
The trivacant heteropolyanion A-α[PMo9O31(OH2)3]3− spontaneously dimerizes in acetonitrile to form the Dawson complex [P2Mo18O62]6−. A rapid new quantitative preparation of the sodium salt of this Dawson complex is described. In addition, the structure of TBA5[HP2Mo18O62] is given and the featuring data compared with the isomorphous sodium salt (space group C2/c).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Lacunary polyoxometalates are useful precursors for reactions with transition metals and for polycondensations, especially for tungsten based species. For molybdenum derivatives, the sodium salt of the trivacant compound A-α[PMo9O31(OH2)3]3− has been isolated and characterized for a long time [1–3]. The synthesis and structure of Na3[H6PMo9O34] · xH2O were first described by Strandberg [1]. The structures of the hydrate Na3[H6PMo9O34]· 7H2O and Na4[H2P2Mo18O62] · 20H2O were solved by D’amour [2] and compared to that of K6[P2W18O62] · 14H2O. The coordination of three water molecules to three molybdenum atoms has been determined later by neutron diffraction on Na3[PMo9O31(OH2)3] · 12–13H2O [3]. This type of coordination could explain the preference for forming at lower pH [H6PMo9O34]3− rather than the higher charged monovacant [PMo11O39]7− as it was reported in a study relative to the speciation of the aqueous H+–MoO 2–4 –HPO 2−4 system [4]. This behaviour is very different from that of the more acidic tungsten species [5]. While equilibria related to the molybdenum system are rapid, the condensation reaction of two PMo9 precursors to form the P2Mo18 Dawson is very slow, even in high Mo concentration. The formation of the P2Mo18 dimer in aqueous acid solution was studied by Himeno et al. [6] who reported that if the Mo/P ratio has influence on the reaction, the temperature is also an important parameter [7].
Results dealing with the reactivity of [H6PMo9O34]3− in organic solvent are not so numerous. Acetonitrile was chosen here since hydrated acid or some alkaline salts of polyoxometalates are soluble in this medium. The tetrabutyl ammonium salt of [H6PMo9O34]3− was also prepared as a precursor for the study of the reactivity in other organic solvents.
The originality of this communication lies in the evidence of a rapid dimerization in acetonitrile of [H6PMo9O34]3− into [P2Mo18O62]6−. The crystal structure of the tetrabutylammonium salt TBA5[HP2Mo18O62] is presented.
EXPERIMENTAL SECTION
Syntheses
Na3[H6PMo9O34] was obtained by adaptation of the method previously described by Himeno et al. [6]. An 87 mL aqueous solution of 2.9 mL of 75% H3PO4 and 70 g of Na2MoO4 · 2H2O was cooled down in an ice bath. The pH was adjusted to 1.1 by addition of about 40 mL of concentrated 37% HCl. The pH value was maintained constant throughout the precipitation by addition of acid. The solid formed was filtered off after the pH was stabilized over 30 min. After air drying, 55 g of compound were obtained (yield 93.4% in molybdenum).
TBA3[H6PMo9O34] was prepared by an equivalent procedure. A more diluted solution of 200 mL containing 5 g of Na2MoO4 · 2H2O and 0.1776 mL of 75% H3PO4 was acidified by concentrated HCl until pH 1.5. Then, the addition of 3.7 g of TBABr (TBA=N(C4H9)4) led to an immediate precipitation of a pale yellow solid. After filtration the solid was washed with water and dried at room temperature (84% yield in molybdenum).
TBA5[HP2Mo18O62] has been crystallized as green needles from an solution obtained by dissolution of 2 g of Na3[H6PMo9O34] and 1.6 g of tetrabutylammonium bromide N(C4H9)4Br in 80 mL of CH3CN at 40°C, followed by the addition of 50 mL of water. The yellow precipitate formed was eliminated by filtration. The resulting heavy organic part of the filtrate diluted by acetonitrile was allowed to crystallize at room temperature. Crystals suitable for X-ray diffraction were obtained after 10 days.
Instrumentation
Powder X-ray diffraction patterns (XRD) were recorded at room temperature with a Siemens D 5000 diffractometer using Cu Kα radiation (λ=1.5418 Å). Crystal of dimensions 0.34 × 0.10 × 0.04 mm3 was glued to a glass fibre. X-ray intensity data were collected at room temperature on a Bruker X8-APEX2 CCD area-detector diffractometer using Mo Kα radiation (λ=0.71073 Å). Six sets of narrow data frames (30 s per frame) were collected at different values of θ, for 3 and 3 initial values of ϕ and ω, respectively, using 0.5° increments of ϕ or ω. Data reduction was accomplished using SAINT V7.03 [8]. The substantial redundancy (3.5) in data allowed a semi-empirical absorption correction (SADABS V2.10) [9] to be applied, on the basis of multiple measurements of equivalent reflections. The structure was solved by direct methods, developed by successive difference Fourier syntheses, and refined by full-matrix least-squares on all F 2 data using SHELXTL V6.14 [10]. Hydrogen atoms were included in calculated positions and allowed to ride on their parent atoms. Crystal parameters are listed in Table I, and final atomic coordinates and thermal parameters are listed in Table II and selected bond lengths in Table III, both of which are presented in the supplementary material.
Thermogravimetric analyses (TGA) were carried out in air flow (60 mL/min) with a Perkin–Elmer electrobalance TGA-7 at a heating rate of 5°C/min up to 600°C. Infrared spectra of KBr pellets were recorded on a Fourier transform Nicolet 550 apparatus. Solution 31P NMR spectra were recorded on a Bruker AC 200 apparatus working at 81 MHz. Chemical shifts are referenced to 85% H3PO4.
RESULTS AND DISCUSSION
The characteristic vibrations of the compounds listed in Table I are in agreement with that reported in the literature [6, 11].
Generally, vibrations of the phosphate group are meanly sensitive to the structure of the polyanion and the nature of the counter-ion has little influence. Nevertheless they are modified in the dimer, probably since the distortion of the tetrahedral geometry is more important: the splitting of P–Oa vibration in the monomer is 56 and 75 cm−1 in the Dawson structure. The Mo–O vibrators are also affected by the nature of the counter-ion [12].
TGA analyses on the TBA salt of the Dawson compound revealed that no hydration water is present. The decomposition of the organic cation occurs between 240 and 600°C. At this high temperature, 71% in weight of the solid are remaining, corresponding to 5 TBA per polyoxometalate. Two steps can be distinguished on heating, three molecules are first eliminated at 370°C and a quasi-plateau of 60°C is observed before the release of the two other molecules. Quite the same behaviour has been reported for tert-butylammonium salt [13].
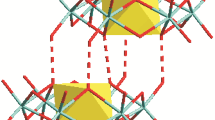
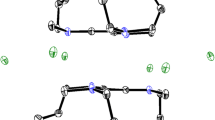
The structureFootnote 1 of TBA5[HP2Mo18O62] has been resolved from needle shaped monocrystals at RT (Figs. 1, 2). The compound crystallized in the monoclinic space group C2/c (no. 15), as for the sodium salt [2], but the unit cell differs by the c unit cell parameter value which increased from 23.15 to 36.41 Å for the same Z=4. Therefore, the density of the TBA salt is lowered from 3.01 to 1.93. The structure of the polyoxoanion is quite similar to that of the sodium salt and is represented in Fig. 1. The effective symmetry of the anion is D3 [7]. Nevertheless, some differences can be pointed out. It seems quite surprising to observe that the geometry of the central PO4 entity has been modified. Two distances are shortened, but the angle are more regular: three of them lie about 107° and three others around 111°, instead of two at 112°, one at 110° and three at 107°. The Mo–O distances at the bridging oxygen atoms are globally shortened. The six “belt” molybdenum atoms in the ring are in the same plane (Δz=0.177(1) Å) while a more important distortion was reported for the sodium salt (Δz=0.235(1) Å): the structure of the TBA-polyanion is less constrained than that of the sodium salt. Interactions with cations are probably responsible for this effect. In the sodium salt, a terminal oxygen atom attached to a belt molybdenum atom has a direct interaction with a Na+ cation (dO–Na =2.55(1) Å) or with protons of a water molecule coordinated to a sodium atom. The shortest O–Ow distances are about 3.2(1) Å. In the TBA salt, interactions with protons of the methyl group are weaker. In alkyl ammonium cations three alkyl chains develop in a zig–zag manner, for the others, the angle at the terminal carbon atoms are quite short (95(2)°, 98(3)° and 104(2)°). These methyl groups interact with two other methyls of two neighbouring alkyl ammoniums. The shortest distance between the carbon of a methyl group and terminal oxygen of a belt molybdenum atom is 3.33(1) Å. Another alkyl ammonium structure has been previously determined, that of the tert-butylammonium-salt [(CH3)3CNH3]6[P2Mo18O62] · 6H2O [13] which crystallizes in the different space group P-1, with a higher density of 2.53. Hydrogen bounding are observed mainly with water molecules and protons of the ammonium group (C–H⋯O (3.18–3.46 Å), N–H⋯O (2.86–3.12 Å), N–H⋯Ow (2.83–3.11 Å), Ow–H⋯O (2.96–3.59 Å), Ow–H⋯Ow (2.75–2.96 Å).

(a) Representation of [P2Mo18O62]6−, thermal ellipsoids are drawn at 50% probability level, mean plans based on molybdenum and oxygen atoms; (b) distribution of tetrabutyl ammonium cations around the Dawson anion.

Powder X-ray pattern of TBA5[HP2Mo18O62] (top) and simulation (bottom) based on single crystal data.
31P NMR spectroscopy revealed the dimerization of [H6PMo9O34]3− is rapid in acetonitrile as represented in Fig. 3. Solutions of crystals of TBA5[HP2Mo18O62] dissolved in acetonitrile exhibit spectra containing a single peak at −2.07 ppm. Two lines located at −0.91 and −1.99 ppm, respectively, are observed after the addition of A-αNa3[H6PMo9O34] to a solution of TBA5[HP2Mo18O62] (equal concentration of both the polyanions, 6 × 10−3 M). The −0.91 ppm signal can be attributed without any ambiguity to A-[H6PMo9O34]3− (−0.93 ppm in 0.5 M HCl solution [6]) while the peak at −1.99 ppm corresponds to [P2Mo18O62]6− (−2.45 ppm in 0.5 M HCl solution [6]). The relative intensity of the signals rises an interesting point. Theoretically, a 1/2 ratio is expected, but the experimental values lead to 1/4. This result reflects the easiness of the PMo9 dimerization, even in the presence of P2Mo18. Furthermore, the spectrum of a solution of Na3[H6PMo9O34] in acetonitrile exhibits a single peak located at −1.94 ppm. For solutions of TBA3[H6PMo9O34], a weak peak at −2.34 ppm and an intense peak at −1.96 ppm are systematically observed. In order to identify the new signal at −2.34 ppm, the spectrum of a solution of TBA3[PMo12O40] was recorded in the same conditions resulting in a peak at −2.32 ppm. In addition, it was observed that the proportion of [PMo12O40]3− increased with time in the solution of TBA3[H6PMo9O34] in acetonitrile, and crystals of TBA3[PMo2O40] were obtained in few days from this solution. On the other hand, a solution of the sodium salt in acetonitrile is stable for almost one day; IR spectra and XRD of the solid recovered as a powder after evaporation of the solvent is characteristic of the Dawson structure [P2Mo18O62]6−.

31P NMR spectra of freshly prepared acetonitrile solutions at 28°C of TBA5[HP2Mo18O62] (10−3 M), TBA5[HP2Mo18O62] and Na3[H6PMo9O34] (equal concentration 0.6 × 10−3 M), Na3[H6PMo9O34] (10−3 M), TBA3[H6PMo9O34] (10−3 M) and TBA3[PMo12O40] (10−3 M).
Thus a new way can be proposed for the preparation in high yield (90%) of the Dawson polyanion without strong heating: A-αNa3[H6PMo9O34] (prepared in 90% yield from molybdate) is dissolved (10−3 M) in acetonitrile at 30°C. Na6[P2Mo18O62] is rapidly formed and isolated as a glassy film by evaporation of the solvent in a rotating evaporator.
CONCLUSIONS
The trivacant heteropolyanion A-α[PMo9O31(OH2)3]3−, easily obtained as sodium or TBA salts, is not stable when dissolved in acetonitrile. Dissociation of aquo ligand from molybdenum atoms leads to the rapid dimerization in [P2Mo18O62]6−. Some Keggin structures are formed as impurity when using tetrabutyl ammonium salt as precursor. In contrast, pure Dawson complex is quantitatively obtained starting from sodium salt A-Na3[PMo9O31(OH2)3].
SUPPLEMENTARY MATERIALS
X-ray crystallographic data in CIF format for TBA5[HP2Mo18O62] have been deposited with the depository number CCDC 291596 at the Cambridge Crystallographic Data Centre.
Crystal parameters are listed in Table I. Final atomic coordinates and thermal parameters are listed in Table II and selected bond lengths in Table III given in the supplementary material.
Notes
Crystal structure analysis: monoclinic, space group C2/c; dimensions a = 25.4115(10) Å, b=15.3850(5) Å, c=36.4090(17) Å, β=105.471(2)°, V = 13718.5(9) Å3; Z=4; total reflections collected: 74,730; independent reflections: 19,603 (10,365 Fo>4σ(Fo)); data were collected up to a 2Θmax value of 59.92° (98.3% coverage). Number of variables: 714; R1=0.0708, wR2=0.1825, S=1.044; highest residual electron density 2.709 e·Å−3.
References
Strandberg R. (1974). Acta Chem. Scand. A28:217
D’amour V. H. (1976). Acta Cryst. B32:729
Hedman B. (1978). Acta Chem. Scand. A32:439
Petterson L., Anderson I., Öhman L.-O. (1986). Inorg. Chem. 25:4726
G. Hervé, A. Tézé, R. Contant, in J. J. Borras-Almenar et al. (eds.), Polyoxometalate Molecular Science (Kluwer Academic Publishers, Netherland, 2003), pp. 33–54
Himeno S., Hashimoto M., Ueda T. (1999). Inorg. Chim. Acta 284:237
Pope M. T. (1976). Inorg. Chem. 15:2008
APEX2 Version 1.0-8; Bruker AXS: Madison, WI, 2003
G. M. Sheldrick, SADABS; Program for Scaling and Correction of Area Detector Data, University of Göttingen, Germany, 1997
SHELXTL Version 6.14; Bruker AXS: Madison, WI, 2001
Garvey J. F., Pope M. T. (1978). Inorg. Chem. 17:1115
Rocchiccioli-Deltcheff C., Fournier M., Franck R., Thouvenot R. (1983). Inorg. Chem. 22:207
Wéry A. S. J., Gutiérrez-Zorrilla J. M., Luque A., Ugalde M., Romàm P. (1997). Polyhedron 16:2589
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is dedicated to Professor M. T. Pope on the occasion of his retirement.
Rights and permissions
About this article
Cite this article
Marchal-Roch, C., Ayrault, E., Lisnard, L. et al. Dimerization in Acetonitrile of [H6PMo9O34]3− into [P2Mo18O62]6−: Structural Characterization of the Tetrabutyl Ammonium Salt. J Clust Sci 17, 283–290 (2006). https://doi.org/10.1007/s10876-006-0051-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-006-0051-3