. In particular, the structure most commonly studied was the so-called “cubane-like” cluster of the general formulas L4Cu4X4 (L = monodentate ligand). The distances between the four tetrahedrally coordinated copper centers are functions of both the halide X and the ligand L. For a given L, for example, the average Cu–Cu distance tends to follow the trend of Cl > Br > I, the shortest ones (2.69 Å) being commonly observed for the I analogs and the longest ones (longer than ~ 2.8 Å) being the Cl analogs. Notably, the iodide complex is strongly luminescent at ambient temperature as a solid and moderately so in solution, while the chloride derivative shows no luminescence in solution and only weak emission as a solid at room temperature. This difference between I and Cl analogs was attributed to the shorter Cu–Cu distance (2.69 Å) of the I analogs, which is less than the sum of the van der Waals radii (2.8 Å), a condition argued by Holt to be a necessary condition for the emission. Specifically, the luminescence spectrum of the Cu4I4py4 cluster displays two distinct emission bands with different emission lifetimes. The strong lower energy (LE) emission was assigned to a triplet cluster-centered (3CC) ES, a combination of iodide to copper charge transfer (XMCT) and d–s transitions, and the weaker higher energy (HE) band to a triplet halide to ligand charge transfer (3XLCT) ES.
In contrast to these observations, clusters 1 and 2 have two short Cu–Cu distances of 2.64–2.66 Å (cf. Table 2), well below the 2.69 Å expected for emission. These short Cu–Cu distances can be attributed, albeit in part, to the existence of the encapsulated hydride at the center of the Cu4 tetrahedron. As a result, strong photoluminesecence was observed even in solution and at room temperature. In the solid state (polycrystalline samples), the emission was orders of magnitude greater at room temperature (cf. Fig. S5).
The measured lifetimes of 93.3 and 91.9 ns reported here for 1 and 2 also strongly suggest that the photoluminescence is a fluorescence emission rather than phosphorescence (as for Cu4I4py4). The main cause of this difference is believed to be the existence of the encapsulated hydride which shortens two of the six Cu–Cu distances of the tetrahedral Cu4 core.
Superatomic Model: Electron Counting
Electron counting is useful in the understanding and rationalization of the stereochemistry of molecules or clusters. Extra stability is often achieved for closed-shell atomic and electronic configurations. Jellium model (JM) is an electronic shell model describing the delocalized valence electrons in a metal atom cluster [43,44,45,46,47,48,49].
If the hydrides in 1–3 were considered as bearing a − 1 charge and acting as ligands instead, then these clusters would have been classified as zero-electron jelliumatic clusters: 4 × 1(Cu) − 1(H) − 2(X) − 1(charge) = 0 (X = halides). If, on the other hand, the hydrogen atoms are considered as an integral part of the cluster framework, then clusters 1-3 are best considered as 2e superatomic systems: viz, 4 × 1(Cu) + 1(H) − 2(X) − 1(charge) = 2.
In terms of the Jelliumatic Shell Model (JSM), a variant of the Jellium Model (JM) originally proposed by Teo and Yang [50], clusters 1–3 can be considered as two-shell Jelliumatic systems with superatomic electron counts of 2e@0e corresponding to the two-shell description of H−@[Cu4X2(PPh2Py)4]2+.
We note that the jelliumatic shell model (JSM) was originally developed in an attempt to rationalize the electron counts of a number of metal clusters that violate the conventional jelliumatic model (JM). Here a distinction is explicitly made between the JSM and the conventional jelliumatic model (JM) in that atomic shells of a given cluster are grouped together to form electronic shells. If each and every electronic shell, which may comprise one or more neighboring atomic shells, can be made to conform to the conventional jelliumatic model (JM) with an electronic configuration and an jellium shell electron (jse) count consistent with the shape of the particular shell, then such a system is said to be in compliance with the JSM. It is assumed that each electronic shell has an “identity” and possesses certain characteristics or attributes (such as possessing an independent electron count of jse, morphology of the shell, size of the radial function and the directionalities (angular momentum) of the jelliumatic orbitals, etc.) expected from a JM but confined to the specific electronic shell under consideration. In this context, the core of 1–3 has a jse of 2e and the surface shell has a jse of 0e. The former is consistent with the spherical shape of the encapsulated hydride (H−) having the superatomic electronic configuration of 1S2.
Conclusion
Three tetrahedral copper hydride clusters, [Cu4(μ4-H)(μ2-X)2(PPh2Py)4]+ clusters (X = Cl, Br; Py = pyridyl), which represent first examples of a standalone tetranuclear copper hydride [Cu4(μ4-H)] unit having a μ4-H encapsulated in the tetrahedral Cu4 cavity, were synthesized and structurally characterized. It is conjectured that the hydride acts as a template in the formation of these tetrahedral clusters. A detailed analysis of the structures suggests that the chelating ligand PPh2Py not only plays an important role in the assembly of these clusters, but also causes different senses or modes of distortions of the tetrahedral metal core. A detailed analysis of the Cu–Cu distances in the title clusters revealed that the Cu4 cages deviate substantially from Td point group symmetry. The six Cu–Cu distances can be divided into three groups (2.65, 2.85, and 2.95 Å), lowering the idealized point group of the Cu4 core to D2 symmetry, thereby resulting in intrinsically chiral metal clusters which exist as racemic pairs in the centrosymmetric crystal structures.
Interestingly, the [Cu4(μ4-H)(μ2-Cl)2(PPh2Py)4]+ clusters display strong photoluminescence upon near-UV irradiation, both in solution and in the solid state at ambient temperature. The unusually strong emissions can be attributed to the existence of the two short Cu–Cu distances of 2.64–2.66 Å, well below the 2.69 Å expected for emission. These short Cu–Cu distances can be traced, albeit in part, to the existence of the encapsulated hydride at the center of the Cu4 tetrahedron.
Finally, the title clusters can be considered as two-shell Jelliumatic systems with superatomic electron counts of 2e@0e corresponding to the two shells of H−@[Cu4X2(PPh2Py)4]2+ (X = Cl, Br). The former is consistent with the spherical shape of the encapsulated hydride (H−) having the superatomic electronic configuration of 1S2.
References
W. S. Mahoney, D. M. Brestensky, and J. M. Stryker (1988). J. Am. Chem. Soc. 110, 291.
A. J. Hoskin and D. W. Stephan (2002). Coord. Chem. Rev. 233, 107.
M. A. Esteruelas and L. A. Oro (1998). Chem. Rev. 98, 577.
M. Konkol and J. Okuda (2008). Coord. Chem. Rev. 252, 1577.
Y. Ren, X. Xu, K. Sun, and J. Xu (2005). Tetrahedron Asymmetry 16, 4010.
G. S. McGrady and G. Guilera (2003). Chem. Soc. Rev. 32, 383.
C. Deutsch, N. Krause, and B. H. Lipshutz (2008). Chem. Rev. 108, 2916.
S. K. Brayshaw, M. J. Ingleson, J. C. Green, J. S. McIndoe, P. R. Raithby, G. Kociok-Köhn, and A. S. Weller (2006). J. Am. Chem. Soc. 128, 6247.
J. Graetz (2009). Chem. Soc. Rev. 38, 73.
J. Yang, A. Sudik, C. Wolverton, and D. J. Siegel (2010). Chem. Soc. Rev. 39, 656.
R. S. Dhayal, J.-H. Liao, Y.-R. Lin, P.-K. Liao, S. Kahlal, J.-Y. Saillard, and C. W. Liu (2013). J. Am. Chem. Soc. 135, 4704.
N. P. Mankad, D. S. Laitar, and J. P. Sadighi (2004). J. Organomet. Chem. 23, 3369.
C. M. Wyss, B. K. Tate, J. Bacsa, T. G. Gray, and J. P. Sadighi (2013). Angew. Chem. Int. Ed. 52, 12920.
Z. Mao, J.-S. Huang, C.-M. Che, N. Zhu, S. K.-Y. Leung, and Z.-Y. Zhou (2005). J. Am. Chem. Soc. 127, 4562.
J. Li, J. M. White, R. J. Mulder, G. E. Reid, P. S. Donnelly, and R. A. J. O’Hair (2016). Inorg. Chem. 55, 9858.
K. Nakamae, B. Kure, T. Nakajima, Y. Ura, and T. Tanase (2014). Chem. Asian J. 9, 3106.
G. Ma, M. J. Ferguson, R. McDonald, and R. G. Cavell (2010). J. Organomet. Chem. 29, 4251.
M. S. Eberhart, J. R. Norton, A. Zuzek, W. Sattler, and S. Ruccolo (2013). J. Am. Chem. Soc. 135, 17262.
P.-K. Liao, C.-S. Fang, A. J. Edwards, S. Kahlal, J.-Y. Saillard, and C. W. Liu (2012). Inorg. Chem. 51, 6577.
C. Latouche, S. Kahlal, Y.-R. Lin, J.-H. Liao, E. Furet, C. W. Liu, and J.-Y. Saillard (2013). Inorg. Chem. 52, 13253.
P.-K. Liao, B. Sarkar, H.-W. Chang, J.-C. Wang, and C. W. Liu (2009). Inorg. Chem. 48, 4089.
R. S. Dhayal, J.-H. Liao, H.-N. Hou, R. Ervilita, P.-K. Liao, and C. W. Liu (2015). J. Chem. Soc. Dalton Trans. 44, 5898.
T. A. Nguyen, B. R. Goldsmith, H. T. Zaman, G. Wu, B. Peters, and T. W. Hayton (2015). Chem. Eur. J. 21, 5341.
M. A. Huertos, I. Cano, N. A. G. Bandeira, J. Benet-Buchholz, C. Bo, and P. W. N. M. van Leeuwen (2014). Chem. Eur. J. 20, 16121.
J.-H. Liao, R. S. Dhayal, X. Wang, S. Kahlal, J.-Y. Saillard, and C. W. Liu (2014). Inorg. Chem. 53, 11140.
A. J. Edwards, R. S. Dhayal, P.-K. Liao, J.-H. Liao, M.-H. Chiang, R. O. Piltz, S. Kahlal, J.-Y. Saillard, and C. W. Liu (2014). Angew. Chem. Int. Ed. 53, 7214.
R. S. Dhayal, J.-H. Liao, S. Kahlal, X. Wang, Y.-C. Liu, M.-H. Chiang, W. E. van Zyl, J.-Y. Saillard, and C. W. Liu (2015). Chem. Eur. J. 21, 8369.
V. Schramm (1978). Inorg. Chem. 17, 714.
A. Bonnot, M. Knorr, F. Guyon, M. M. Kubicki, Y. Rousselin, C. Strohmann, D. Fortin, and P. D. Harvey (2016). Cryst. Growth Des. 16, 774.
J. C. Dyason, P. C. Healy, L. M. Engelhardt, C. Pakawatchai, V. A. Patrick, C. L. Raston, and A. H. White (1985). J. Chem. Soc. Dalton Trans. 4, 831.
J. S. Filippo Jr., L. E. Zyontz, and J. Potenza (1975). Inorg. Chem. 14, 1667.
L. Naldini, F. Demartin, M. Manassero, M. Sansoni, G. Rassu, and M. A. Zoroddu (1985). J. Organomet. Chem. 279, C42.
P. D. Harvey and M. Knorr (2015). J. Cluster Sci. 26, 411.
C. Latouche, C. W. Liu, and J.-Y. Saillard (2014). J. Cluster Sci. 25, 147.
B. K. Teo and D. M. Barnes (1976). Inorg. Nucl. Chem. Lett. 7, 681.
B. K. Teo and J. C. Calabrese (1976). Inorg. Chem. 15, 2474.
C. K. Ryu, M. Vitale, and P. C. Ford (1993). Inorg. Chem. 32, 874.
E. Lindsayz and P. C. Ford (1996). Inorg. Chim. Acta 242, 5l.
F. Angelis, S. Fantacci, A. Sgamellotti, E. Cariati, R. Ugo, and P. C. Ford (2006). Inorg. Chem. 45, 10576.
M. Vitale and P. C. Ford (2001). Coord. Chem. Rev. 219, 3.
P. C. Ford, E. Cariati, and J. Bourassa (1999). Chem. Rev. 99, 3625.
P. Roesch, J. Nitsch, M. Lutz, J. Wiecko, A. Steffen, and C. Müller (2014). Inorg. Chem. 53, 9855.
W. D. Knight, K. Clemenger, W. A. de Heer, W. A. Saunders, M. Y. Chou, and M. L. Cohen (1984). Phys. Rev. Lett. 52, 2141.
S. J. Bjjzfrnholm, O. Borggreen, K. Echt, J. Hansen, J. Pedersen, and H. D. Rasmussen (1991). Z. Phys. D 19, 47.
K. Clemenger (1985). Phys. Rev. B 32, 1359.
W. A. de Heer (1993). Rev. Mod. Phys. 65, 611.
M. Brack (1993). Rev. Mod. Phys. 65, 677.
B. K. Teo and H. Zhang (1991). Proc. Natl. Acad. Sci. 88, 5067.
B. K. Teo and H. Zhang (1995). Coord. Chem. Rev. 143, 611.
B. K. Teo and S.-Y. Yang (2015). J. Cluster Sci. 26, 1923.
Acknowledgements
Financial supports from NNSFC (Grant Nos. 21071117, 21471125) and iChEM, Xiamen University, are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nie, HH., Han, YZ., Tang, Z. et al. Hydride Induced Formation and Optical Properties of Tetrahedral [Cu4(μ4-H)(μ2-X)2(PPh2Py)4]+ Clusters (X = Cl, Br; Py = pyridyl). J Clust Sci 29, 837–846 (2018). https://doi.org/10.1007/s10876-018-1359-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-018-1359-5