
Abstract
Two new photoluminescent multinuclear Cu(I) cluster complexes supported by monoanionic bidentate ligand N-methylbenzimidazolethiolate (Me-bimt–), [Cun(Me-bimt)n] with n = 4 (1) and 6 (2), have been synthesized and structurally characterized by single-crystal X-ray diffraction analysis. For 1 and 2, the Cu(I) ions and the Me-bimt– ligands construct a cubane-type {Cu4S4} and a hexagonal-prism {Cu6S6} frameworks, respectively. In the crystalline state, complexes 1 and 2 exhibit green (λem = 500 nm) and near-infrared (λem = 876 nm) emission, respectively, under UV irradiation (λex = 365 nm) at room temperature. Both crystals reveal temperature-dependent dual emission below 200 K: complex 1 emits in the visible wavelength region (λem = 493 and 542 nm) and complex 2 in the visible to near-infrared wavelength region (λem = 752 and 973 nm) which are attributed to multiple photoexcited states at the cluster frameworks with distinct metal nuclearity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Multinuclear transition-metal complexes with d10 electronic configuration, such as copper(I) and silver(I), show rich photoluminescent behavior associated with multiple photo-excited states such as metal-to-ligand charge transfer (MLCT), multi-metal cluster-centered (CC) charge transfer, and inter-ligand (IL) transition (Yam and Lo 1999). In some cases, radiative decay from two distinct photoexcited states simultaneously occurs, which represents the “dual emission” phenomenon (Ford et al. 1999).
Monoanionic iminothiolato (N = C–S–) bidentate ligands such as pyridinethiolate and imidazolethiolate are able to link two or more metal centers to form multinuclear cluster complexes (Ford and Vogler 1993). Many literature studies are available for crystal structures of the iminothiolato multicopper(I) complexes with a tetrameric {Cu4S4} core and a hexameric, hexagonal pillar-type {Cu6S6} core, but only a few studies have been reported so far on their photophysical properties (Kundu et al. 2014; Xie et al. 2005). Such multicopper(I) complexes are typically formed via spontaneous self-assembly of the corresponding metallic sources and ligands mixed together with the appropriate molar ratio in solution, but the synthetic methodology to precisely control the metal nuclearity (e.g., the number of metal centers in a single molecule) of products remains yet, in general, to be established for transition-metal coordination complexes. Herein we describe the synthesis, molecular structures, and solid-state photoluminescence properties of two new Cu(I) iminothiolate clusters formulated [Cun(Me-bimt)n] with n = 4 (1) and 6 (2), where Me-bimt– is a monoanionic bidentate ligand N-methylbenzimidazolethiolate(1–). The synthetic conditions employed gave initially a crude solid containing both tetramer 1 and hexamer 2 but the individual clusters were subsequently transformed to either form by adopting appropriate solvent combinations for recrystallization. Selective isolation of 1 and 2 has been unambiguously confirmed by single-crystal X-ray diffraction analyses. We show that both compounds exhibit temperature-dependent dual emission in the solid state upon UV excitation, wherein 1 exhibits thermochromic dual emission in the visible region and 2 from visible to near-infrared (NIR) region.
Experimental
Materials and methods
Chemical reagents and solvents used in this study were purchased from TCI and WAKO and used without further purification unless otherwise stated. All of the synthetic reactions in this study were performed under Ar atmosphere.
Apparatus and equipment
IR measurements were performed on a JASCO FT/IR-4000 at 298 K by an attenuated total reflectance (ATR) method. 1H NMR spectroscopy was performed at 600 MHz on a JEOL ECA-600 and a JEOL ECZ-600R/S1 spectrometers using CDCl3 as solvents, and TMS was employed as an internal standard to determine the chemical shift. Elemental analysis was carried out by the Laboratory for Organic Elemental Microanalysis, Kyoto University. UV–Vis diffuse reflectance spectra (DRS) were measured by a JASCO V-670 UV–Vis–NIR spectrometer with an ISN-723 integrating sphere attachment. Luminescence spectra of the single crystals were measured by a Hamamatsu PMA-11 (C7473-46) spectrometer (190–900 nm) and an Ocean Optics FLAME-S spectrometer (600–1100 nm). The crystalline samples were placed in a Linkam THMS600 temperature-controlled (78–293 K) microscope stage. A Panasonic UJ-20 UV-LED system was used as an excitation light source (λex = 365 nm). The emission from the solid samples was introduced to the optical fibers of the spectrometer through a microscope. Lifetimes and quantum yields of solid-state photoluminescence were measured on a Hamamatsu Quantaurus-Tau C11367-02 fluorescence lifetime spectrometer and Quantaurus-QY C11347 absolute PL yield spectrometer, respectively.
Synthesis and characterization
Synthesis of (triphenylmethyl)sulfanylbenzimidazole (Tr-bimt).
This compound was prepared according to the literature procedure (Doerge and Cooray 1991). A solution of benzimidazole-2-thiol (bimtH) (4.2 g, 28 mmol), trityl chloride (8.0 g, 28 mmol), and Et3N (3.1 g, 28.5 mmol) in dry THF (150 mL) was stirred for 8 h at 293 K. The resulting suspension was filtered to remove triethylammonium salt which was washed thoroughly with THF. The combined filtrate was concentrated by evaporation to dryness under reduced pressure. The white solid of Tr-bimt was identified by 1H NMR spectroscopy as a mono-THF adduct. Yield: 10.1 g (92%). The obtained product was used in the following synthesis without further purification. 1H NMR (600 MHz, CDCl3, 298 K) δ 7.70 (d, 1H, J = 8.22 Hz, benzimidazole 7-H), 7.64 (s, 1H, NH), 7.47 (d, 6H, J = 6.9 Hz, trityl group ortho H), 7.33–7.27 (m, 9H, trityl group meta and para H), 7.19–7.12 (m, 2H, benzimidazole 5,6-H), 6.99 (d, 1H, J = 7.6 Hz, benzimidazole 4-H), 3.75 (t, 4H, THF –O–CH2–CH2–), 1.85 (m, 4H, THF –O–CH2–CH2–).
Synthesis of methyl-2-(triphenylmethyl)sulfanylbenzimidazole (Tr–Me-bimt).
This compound was obtained by a modified method of the literature (Doerge and Cooray 1991). Ground powder of KOH (7.0 g, 125 mmol) was placed in a round-bottom flask (500 mL) and dried under vacuum for 3 h. To this was added a solution of Tr-bimt–THF adduct (10.0 g, 25.5 mmol) in dry acetone (170 mL) followed by MeI (5.9 g, 41 mmol), and the mixture was stirred for 1 h at 293 K. The resulting pale-yellow solution was filtered to remove a suspended solid of KOH. The filtrate was added to toluene (700 mL), and the mixture was divided into two portions which were washed first with water (150 mL) and second with a saturated aqueous solution of NaCl (150 mL). After combining the individual solutions, the solution was dried over anhydrous Na2SO4 and was concentrated by evaporation to dryness under reduced pressure. Yield: 7.1 g (68%). The yellow solid of Tr–Me-bimt as a crude product was used in the following synthesis without further purification. 1H NMR (600 MHz, CDCl3, 298 K) δ 7.31–7.27 (m, 15H, trityl group), 7.23–7.12 (m, 4H, benzimidazole 4,7-H), 3.75 (s, 3H, CH3).
Synthesis of N-methyl-benzimidazolethiol (Me-bimtH).
A solution of Tr–Me-bimt (7.1 g, 16.9 mmol) in acetic acid (7.5 mL)–MeOH (142.5 mL) was heated under contentious stirring for 5 h at 358 K. The resulting yellow solution was concentrated by evaporation to dryness under reduced pressure. The yellow solid was dissolved in CH2Cl2 (150 mL) and was washed with an aqueous solution of 10% sodium bicarbonate (100 mL × 3). The organic layer was dried over anhydrous Na2SO4 and concentrated by evaporation to dryness under reduced pressure. The light-yellow crude product was collected by filtration and washed with toluene to remove unreacted Tr–Me-bimt. The obtained white powder was identified by 1H NMR spectroscopy. Yield: 1.59 g (58%). 1H NMR (CDCl3, 600 MHz, 298 K) δ 10.68 (s, br, 1H, SH), 7.27–7.20 (m, 1H, benzimidazole 7-H), 7.23 (ddd, 2H, J = 6.9, 6.5, and 1.4 Hz, benzimidazole 5,6-H), 7.17–7.15 (m, 1H, benzimidazole 4-position), 3.79 (s, 3H, CH3).
Synthesis of N-methyl-benzimidazolethiolatocopper(I) [Cun(Me-bimt)n] (n = 4 and 6).
A solution of Me-bimtH (0.092 g, 0.6 mmol) with Et3N (0.151 g, 1.5 mmol) in CH3CN (10 mL) was added dropwise to a solution of [Cu(CH3CN)4]CF3SO3 (0.196 g, 0.5 mmol) in CH3CN (10 mL) under continuous stirring at 293 K. The resulting pale-yellow suspension was heated under continuous stirring for 2 h at 323 K. A white solid formed was separated by filtration, washed with CH3CN, and dried under vacuum. This preparative procedure gave a mixture of two species [Cun(Me-bimt)n] with n = 4 and 6 on the basis of elemental analysis and 1H NMR spectroscopy. Yield: 0.102 g (87%). Anal. Calcd. for C8H7CuN2S: C, 42.37; H, 3.11; N, 12.35%. Found: C, 42.45; H, 3.01; N, 12.18%.
Separated recrystallization and single-crystal X-ray structure analysis for [Cu4(Me-bimt)4] (1) and [Cu6(Me-bimt)6] (2).
Single crystals of 1 were grown at 293 K as colorless block-shaped crystals from a toluene solution of the above-mentioned mixture by layering and slowly diffusing Et2O. Single crystals of 2 were obtained at 303 K as red platelet-shaped crystals from a CH2Cl2 solution of the mixture by layering and slowly diffusing CH3CN. Single-crystal X-ray diffraction measurements were performed at 290 and 150 K for both crystals on a Rigaku Rapid image plate diffractometer using Mo Kα (λ = 0.7107 Å) radiation. The crystal was attached to a thin Nylon loop and kept under a cold N2 gas stream from a He gas-expansion cryostat. All non-hydrogen atoms were refined anisotropically, and hydrogen atoms were placed at the calculated positions and treated according to the riding model during refinements. All the calculations for structure determination and refinements were carried out using WinGX (Farrugia 2012) crystallographic software suite with SHELXL-2016/6 refinement program (Sheldrick 2015). For the refinement of 1, the structure was treated as a two-component twin using TWIN and BASF commands in the SHELXL program. It was found that the final R value for 1 was lower at high temperature (290 K, R = 0.091) than low temperature (150 K, R = 0.119), allowing us the high-temperature data to use for structural discussion. In contrast, free from the twin problem, the results for structural analysis of 2 were satisfactory at both temperatures (150 K, R = 0.052; 290 K, R = 0.064). Under these circumstances, the 290-K data were employed for both complexes for their structural description and comparison.
Crystallographic data of 1 (T = 290 K): C32H28Cu4N8S4, Mr = 907.02, colorless prism, 0.34 × 0.34 × 0.28 mm3, monoclinic, space group C2/c (No. 15), a = 20.307(2), b = 15.107(1), c = 15.185(1) Å, β = 132.511(2)°, V = 3434.0(4) Å3, µ(λ = 0.7107 Å) = 2.73 mm–1, Z = 4, θmax = 30.0°, 20,220 reflections measured, 4976 unique (Rint = 0.07) which were used in all calculations. The final R = 0.091, wR(F2) = 0.318, and GOF = 1.09 (all data).
Crystallographic data of 2 (T = 290 K): C48H42Cu6N12S6, Mr = 1360.53, red platelet, 0.20 × 0.15 × 0.07 mm3, trigonal, space group R-3 (No. 142), a = 13.479(2), c = 24.061(3) Å, V = 3786.0(10) Å3, µ(λ = 0.7107 Å) = 2.78 mm–1, Z = 3, θmax = 30.0°, 15,063 reflections measured, 2445 unique (Rint = 0.073 which were used in all calculations. The final R = 0.064, wR(F2) = 0.233 and GOF = 1.17(all data).
CCDC reference numbers for 1 are 1,981,286 (290 K) and 1,981,287 (150 K) and those for 2 are 1,981,293 (290 K) and 1,981,294 (150 K).
Density functional theory (DFT) calculations
DFT calculations were carried out with the ADF2019 suite of programs (Baerends et al. 2019; te Velde et al. 2001). The geometrical parameters for 1 and 2 were taken from the crystal structures. Geometry optimizations were performed with applying S4 and D3d molecular symmetry to 1 and 2, respectively. Exchange correlation function (XC) of PW91 method was applied for calculations. The valence shell atomic orbitals of Cu, S, N, C, and H atoms were described by triple-zeta Slater-type basis sets with two polarization functions (ADF database TZ2P).
Results and discussion
Synthesis and crystallization
We have synthesized and isolated single crystals of tetranuclear complex [Cu4(Me-bimt)4] (1) and hexanuclear complex [Cu6(Me-bimt)6] (2), as shown in Fig. 1. The reaction of [Cu(CH3CN)4]CF3SO3 with a slight excess of Me-bimtH in CH3CN in the presence of Et3N (2 h, 323 K) gave an off-white solid which was identified as mixture of two species [Cun(Me-bimt)n] with n = 4 and 6 (87% yield). Selective isolation of either form was successfully achieved by recrystallization of the mixture by the use of appropriate solvent combination. Thus, complexes 1 and 2 were crystallized separately at room temperature from toluene/Et2O and CH2Cl2/CH3CN systems, respectively. Interestingly, the color of the crystals is markedly different, e.g., colorless and deep red for 1 and 2, respectively. It is noted that when lower temperature conditions (such as 273 K) were applied complex 2 was contaminated with 1, implying an equilibrium between 1 and 2 in solution. The formation of the off-white solid during the synthetic reaction may infer that complex 1 is a kinetic product.

Schematic of preparation and crystallization procedures of 1 and 2. Photographic images of the crystalline solid are also shown
We have also found a reversible transformation between 1 and 2 upon recrystallization by appropriate solvent choice mentioned above. Thus, recrystallization of red crystals 2 from toluene/Et2O gave colorless crystals 1 to be deposited from the solution. Similarly, recrystallization of colorless crystals 1 from CH2Cl2/CH3CN results in the deposition of red crystals 2. The solvent combination thus leads to a complete control over metal nuclearity of products as a crystal. There are only a few previous reports for such a separation of tetrameric and hexameric Cu(I) complexes in preparative procedures (Chivers et al. 2001; Yue et al. 2009). In the current study, we have shown not only a complete separation but also a reversible conversion between tetramer and hexamer through recrystallization.
A qualitative insight has been gained for solution equilibrium between 1 and 2 by 1H NMR spectroscopy. Dissolution of either crystal, 1 or 2, in CDCl3 at room temperature was found to give, within minutes, a light-yellow clear solution. Figure 2 shows 1H NMR spectrum of a CDCl3 solution prepared by dissolution of 1 at 295 K, wherein two groups of resonances were observed for the benzimidazole moiety with labels (a–e) for the larger intensity group (defined as group A) and (a’–e’) for the smaller (defined as group B). Here, either group is tentatively assigned to 1 or 2. The chemical shift data (CDCl3, 600 MHz, 295 K) and the assignment are as follows. Group A: δ 7.32, 7.04, 6.93, 6.63 (s, 1H × 4, aromatic protons of the benzimidazole moiety), and 3.46 (s, 3H, CH3). Group B: δ 7.35, 6.96, 6.80, 6.38 (s, 1H × 4, aromatic protons of the benzimidazole moiety), and 3.23 (s, 3H, CH3). The observed B/A ratio in intensity was 0.4. Slightly broadened resonances may infer the presence of exchange between two species on the NMR timescale. Further study, including variable-temperature NMR spectroscopy, is necessary to describe in more detail the dynamics and kinetics of these clusters in solution.

A 1H NMR spectrum (600 MHz, 295 K) of a CDCl3 solution prepared upon dissolution of crystals of 1. Note that two different species labeled as (a–e) and (a’–e’) were readily formed in the room-temperature solution; these are described in the main text as groups A and B, respectively. Inset: the proton labeling scheme for the benzimidazolate moiety
Single-crystal X-ray diffraction study
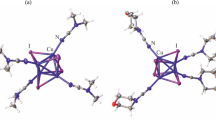
The molecular structures of 1 and 2 were unambiguously determined by single-crystal X-ray diffraction analysis. The molecular structure and the {Cu4S4} framework of 1 at 290 K are depicted in Fig. 3. Selected interatomic distances and bond angles are listed in Table 1. Complex 1 crystallizes in the monoclinic space group C2/c and has a cuboidal {Cu4S4} framework, wherein four Cu(I) centers are bridged by N and S atoms from the bidentate Me-bimt– ligands (Fig. 3a). In the crystalline lattice, a twofold rotation axis parallel to the b-axis penetrates the molecule and half of the molecule is crystallographically independent. The whole molecule has virtually an S4 symmetry. The Cu(I) centers adopt a highly distorted, three-coordinate geometry with an “NS2” donor set. Four Cu(I) and four S atoms provided from Me-bimt– form a cuboidal {Cu4S4} framework (Fig. 3b) with a tub-shaped, eight-membered {Cu4S4} ring (Fig. 3c). Interatomic separations between two Cu(I) centers perpendicular to the molecular S4 axis (Cu1•••Cu1* and Cu2•••Cu2*) are 3.158(2) and 3.169(2) Å (Fig. 3b). In contrast, four other Cu•••Cu contacts (Cu1•••Cu2, Cu1•••Cu2*, Cu1*•••Cu2, and Cu1*•••Cu2*) are much shorter, 2.653(2) and 2.656(2) Å (Fig. 3c) and these are also shorter than the summation of van der Waals radii (2.8 Å). The observed Cu•••Cu separations and tetragonal compression (along the b-axis) of the {Cu4} core in 1 are typical of those found in imidazolethiolate tetracopper(I) complexes reported earlier (Raper et al. 1991; Venkatesh et al. 2014).

ORTEP diagrams of 1 at 290 K with thermal ellipsoids at a 50% probability level. Hydrogen atoms are omitted for clarity. a The molecular structure. b The {Cu4S4} framework projected along the c-axis and c that along the b-axis. For b and c, Cu•••Cu distances (Å) are given. Symmetry code: (*) –x, y, 1/2–z
Crystal packing of 1 is illustrated in Fig. 4. Selected intermolecular contacts are listed in Table S1 (Supplementary data). The molecules are aligned along the b-axis to form 1D columns via intermolecular van der Waals contacts between benzene ring moieties in Me-bimt– (C16•••C5′, 3.86(2) Å) (Fig. 4a). In addition, weak π–π contacts (C6•••C6′, 3.31(3); C16•••C16, 3.33(2) Å) and C–H•••S contacts (S1•••C3′, 3.69(1); C13•••S2′, 3.67(2) Å) are observed among molecules in the ac-plane (Fig. 4b).

Stick-model representations for crystal packing of 1 at 290 K. a Projected along the a*-axis (on the bc plane). b Projected along the b-axis. Two sets of symmetry-equivalent ligand moieties are drawn as pink and light-green sticks. Interatomic contacts within the sum of the van der Waals radii are indicated as broken cyan lines
The molecular structure and {Cu6S6} framework of 2 at 290 K are depicted in Fig. 5. Selected interatomic distances and bond angles are listed in Table 2. Complex 2 crystallizes in the trigonal space group R-3. Two hexagonal {Cu3S3} moieties are located mutually in an anti-parallel fashion to form the {Cu6S6} framework, and six planar Me-bimt– ligands support the distorted {Cu6} octahedron to form a six-bladed paddle-like shape (Fig. 5a). The whole molecule has a threefold rotoinversion axis with an exact S6 symmetry, in which one Cu(I) and one Me-bimt– are crystallographically independent. The Cu•••Cu distances are 3.137(2) and 2.918(2) Å (Fig. 5b), indicating trigonal elongation of the {Cu6} core (Fig. 5c). Each Cu(I) adopts three-coordinate geometry with an “N2S” donor set.

ORTEP diagrams of 2 at 290 K with thermal ellipsoids at a 50% probability level. Hydrogen atoms are omitted for clarity. a The molecular structure. b The {Cu6S6} framework along the c-axis and c that along the a-axis. For b and c, Cu···Cu distances (Å) are given. Symmetry codes: (i) 2/3 + x–y, 1/3 + x, 4/3–z; (ii) 1–y, 1+x–y, z; (iii) 2/3–x, 4/3–y, 4/3–z; (iv) –x + y, 1–x, z; (v) 1/3 + y, 1/3–x + y, 4/3–z
Crystal packing of 2 is shown in Fig. 6. Selected interatomic contacts are listed in Table S2 (Supplementary data). The molecules are engaged each other like “gears” with π•••π contacts between neighboring ligands (C6•••C7′, 3.40(1) Å) to form a 2D layer (Fig. 6a) which is perpendicular to the c-axis (Fig. 6b).

Stick- and space-fill model representations of the crystal packing of 2 at 290 K. a Projected along the c-axis. b Projected along the a-axis. Intermolecular contacts within the sum of the van der Waals radii are indicated as broken cyan lines
Diffuse reflectance spectra (DRS) and density functional theory (DFT) calculations
UV–Vis DRS of colorless crystals of 1 display a single band centered at 335 nm and is transparent in the visible region, as shown in Fig. S1 (Supplementary data). In contrast, deep red crystals of 2 exhibit a low-energy, weaker shoulder at 460 nm in addition to a high-energy, stronger band at 330 nm as shown in Fig. S2 (Supplementary data), the former of which causes the coloration of 2.
To gain insights into absorption properties of 1 and 2, especially to identify the nature of visible coloration in 2, DFT calculations have been carried out. The results are shown in Figs. S3 and S4 (Supplementary data). Irrespective of the metal nuclearity, the highest occupied molecular orbital (HOMO) is assigned to an admixture of (Cu 3d, S 3p) orbitals in the {CunSn} cores (n = 4 or 6) which are non-bonding in character. In contrast, the nature of the lowest unoccupied molecular orbital (LUMO) is different between the two complexes. The LUMO for 1 is localized to the benzimidazole moiety (ligand-π* orbital) (Fig. S3) whereas that of 2 is ascribed to an admixture of (Cu 4 s/4p, S 4 s/4p) orbitals in the {Cu6S6} core which is bonding in character (Fig. S4). The LUMO level in 2 becomes lower than the ligand-π* orbital (second LUMO) in energy. The decrease in the LUMO level of 2 relative to 1 is ascribable to electronic delocalization over the {Cu6S6} core (Ford et al. 1999; Sabin et al. 1992; Yue et al. 2009). The high-energy bands in DRS for both complexes (330 and 335 nm for 1 and 2, respectively) are attributed to “{CnSn} cluster-to-ligand” charge-transfer transitions. The low-energy band for 2, corresponding to the narrower HOMO–LUMO gap, is attributable to “{C6S6} cluster-centered” transitions in the {Cu6S6} core.
Solid-state photoluminescence
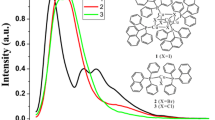
In the crystalline state, both compounds are emissive under UV illumination (λex = 365 nm). The photoluminescence spectra of 1 and 2 at various temperatures are shown in Fig. 7, which reveals a markedly contrast in luminescence depending on the metal nuclearity and temperature.

Temperature-dependent solid-state photoluminescence spectra (λex = 365 nm) of a 1 (T = 78 to 298 K) and b 2 (T = 78 to 293 K)
At 293 K, complex 1 emits in greenish-blue with a broad emission band centered at 502 nm (Fig. 7a). The emission quantum yield was Φ = 0.16 at 293 K. The emission lifetime of τ = 11 µs at 293 K suggests phosphorescence from a triplet state. Intensity of this emission increased as the temperature was decreased. Below 200 K, dual-emissive character became apparent with the higher energy (HE) peak at 493 nm and the lower energy (LE) peak at 542 nm. Further decrease in temperature led to the decrease in the LE intensity with concomitant growth of the HE band. At 78 K, single emission, derived from the HE band, is observed with a complete lack of the LE band. In the dual-emissive temperature region, the energy of LE and HE bands remains less affected but the intensity of both is highly temperature dependent. On the basis of the emission lifetime and the DFT analysis for 1 described above, we tentatively assign the LE (λem = 542 nm) and HE emissions (λem = 493 nm) to “{Cu4S4} cluster-to-ligand” charge-transfer and “{Cu4S4} cluster-centered” excited states, respectively. We note that the thermochromic dual-emissive character for iminothiolate tetracopper(I) clusters is reported herein for the first time, though it has been of extensive interest for cubane-type [Cu4I4L4] clusters (Benito et al. 2015; De Angelis et al. 2006; Ford et al. 1999; Huitorel et al. 2018; Kitagawa et al. 2010; Nagaoka et al. 2018; Perruchas et al. 2010; Yang et al. 2016).
The thermochromic dual luminescence is also observed for 2 but occurs in the NIR region (Fig. 7b). The emission intensity of 2 is much weaker than that of 1. The temperature-dependent spectral change for 2 appears more complex than that in 1. At 293 K, complex 2 gives a single, broad emission band centered at 876 nm. Similar to 1, the emission lifetime of 2 is of microsecond order, τ = 1.7 µs at 293 K, indicating phosphorescence from a triplet state. The emission quantum yield is Φ = 0.01 at 293 K. Upon cooling, the emission band shifts to a lower energy side with maintaining the single emission feature. At 218 K, an additional HE emission band begins to grow at ca. 720 nm. Further cooling leads to dual emission with LE (NIR region) and HE (visible region). At 78 K, the LE peak reaches 973 nm and the HE peak 752 nm. According to DFT calculations for 2, the LE emission is tentatively assigned to “{Cu6S6} cluster-centered” triplet excited state and the HE emission to “{Cu6S6} cluster-to-ligand” charge-transfer triplet excited state. In the literature, solid-state (single) NIR emission is not unusual for transition-metal complexes, including precedents for copper complexes like [Cu6(mtc)6] (mtc– = di-n-propylmonothiocarbamate) (Sabin et al. 1992), [Cu6(btt)6] (btt– = benzothiazolethiolate) (Yue et al. 2009), [Cu6(Hmna)6] (H2mna = 2-mercapto-nicotinic acid) (Kundu et al. 2014), and [Cu4I4(SbR3)4] (R = isopropyl and cyclohexyl) (Taylor et al. 2019, 2016). However, report for dual-emission spanning to NIR, like 2, remains much scarce (Xie et al. 2005).
Conclusions
In this paper, we described the synthesis and successful crystallization of two new Cu(I) complexes supported by monoanionic iminothiolate ligands, N-methylbenzimidazolethiolate (Me-bimt–) and formulated [Cun(Me-bimt)n], which comprises different metal nuclearity (n = 4 and 6). The crystal structures for both clusters were unambiguously determined by single-crystal X-ray diffraction analysis. Interestingly, the color of the crystals was highly nuclearity-dependent, colorless and deep red for 1 and 2, respectively, which was rationalized by HOMO–LUMO gaps determined by DFT calculations. Upon UV excitation at room temperature, complexes 1 and 2 gave solid-state photoluminescence with lifetimes of microsecond order. Both complexes displayed temperature-dependent dual emission (78–298 K) in the solid state. The thermochromic dual emission especially found in the entire optical region from visible to NIR is unusual for molecular compounds. The iminothiolate tetra- and hexacopper(I) system reported herein is thus of considerable interest in a new class of optical molecular thermometers covering a wide range of wavelength. Many other applications may also be anticipated for this class of compounds with a strong advantage for utilizing a naturally-abundant, low-cost transition-metal, copper.
References
Baerends EJ et al (2019) ADF 2019.3, SCM, theoretical chemistry. Vrije Universiteit, Amsterdam
Benito Q et al (2015) Geometry flexibility of copper iodide clusters: variability in luminescence thermochromism. Inorg Chem 54:4483–4494. https://doi.org/10.1021/acs.inorgchem.5b00321
Chivers T, Downard A, Parvez M, Schatte G (2001) Hexameric and tetrameric copper(I) thioamidates. Organometallics 20:727–733. https://doi.org/10.1021/om000809k
De Angelis F, Fantacci S, Sgamellotti A, Cariati E, Ugo R, Ford PC (2006) Electronic transitions involved in the absorption spectrum and dual luminescence of tetranuclear cubane [Cu4I4(pyridine)4] cluster: a density functional theory/time-dependent density functional theory investigation. Inorg Chem 45:10576–10584. https://doi.org/10.1021/ic061147f
Doerge DR, Cooray NM (1991) Synthesis of N-substituted benzimidazole-2-thiones. Synth Commun 21:1789–1795. https://doi.org/10.1080/00397919108021578
Farrugia L (2012) WinGX and ORTEP for Windows: an update. J Appl Crystallogr 45:849–854. https://doi.org/10.1107/S0021889812029111
Ford PC, Vogler A (1993) Photochemical and photophysical properties of tetranuclear and hexanuclear clusters of metals with d10 and s2 electronic configurations. ACC Chem Res. https://doi.org/10.1021/ar00028a013
Ford PC, Cariati E, Bourassa J (1999) Photoluminescence properties of multinuclear copper(I) compounds. Chem Rev 99:3625–3647. https://doi.org/10.1021/cr960109i
Huitorel B et al (2018) Evaluation of ligands effect on the photophysical properties of copper iodide clusters. Inorg Chem 57:4328–4339. https://doi.org/10.1021/acs.inorgchem.7b03160
Kitagawa H, Ozawa Y, Toriumi K (2010) Flexibility of cubane-like Cu4I4 framework: temperature dependence of molecular structure and luminescence thermochromism of [Cu4I4(PPh3)4] in two polymorphic crystalline states. Chem Commun 46:6302–6304. https://doi.org/10.1039/c0cc01434f
Kundu T, Jana AK, Natarajan S (2014) Stepwise crystallization: illustrative examples of the use of metalloligands [Cu6(mna)6]6– and [Ag6(Hmna)2(mna)4]4– (H2mna = 2-mercapto nicotinic acid) in the formation of heterometallic two- and three-dimensional assemblies with brucite, pcu, and sql topologies. Cryst Growth Des 14:4531–4544. https://doi.org/10.1021/cg500632d
Nagaoka S, Ozawa Y, Toriumi K, Abe M (2018) A Dual-emission strategy for a wide-range phosphorescent color-tuning of a crystalline-state molecular cluster [Cu4I4(2-Bzpy)4] (2-Bzpy = 2-Benzylpyridine). Chem Lett 47:1101–1104. https://doi.org/10.1246/cl.180435
Perruchas S et al (2010) Mechanochromic and thermochromic luminescence of a copper iodide cluster. J Am Chem Soc 132:10967–10969. https://doi.org/10.1021/ja103431d
Raper ES, Creighton JR, Clegg W (1991) tetrahedro-[Tetrakis {(1-methylimidazoline- 2(3H) -thionato) copper(I)}]: electrochemical synthesis, thermal analysis, cyclic voltammetry and crystal structure. Inorg Chim Acta 183:179–187. https://doi.org/10.1016/S0020-1693(00)83012-1
Sabin F, Ryu CK, Ford PC, Vogler A (1992) Photophysical properties of hexanuclear copper(I) and silver(I) clusters. Inorg Chem 31:1941–1945. https://doi.org/10.1021/ic00036a040
Sheldrick G (2015) Crystal structure refinement with SHELXL. Acta Crystallogr Sect C 71:3–8. https://doi.org/10.1107/S2053229614024218
Taylor WV, Soto UH, Lynch VM, Rose MJ (2016) Antimony-supported Cu4I4 cuboid with short Cu–Cu bonds: structural premise for near-infrared thermoluminescence. Inorg Chem 55:3206–3208. https://doi.org/10.1021/acs.inorgchem.5b02933
Taylor WV, Cammack CX, Shubert SA, Rose MJ (2019) Thermoluminescent antimony-supported copper-iodo cuboids: approaching NIR emission via high crystallographic symmetry. Inorg Chem 58:16330–16345. https://doi.org/10.1021/acs.inorgchem.9b00229
te Velde G, Bickelhaupt FM, Baerends EJ, Guerra CF, van Gisbergen SJA, Snijders JG, Ziegler T (2001) Chemistry with ADF. J Comput Chem 22:931–967. https://doi.org/10.1002/jcc.1056
Venkatesh V, Pachfule P, Banerjee R, Verma S (2014) Evolution of an adenine-copper cluster to a highly porous cuboidal framework: solution-phase ripening and gas-adsorption properties. Chem Eur J 20:12262–12268. https://doi.org/10.1002/chem.201403115
Xie H, Kinoshita I, Karasawa T, Kimura K, Nishioka T, Akai I, Kanemoto K (2005) Structure study and luminescence thermochromism in hexanuclear 6-methyl-2-pyridinethiolato copper(I) crystals. J Phys Chem B 109:9339–9345. https://doi.org/10.1021/jp0446985
Yam VW-W, Lo KK-W (1999) Luminescent polynuclear d10 metal complexes. Chem Soc Rev 28:323–334. https://doi.org/10.1039/a804249g
Yang K, Li S-L, Zhang F-Q, Zhang X-M (2016) Simultaneous luminescent thermochromism, vapochromism, solvatochromism, and mechanochromism in a C3-symmetric cubane [Cu4I4P4] cluster without Cu–Cu interaction. Inorg Chem 55:7323–7325. https://doi.org/10.1021/acs.inorgchem.6b00922
Yue C, Yan C, Feng R, Wu M, Chen L, Jiang F, Hong M (2009) A polynuclear d10–d10 metal complex with unusual near-infrared luminescence and high thermal stability. Inorg Chem 48:2873–2879. https://doi.org/10.1021/ic801840g
Acknowledgements
This work was supported by JSPS KAKENHI Grant no. JP16H06514 (Coordination Asymmetry) and the special research grant from University of Hyogo (2016, 2017). Prof. Morifumi Fujita (University of Hyogo) is acknowledged for useful discussion on 1H NMR spectroscopy.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Additional information
This paper is dedicated to Professor Milan Melník for his outstanding contribution to coordination and bioinorganic chemistry.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ozawa, Y., Mori, M., Kiyooka, H. et al. Tetra- and hexanuclear copper(I) iminothiolate complexes: synthesis, structures, and solid-state thermochromic dual emission in visible and near-infrared regions. Chem. Pap. 74, 3717–3725 (2020). https://doi.org/10.1007/s11696-020-01251-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-020-01251-w