
Abstract
The ground-state geometrical and electronic properties of neutral and charged M n C2 (M = Fe, Co, Ni, Cu; n = 1–5) clusters are systematically investigated by density-functional calculations. The growth evolution trends of neutral and charged Fe n C2, Co n C2, Ni n C2 and Cu n C2 (n = 1–5) clusters are all from lower to higher dimensionality, while it is special for Cu n C ±2 (n = 1–5) clusters which favor planer growth model. The space directional distributions of Co and Ni indicate stronger magnetic anisotropy than that in Cu atoms. Compare with experimental data (photoelectron spectroscopy), our results are in good agreement. The interaction strengths between metal and carbon atoms in TM–C (TM = Fe, Co, Ni) clusters are comparable and are obviously larger than that in Cu–C clusters, and this interaction strengths also decrease through the sequence: cation > neutral > anion, which may be crucial in exploring the differences in the growth mechanisms of metal–carbon nano-materials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cluster system has unique physical and chemical properties in comparison with the conventional behavior of free atoms and bulk atoms [1]. In recent years, much effort has been made to study clusters both theoretically and experimentally [1]. Transition metal clusters is of special research interest for many researchers as it has important magnetic properties and catalytic function in the process of growing carbon nano-materials. Typical transition metal clusters, such as iron, cobalt and nickel, are commonly used as catalysts in the fabrication of Single-walled carbon nanotubes (SWCNTs) and copper is good catalyst in the growth of carbon fiber.
Recently the most frequently studied cluster is transition-metal carbide clusters [2–5]. Gustev et al. [6] investigated the geometries and electronic structures of Fe n C and Fe n C± (n = 1–6) clusters by density-functional theory (DFT) and found rather high metal–carbon bond energies. Joswig et al. [7] calculated the electronic and geometrical structures of titanium carbohedrene clusters Ti m C n (m = 7, 8; n = 10–14) using a newly developed genetic algorithm combined with a DFT tight-binding method. In experiment, Li et al. reported the photoelectron spectra of MC2 − (M = Sc, V, Cr, Mn, Fe and Co) clusters through a magnetic bottle anion photoelectron spectroscopy (PES). They found that the trends of the electron affinities and vibrational frequencies for MC2 species are well correlated with the corresponding monoxides [8]. Tono et al. [9] measured the photoelectron spectra of Co2C − n (n = 2, 3) and V2C − n (n = 2–4) clusters using laser ablation technique. The carbide-formation processes of the late 3d transition metals and the carbide-formation of the early 3d transition metals is different. The early 3d transition metals forms large metal-carbide networks, such as metallocarbonhedrenes and metal carbide compounds [9], while the late 3d transition metal does not [9].
We studied the geometrical and electronic properties of neutral M n C (M = Fe, Co, Ni, Cu; n = 1–6) clusters [10]. Especially we addressed the critical influence of the interaction strength between the carbon and metal clusters in neutral systems for growing the SWCNTs. In experiment, the arc discharge (AD), laser ablation (LA) and chemical vapor deposition (CVD) are used to fabricate the SWCNTs. The carbon nano-metals are produced at high temperature in the experiments of AD and LA, the systems involved there are most probably charged. In order to clarify the growth mechanism of metal–carbon systems, it is necessary to investigate the neutral and charged transition metal carbide clusters systemically. In this work, the ground-state properties of the neutral and charged M n C2 (M = Fe, Co, Ni, Cu, n = 1–5) clusters were investigated by DFT calculations.
Computational Details
All calculations were carried out within the gradient corrected DFT framework using the Quantum-ESPRESSO package [11]. The potential between the ion core and the valence was expressed in terms of the Vanderbilt-type ultrasoft pseudopotential. The valence electronic configurations of Fe, Co, Ni and Cu are 3s23p63d64s2, 3d84s1, 3d94 s1 and 3d104s1. The exchange–correlation interaction was calculated within the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) functional. Wave functions were expanded in a plane-wave basis set within 30 Ry kinetic energy. For the finite sizes, clusters were placed in a cubic supercell with an edge of 12–16 Å for different cluster sizes. This length scale is large enough to avoid interactions between clusters in neighboring cells.
The geometries of clusters were optimized by Broyden–Fletcher–Goldfrab–Shanno algorithm (BFGS). The initial structures were generated as our previous work [10] from one dimension to three dimension following two steps : firstly, using two C atoms to substitute two metal atoms on different potential energy surface sites of the pure metal clusters (M n+2, n = 5) [12–15]; secondly, these clusters were constructed according to different symmetry. The convergence criteria for the total energy and inter-atomic force were 10−6 ryd and 0.01 eV/Å.
Results and Discussion
Geometrical Structures of the Ground State
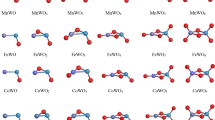
To check the accuracy of the method we used, several available experimental and theoretical (vs. LDA, PW91 and BP method) results are shown in Table 1. The calculated binding energy (Eb), bond length (R), and magnetic moments (M) of these dimers (Fe2, Co2, Ni2, Cu2, C2, FeC) are listed in Table 2. Using the method described in Sect. 2, we found the data of Table 2 being in good agreement with the experimental and theoretical reports. The ground-state geometries of the neutral and charged M n C2 (M = Fe, Co, Ni, Cu; n = 1–5) clusters are shown in Fig. 1. All atoms were relaxed without any constraints in the process of structural optimization (Table 3).

The geometrical structures of the ground state for neutral and charged M n C2 (M = Fe, Co, Ni, Cu; n = 1–5) clusters (Where the big balls with plum purple stand for metal atoms, and the small balls with purple stand for C atoms) (Color figure online)
Cationic Clusters
The most stable structures of Fe2C2 +, Ni2C2 + and Cu2C2 + are linear chain under the transformation of Dh group. Co2C2 + is a rhombus with D2h symmetry. The ground state structure of Fe3C2 + and Co3C2 + are both trigonal bipyramids but with different symmetries, Fe3C2 + has Cs symmetry and Co3C2 + has D3h symmetry. It is interesting to note that, the ground-state geometry of Ni3C2 + and Cu3C2 + clusters are still two-dimensional configurations. The Ni3C2 + cluster has a sector with C2ν symmetry. The Cu3C2 + cluster is formed by adding a metal atom on Cu2C2 + cluster with C2ν symmetry. The ground-state geometry of Fe4C2 + cluster is a pentacle pyramid with Cs symmetry. The ground-state geometry of Co4C2 + and Ni4C2 + clusters are distorted octahedra with C2ν symmetry. The lowest energy structure of Cu4C2 + is a butterfly-like structure with D2h symmetry. For n = 5, Fe5C2 +, Co5C2 + and Ni5C2 + all are pentacle bipyramids with C2ν symmetry. Cu5C2 + is still a two-dimensional structure which could be seen as one Cu atom added on the distorted Cu4C2 + with Cs symmetry.
Neutral Clusters
The ground state structures of neutral MC2 (M = Fe, Co, Ni, Cu) clusters are all isosceles triangles with C2ν symmetry. The ground-state geometry of the FeC2 cluster is identical with the results reported before [19, 25]. All of the ground-state geometries for the Fe2C2, Co2C2, Ni2C2 clusters are the same fanlike structures with Cs symmetry. For Cu2C2, two copper atoms locate far away from each other. The lowest energy structures of Fe3C2, Co3C2 are all trigonal bipyramid with different symmetries. Ni3C2 and Cu3C2 employ fanlike structures as the ground-state structures with C2ν symmetry. The ground-state geometry of the Fe4C2 (Cs), Co4C2 (C2v) clusters are the same as those of Fe4C2 + and Co4C2 + clusters. Ni4C2 shows a similar structure as Fe4C2. For Cu4C2, the lowest energy structure is evoluting as a three dimensional structure with Cs symmetry. Fe5C2, Co5C2 and Cu5C2 are all Pentagonal bipyramids. Ni5C2 is formed by adding a nickel atom to a quadrilateral bipyramids structure on one side.
Anionic Clusters
The lowest energy structures of MC2 − (M = Fe, Co, and Ni) are all isosceles triangles with C2ν symmetry. CuC2 − prefers a linear chain with the Cu atom at the edge site which is in good agreement with the discussions by Tono [9] and Alexandrova [26]. In Ref. [26], the global minimum structure of CuC2 − is linear chain. The energy of triangular isomer is just 3.82 kcal/mol (0.166 eV) higher than the B3LYP/6-311 + G* level theory. The most interesting thing is that these two isomers are both observed by experimental spectra. In this work, we also computed the triangular isomer as the first low-lying structure. The energy difference of linear isomer is only 0.20 eV. This is identical to Alexandrova’s work. The ground-state structures of Fe2C2 −, Co2C2 − and Ni2C2 − all are quadrangles with Cs symmetry. CoC2 − and Co2C2 − show geometric structures similar to those of the corresponding neutral ones, which are in good agreement with Tono’s results [9]. Cu2C2 − shows the similar ground state structure and symmetry with Cu2C2 +. The ground-state geometry of the Co3C2 − cluster is trigonal bipyramid with the C2ν symmetry. All the lowest energy structures of Fe3C2 −, Ni3C2 −and Cu3C2 − are planar with Cs, Cs and C2ν, respectively. Fe4C2 − is a pentacle pyramid with Cs symmetry. Distorted octahedra with C2ν symmetry are found to be the lowest-energy structures of Co4C2 − and Ni4C2 −. Cu4C2 − was a planar with C2ν symmetry. Fe5C2 − is a pentacle bipyramid with C2ν symmetry. Co5C2 − and Cu5C2 − have the same Cs symmetry. Co5C2 − is hexagonal pyramid with a Co atom on the peak. Cu5C2 − adds a Cu atom on the base of Cu4C2 −. Ni5C2 − with C2ν symmetry is formed by adding a C atom on one side of a octahedra.
As described above, the ground-state structures of neutral M n C2 (M = Fe, Co, Ni; n = 1–5) clusters evolve from planar to stereoscopic. Similar growth model are also found for the charged M n C ±2 (M = Fe, Co, and Ni, n = 1–5) clusters. However the ground state structures of the neutral Cu n C2 and charged Cu n C ±2 (n = 1–5) clusters always have planar geometry except Cu5C2. It’s interesting to note that the pure Cu n clusters still present planer configurations as the lowest-energy geometry up to n = 6. As we all known the radius of C atom is much more smaller than that of the metal (Fe, Co, Ni, Cu) atoms, adding two C atoms should not obviously change the main growth model of the host metal clusters M n .
The extent of C–C coupling in those metal–carbon clusters has many interesting behaviors. The two carbon atoms bond together in the ground-state for all neutral and charged Fe n C2 and Cu n C2 (n = 1–5) clusters. The C–C bond is much stronger than Fe–C bond [27, 28]. So Fe would not insert into the C–C chain since that would break the stronger C–C bonds and form the weaker Fe–C bonds [29]. While for the neutral and charged Co n C2 and Ni n C2 (n = 1–5) clusters the two C atoms are uncoupled in most cases. These results suggest that the difference of bond behavior within M–C plays the most important role in building carbon nanometer materials.
Electronic Properties
The density of states (DOS) of CuC2 was calculated to compare with reported experimental data (PES of CuC2 − [26]). In Fig. 2, the fluctuating trend of the two graphs are similar. The main peaks in PES also present in curve of DOS. The HOMO–LUMO energy gap (1.09 eV) in Fig. 2a is identical with the energy gap between the first sharp peak * and X (about 1.05 eV) in Fig. 2b.

a Density of state for CuC2 cluster. b Photoelectron spectra of CuC2 − at 193 nm [25]
The magnetic properties of transition metal clusters attract many research interests in recent years. The magnetic moments are enhanced in clusters of the 3d transition metal Fe, Co and Ni as compared to their bulk values [30–32]. Variations of the magnetic moments of M n C2 and M n C ±2 (M = Fe, Co, Ni, Cu; n = 1–5) clusters are shown in Fig. 3. The magnetic moment decreases following the sequence of (Fe n C2–Fe n C ±2 cluster, Co n C2–Co n C ±2 cluster, Ni n C2–Ni n C ±2 cluster) with the same size, this is similar to the case of pure transition metal (Fe, Co and Ni) clusters observed in experiment [32]. Among all the neutral and charged clusters, the magnetic moment of Fe n C2 and Fe n C ±2 (n = 1–5) clusters are almost the largest especially for the clusters with n = 5. The magnetic moment of the Fe n C2 and Fe n C ±2 (n = 1–5) clusters increases monotonically as the cluster size increases. While the magnetic moment of Cu n C2 and Cu n C ±2 (n = 1–5) clusters exhibits oscillation behavior between 0 and 1 except CuC2 +.

Magnetic moments of neutral and charged M n C2 (M = Fe, Co, Ni, Cu; n = 1–5) clusters
Chemical bond can sensitively impact the magnetic feature of isomers. The charge density of M n C2 and M n C2 − (M = Co, Ni, Cu; n = 1, 2) were calculated and plotted in Fig. 4. As seen in Fig. 4, there is a strong covalent bond between two C atoms. The metal-C2 is consistent with their ionic bonding character. The anisotropic spatial distribution of Co and Ni atoms are obviously stronger than that of Cu atoms. This is an interesting result for the neutral and charged M n C2 (M = Co, Ni, Cu; n = 1–5) clusters. Co and Ni atoms has strong magnetic anisotropy. While Cu n C2 clusters has a more muted effect on magnetic moment.

Charge density of M n C2 and M n C − 2 (M = Co, Ni, Cu; n = 1–2) clusters
The deformation degree in charge density measures the strength of interaction between atoms. The larger the deformation is, the stronger the interaction should be. Ding and co-workers [33] used ab initio and molecular dynamics simulations to investigate the catalytic growth mechanisms of SWCNTS. They found that the adhesion strength between the catalyst and the ending of growing SWCNTS was crucial, which must be larger or comparable to the carbon dangling bond formation energy of the SWCNTS. So it is necessary to investigate the interaction strength between metal–carbon atoms. The fragmentation energy of neutral and charged M n C2 (M = Fe, Co, Ni, Cu; n = 1–5) clusters are calculated by the following formulas:
Where E(M n ), E(C2) and E(M n C2) represent the total energy of the corresponding cluster M n , C2 and M n C2, respectively. The ground-state energies of M n , M ± n , C2, C ±2 , M n C2 and M n C ±2 (M = Fe, Co, Ni, Cu; n = 1–5) clusters have been listed in Table 2.
Figure 5 shows the fragmentation energies of neutral and charged M n C2 (M = Fe, Co, Ni, Cu; n = 1–5) clusters, which increase with increasing the cluster size. The interaction strength in TM–C2 of neutral and charged TM n C2 (TM = Fe, Co, Ni; n = 1–5) clusters are comparable and are both larger than of Cu–C clusters with the same cluster size.

The fragmentation energies of neutral and charged M n C2 (M = Fe, Co, Ni, Cu; n = 1–5) clusters
There are many interesting results to compare the fragmentation energy of the neutral M n C2 following the sequence of different strength: cation > neutral > anion. According to the metal–carbon interaction strength model [33], the interaction strength between the catalytic particle and the SWCNT’s open end is critical for growing the SWCNTs. Now we see that the interaction between the metal cluster and the positively charged carbon atoms are enhanced at the positively charged background. This is why Fe, Co, Ni are more efficient catalysts than other transition metals like Cu, Pd and Au for SWCNTs growth. Recent experiments [34, 35] have shown that the enhanced growth of the SWCNTs occurs at the negatively biased electrode. To explain the experiment, the authors [34] proposed a model considering the charge transport, which depended on a positively charged background at the SWCNT-metal nanoparticle contact region (the positively charged nanotube base, see Fig. 5 in Ref. [34]). They proposed that the condition- and/or temperature-induced charge current along the SWCNT can make enhancement of the SWCNT growth. We believe the enhanced interaction strength at the metal–carbon contact region is also an important factor to promote the growth of the SWCNTs observed in experiment.
Conclusions
We perform spin-polarized DFT calculations to investigate the ground-state geometrical and electronic properties of neutral and charged M n C2 (M = Fe, Co, Ni, Cu; n = 1–5) clusters. The four key points are summarized as follows: (1) The ground-state geometry of neutral clusters could be influenced by adding or losing an electron. TM n C2 (TM = Fe, Co, Ni; n = 1–5) clusters liken to grow from lower dimensional structure to higher dimensional structure. Planar structures dominates the Cu–C clusters. (2) In the ground-states, two carbon atoms bond together in all neutral and charged Fe n C2 and Cu n C2 (n = 1–5) clusters. But they could be separated for the neutral and charged Co n C2 and Ni n C2 (n = 1–5) clusters. This may cause different carbon fabrications. (3) Chemical bond can sensitively impact the magnetic feature of isomers. The spatial distribution of Co and Ni indicate stronger magnetic anisotropy than that of Cu atoms. (4) The interaction strength between metal and carbon atoms is comparable with TM–C (TM = Fe, Co and Ni) clusters and is obviously larger than that in Cu–C clusters with the same cluster size. The interaction strength decreases following the sequence: cation > neutral > anion. The interaction between the metal cluster and the positively charged carbon atoms are enhanced at the positively charged background. The enhanced interaction strength at the metal–carbon contact region is an important factor to study the growth mechanism of metal–carbon nano-material.
References
A. W. Castleman Jr and P. Jena (2006). PNAS. 103, 10554.
E. G. Noya, R. C. Longo, and L. J. Gallego (2003). J. Chem. Phys. 119, 11130.
A. C. Borin, J. P. Gobbo, and B. O. Roos (2006). Chem. Phys. Lett. 418, 311.
D. Tzeli and A. Mavridis (2007). J. Chem. Phys. 126, 194304.
D. J. Brugh and M. D. Morse (2002). J. Chem. Phys. 117, 10703.
G. L. Gutsev and C. W. Bauschlicher Jr (2003). Chem. Phys. 291, 27.
J. O. Joswig, M. Springborg, and G. Seifert (2001). Phys. Chem. Chem. Phys. 3, 5130.
X. Li and L. S. Wang (1999). J. Chem. Phys. 111, 8389.
K. Tono, A. Terasaki, T. Ohta, and T. Kondow (2002). J. Chem. Phys. 117, 7010.
Z. X. Zhang, B. B. Cao, and H. M. Duan (2008). J. Mol. Struct. (Theochem). 863, 22.
S. Baroni et al, http://www.quantum-espresso.org/.
O. Diéguez, M. M. G. Alemany, C. Rey, P. Ordejón, and L. J. Gallego (2001). Phys. Rev. B. 63, 205407.
M. Deshpande, D. G. Kanhere, and R. Pandey (2005). Phys. Rev. A. 71, 063202.
S. Li, M. M. G. Alemany, and J. R. Cheliknowsky (2006). J. Chem. Phys. 125, 034311.
S. Datta, M. Kabir, S. Ganguly, B. Sanyal, T. S. Dasgupta, and A. Mookerjee (2007). Phys. Rev. B. 76, 014429.
H. Purdum, P. A. Montano, G. K. Shenoy, and T. Morrison (1982). Phys. Rev. B. 25, 4412.
S. K. Loh, L. Lian, D. A. Hales, and P. B. Armentrout (1988). J. Phys. Chem. 92, 4009.
D. J. Brugh and M. D. Morse (1997). J. Chem. Phys. 107, 9772.
B. K. Nash, B. K. Rao, and P. Jena (1996). J. Chem. Phys. 105, 11020.
G. L. Gutsev and C. W. Bauschlicher (2003). J. Phys. Chem. A. 107, 4755.
A. Kant and B. Strauss (1964). J. Chem. Phys. 41, 3806.
J. C. Pinegar, J. D. Langenberg, C. A. Arrington, E. M. Spain, and M. D. Morse (1995). J. Chem. Phys. 102, 666.
M. D. Morse, G. P. Hansen, P. R. R. Langridge-Smith, L. S. Zheng, M. E. Geusic, D. L. Michalopoulos, and R. E. Smalley (1984). J. Chem. Phys. 80, 5400.
R. S. Ram, C. N. Jarman, and P. F. Bernath (1992). J. Mol. Spectrosc. 156, 468.
Z. Cao (1996). J. Mol. Struct. (Theochem). 365, 211.
A. N. Alexandrova, A. I. Boldyrev, H. J. Zhai, and L. S. Wang (2005). J. Phys. Chem. A. 109, 562.
M. L. McKee (1990). J. Am. Chem. Soc. 112, 2601.
C. W. Bauschlicher, S. R. Langhoff, H. Partridge, and L. A. Barnes (1989). J. Chem. Phys. 91, 2399.
J. W. Fan, L. Lou, and L. S. Wang (1995). J. Chem. Phys. 102, 2701.
S. E. Apsel, J. W. Emmert, J. Deng, and L. A. Bloomfield (1996). Phys. Rev. Lett. 76, 1441.
I. M. L. Billas, J. A. Becker, A. Châtelain, and W. A. de Heer (1993). Phys. Rev. Lett. 71, 4067.
I. M. L. Billas, A. Châtelain and W. A. de Heer (1994) Science. 265, 1682.
F. Ding, P. Larsson, J. A. Larsson, R. Ahuja, H. M. Duan, A. Rosén, and K. Bolton (2008). Nano. Lett. 8, 463.
N. M. Bulgakova, A. V. Bulgakov, J. Svensson, and E. E. B. Campbell (2006). Appl. Phys. A. 85, 109.
M. Keidar and A. M. Waas (2004). Nanotechnology. 15, 1571.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 10864005, 11265015) and the Scientific Research Starting Foundation for Returned Overseas Chinese Scholars, Ministry of Education, China. Thanks Dr. Qun Jing, Dr. Ying Wang in Xinjiang Technical Institute of Phyics & Chemistry(CAS) for their useful help and suggestions.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Zhang, B., Cao, B.B., Chen, C. et al. Density-Functional Theory Study on Neutral and Charged M n C2 (M = Fe, Co, Ni, Cu; n = 1–5) Clusters. J Clust Sci 24, 197–207 (2013). https://doi.org/10.1007/s10876-012-0543-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-012-0543-2