
Abstract
Hypomorphic IKBKG mutations in males are typically associated with anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID). Some mutations cause immunodeficiency without EDA (NEMO-ID). The immunological profile associated with these NEMO-ID variants is not fully documented. We present a 2-year-old patient with suspected immunodeficiency in which a hemizygous p.Glu57Lys IKBKG variant was identified. At the age of 1 year, he had an episode of otitis media that evolved into a bilateral mastoiditis (Pseudomonas spp). Hypogammaglobulinemia, specific (polysaccharide) antibody deficiency, and low switched memory B cell subsets were noticed. The mother was heterozygous for the variant but had no signs of incontinentia pigmenti. Patient peripheral blood mononuclear cells produced low amounts of IL-6 after stimulation with IL-1β, Pam3CSK4, and FSL-1. In patient fibroblasts, IκB-α was degraded normally upon stimulation with IL-1β or TNF-α. Transduction of wild-type and variant NEMO in NEMO−/− deficient SV40 fibroblasts revealed a slight but significant reduction of IL-6 production upon stimulation with IL-1β and TNF-α. In conclusion, we demonstrated that p.Glu57Lys leads to specific immunological defects in vitro. No other pathogenic PID variants were identified through whole exome sequencing. As rare polymorphisms have been described in IKBKG and polygenic inheritance remains an option in the presented case, this study emphasizes the need for thorough functional and genetic evaluation when encountering and interpreting suspected disease-causing NEMO-ID variants.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
NF-κB transcription factors play a role in various processes related to immunity, inflammation, cell growth, and apoptosis. They are involved in signal transduction of a number of surface and cytoplasmic receptors including T cell receptors, B cell receptors, IL-1 receptor, IL-12/IL-18 receptor, CD40, ectodysplasin A receptor, and Toll-like receptors (TLRs) [1,2,3]. NF-κB transcription factors are inactive in the cytoplasm as they are bound to the inhibitor of NF-κB (IκB) [3]. Upon stimulation, IκBs are phosphorylated, which induces ubiquitination and subsequent degradation of IκBs by the proteasome. After degradation of IκBs, free NF-κB translocates into the nucleus and activates its target genes. IκBs are phosphorylated by the “inhibitor of the NF-κB kinase (IKK) complex” which consists of the IKK-α and IKK-β catalytic subunits and the regulatory subunit IKK-γ, also referred to as “NF-κB essential modulator (NEMO) [3].
NEMO is a 419 amino acid protein encoded on the X chromosome and is required for the activation of the canonical NF-κB pathway [3]. Amorphic (loss-of-function) NEMO mutations are typically lethal in males and result in incontinentia pigmenti (IP), a rare X-linked genodermatosis in females [4,5,6]. Affected females survive because of skewed X-inactivation [4,5,6]. In most IP patients (60–80%), an identical DNA rearrangement (exon 4–10 deletion) occurs, resulting in a substantial truncation of the NEMO protein. Hypomorphic (impaired function) mutations in NEMO result in a spectrum of clinical and immunological phenotypes in males (as a result of the full phenotypic expression of the mutations in the absence of a normal X chromosome), and cause a mild IP phenotype in females [5,6,7,8]. Hypomorphic mutations in males are typically associated with anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID). Ectodermal dysplasia is caused by disturbed ectodysplasin A receptor signaling, whereas immunodeficiency is caused by disturbed signaling via immune receptors (e.g., TLRs, BCR, TCR, ...) [5,6,7,8]. Besides NEMO mutations, heterozygous mutations in NFKBIA, encoding IκB-α, also cause EDA-ID through autosomal dominant inheritance [9, 10].
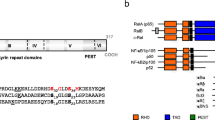
Analysis of 52 patients revealed that in 77% of cases, hypomorphic NEMO mutations were associated with ectodermal dysplasia [7]. Some hypomorphic mutations cause X-linked recessive Mendelian susceptibility to mycobacterial diseases (XR-MSMD) [7,8,9,10,11,12] or immunodeficiency without EDA (NEMO-ID) [7, 13,14,15,16,17,18,19]. At the time of writing, five NEMO-ID patients have been described [7, 13,14,15,16,17,18,19]. The mutations found in these patients are shown in Fig. 1 and include two missense mutations (p.Leu80Pro and p.Asp113Asn), one insertion in exon 2 leading to a frameshift and a premature stop codon (c.110-111insC, p.Met38fs*48), one deletion in exon 7 (c.811_828del, p.∆271–276), and one splice-site mutation causing skipping of exon 9 (c.1056-1G>A, p.∆353–373) [7, 13,14,15,16,17,18,19]. Of note, the NEMO c.110-111insC frameshift mutation was hypomorphic because of residual production of an N-terminally truncated protein created by downstream translation reinitiation (AUG codon) at nucleotide positions 112–114 [13].
In this article, we report a male infant patient with suspected NEMO-ID in whom a p.Glu57Lys IKBKG variant was identified. We set out to determine its pathogenic impact through in vitro studies.

Methods
Immunologic Assays
Serum immunoglobulin concentrations, post-vaccination antibody titers, leukocyte subsets, and T cell proliferative responses were determined in the clinical laboratories of the University Hospitals Leuven or University Hospital Brussels. Results were compared with laboratory specific, age-related reference values. Specific anti-pneumococcal antibodies were measured with an overall assay using the 23-valent pneumococcal polysaccharide vaccine as detecting antigen [20].
Cell Isolation
Peripheral blood mononuclear cells (PBMCs) were prepared from heparinized peripheral blood by Ficoll density separation. Primary skin fibroblasts were derived from a skin biopsy performed on the index patient. Control fibroblasts and PBMCs were donated by healthy, adult volunteers. Fibroblast cell lines were prepared in the Department of Human Genetics at the University Hospitals Leuven. The study was approved by the institutional review board (ethics committee).
In Vitro Evaluation of NF-κB Function in Peripheral Blood
PBMCs were seeded at 2 × 106 cells/ml in a 96-well culture plate and stimulated for 24 h with the following TLR and IL-1R ligands: LPS (TLR4, 37.5 μg/ml), IL-1β (IL-1R, 50 ng/ml), Pam3CSK4 (TLR1/2, 1 μg/ml), FSL-1 (TLR2/6, 100 ng/ml), heat inactivated Streptococcus pneumonia serotype 3 (1.2 × 108 CFU/ml), and Imiquimod (TLR7, 10 μg/ml). The next day, cells were pelleted by centrifugation and the supernatants were frozen at −20 °C and analyzed for IL-6 production. This analysis was also performed on patient and control primary skin fibroblasts. Cells were seeded at a concentration of 2 × 104 cells/stimulation in 96-well culture plates and stimulated for 24 h with IL-1β (1 ng/ml) or TNF-α (10 ng/ml). The supernatants were frozen at −20 °C and analyzed for IL-6 production. An in-house validated ELISA was used to measure serum IL-6 levels. The assay is based on a commercially available antibody pair (BD Biosciences, San Jose, California, USA).
NEMO Sequencing
Sequencing was performed with primer sets to amplify each of the individual IKBKG, IRAK4, and MYD88 exons using Accuprime I Supermix (Thermo Fisher), Accuprime II Supermix (Thermo Fisher), or GoTaq Hot Start Polymerase (Promega). Primer sets are displayed in Table S1. PCR reactions were performed on a Veriti 96-well thermal cycler (Applied Biosystems). IKBKG was sequenced using genomic DNA (exon 1 and exon 2) and cDNA (exon 3–10) to ensure that the identified mutations are in the functional NEMO gene and not in the NEMO pseudogene. gDNA was extracted from EDTA-containing blood tubes using the QIAamp DNA Blood Mini Kit (Qiagen). cDNA was prepared from peripheral blood mononuclear cells (PBMCs) using the Total RNA Isolation Mini Kit (Agilent) and SuperScript VILO cDNA Synthesis Kit (Thermo Fisher). Sanger sequencing was performed on an ABI 3730xl Genetic Analyzer (Applied Biosystems, Foster City, Calif) at the LGC Genomics Facility (Berlin, Germany). Sequencing data were analyzed using FinchTV (Geospiza, Seattle, Washington, USA).
Whole Exome Sequencing (WES)
WES was performed on samples from mother and son. Exome enrichment and sequencing was performed by Macrogen (Seoul, South Korea) using SureSelect XT Library Prep Kit (Agilent Technologies, CA, USA) and Illumina HiSeq 4000 (San Diego, CA, USA). The data analysis was performed as described previously [21]. Briefly, the 101-bp paired-end reads were mapped to the genome reference sequence (b37) using BWA v0.7.12 and further processed using Genome Analysis Tool Kit v3.6 [22, 23]. HaplotypeCaller was run on both samples separately in GVCF mode. Then, the GVCFs of multiple samples were run through a joint genotyping process to produce a multi-sample VCF callset. Variants were annotated, filtered, and prioritized using the in-house developed ExoMiner annotation pipeline based on ANNOVAR.
IκB-α Degradation in Skin-Derived Fibroblasts
Degradation of IκB-α was evaluated by Western blotting. Briefly, 2 × 106 primary skin fibroblasts were incubated for 0, 15, 30, and 60 min with IL-1β (1 ng/ml), TNF-α (10 ng/ml), or left unstimulated. After stimulation, cells were immediately lysed by using NE-PER Nuclear and cytoplasmic extraction reagents (Thermo Scientific, Waltham, Massachusetts). Protein concentration was determined (Qubit 1.0 Fluorometer; Invitrogen, Carlsbad, California) and 15 μg of lysate were subjected to SDS-PAGE separation on 4–12% Bis-Tris Plus gels (Life Technologies, Carlsbad, California, USA). Proteins were transferred to a polyvinylidene difluoride membrane (GE Healthcare, Buckinghamshire, United Kingdom) and immunoblotted with rabbit polyclonal anti IκB-α (BD Biosciences), anti-NEMO (BD Biosciences), anti-pan 14–3–3 (Santa Cruz, Dallas, Texas, USA), HRP-conjugated donkey anti-rabbit (Santa Cruz), and HRP-conjugated goat anti-mouse (Santa Cruz). Bound antibody was detected by enhanced chemiluminescence (Pierce ECL Western blotting substrate, Thermo Scientific) on the ChemiDoc XRS+ system (Bio-Rad. Hercules, California, USA). Densitometry was performed using Image Lab software (Bio-Rad). After detection, membranes were stripped by using 15 g/l glycine pH 2.2, 0.1% Tween, and 0.01 g/l SDS in distilled water at room temperature for 2 × 10 min.
Retroviral Transduction of SV40 NEMO −/− Fibroblast Cell Lines
NEMO-deficient SV40 transformed fibroblasts, obtained from a fetus with incontinentia pigmenti, were kindly provided by Jean-Laurent Casanova [24]. Cells were cultured in DMEM +10%FCS, 100 U/ml Penicillin, 100 μg/ml Streptomycin, and 500 ng/ml Fungizone. HIV-based retroviral pCH-EF1a-3xmyc-Ires-Bsd vectors were developed containing hNEMO WT, p.Glu57Lys, p.Asp113Asn, p.Val146Gly, and p.Cys417Arg. The hNEMO coding sequence was based on RefSeq NM_003639.4. A pCH-EF1a-eGFP-Ires-Bsd retroviral vector was used as a negative control. The respective cDNAs are driven by a universal human EF1α promoter and followed by an internal ribosomal entry site (IRES) and a blasticidin resistance cDNA. All cloning steps were verified by sequencing. Viral vectors were produced as previously described by the Leuven Viral Vector Core [25]. NEMO-deficient SV40 transformed fibroblasts were stably transduced with a dilution series of the respective vectors. Three days after transduction, cells were reseeded and selected (3 μg/ml blasticidin (Invivogen, Toulouse, France)). Following selection, cells were maintained under blasticidin selection. Cells stably transduced with the highest vector dilutions but growing like wild-type cells were chosen for further experiments. These cells displayed comparable levels of NEMO protein expression as determined with Western blot. Stably transduced fibroblasts were seeded at 5 × 104 cells/ml in 96-well culture plates and stimulated for 24 h with IL-1β (50 ng/ml), Poly I:C (12.5 μg/ml), and TNF-α (100 ng/ml). After 24 h, supernatants were analyzed for IL-6 production (BD Biosciences). Statistical analysis was performed using an unpaired Students t test (Graphpad Prism for Windows, version 5.02).
Results
The patient is a boy whose medical history revealed no abnormalities until the age of 1 year when he developed a bilateral acute otitis that evolved into bilateral mastoiditis. He was admitted to the hospital and received intravenous antibiotics. Unilateral mastoidectomy was required because of persistent infection. Pseudomonas species was cultured from the wound. After surgery, targeted antibiotic therapy was given for several weeks, leading to full recovery. A few months later, at the age of 14 months, the child developed an EBV infection with a protracted course. During this time, the child presented with low grade fever but had no other symptoms. He had no signs of EDA. His immunological phenotype is presented in Table 1. Because of hypogammaglobulinemia, immunoglobulin substitution therapy was initiated at the age of 2 years. This prevented further infections. His mother (P1M) had a history of recurrent ear, nose, and throat infections as a child. To this day, she continues to suffer from recurrent throat infections caused by beta-hemolytic Streptococcus species for which she underwent tonsillectomy.
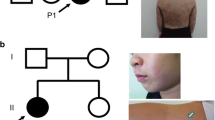
Initially, the patient was evaluated for defects in the IKBKG, IRAK4, and MYD88 genes using Sanger sequencing. A heterozygous polymorphism was detected in the IRAK4 gene (rs4251545). No variants were found in the MYD88 gene. A hemizygous c.169G>A substitution (rs148695964) was identified in the IKBKG gene leading to a p.Glu57Lys missense mutation. His mother was a carrier of this variant and displayed no skewed X-inactivation or signs of IP. Familial segregation and sequencing results are displayed in Fig. 2. Figure 3 shows the results of IL-6 production by PBMCs of the patient compared to IL-6 production by PBMCs of healthy controls after in vitro stimulation with IL-1R and TLR ligands. In the patient, stimulation with IL-1β, Pam3CSK4, or FSL-1 resulted in low IL-6 production on several occasions. NEMO protein expression in fibroblasts was normal (data not shown). IL-6 production (Fig. 3b) and IκB-α degradation (Fig. 4) upon stimulation with IL-1β and TNF-α in fibroblasts of the patient was comparable to IL-6 production and IκB-α degradation in fibroblasts of two controls.

Pedigree and Sanger sequencing results. Variant p.Glu57Lys was identified in P1 and his mother. Sanger sequencing results of IKBKG gDNA exon 2 are displayed

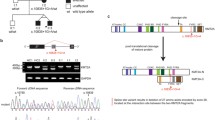
IL-6 production by PBMCs and fibroblasts after IL-1R, TNF-R, or TLR stimulation for 24 h. a IL-6 production by PBMCs for P1 and 20 healthy adult controls. For the patient, each dot represents an IL-6 measurement at a different time point. b IL-6 ratio (= stimulated/unstimulated IL-6 production) of skin-derived fibroblasts upon stimulation with IL-1β and TNF-α for P1 and 2 healthy adult controls. Results of three independent experiments performed in duplicate are displayed

IκB-α degradation in skin-derived fibroblasts from P1 and 2 healthy adult controls. Fibroblasts were stimulated with IL-1β (1 ng/ml) or TNF-α (10 ng/ml) and IκB-α degradation was evaluated by Western blot after 15, 30, 45, and 60 min. a Western blot results are displayed. b For each time point, the IκB-α/pan-14-3-3 ratio normalized to time = 0 (measured by densitometry) is shown. Results are representative for one out of three independent experiments
Finally, the pathogenicity of the variants was evaluated by transduction of retroviral NEMO constructs into SV40-transformed NEMO−/− fibroblasts. NEMO−/− fibroblasts were stably transduced with wild-type NEMO, p.Val146Gly NEMO (a known pathogenic EDA-ID variant in the CC1 domain [26]), p.Gly57Lys NEMO, p.Cys417Arg NEMO (a known pathogenic EDA-ID variant in the ZF domain [7]), and p.Asp113Asn NEMO (a suspected benign NEMO-ID variant in the CC1 domain [16, 27, 28]). As shown in Fig. 5, p.Val146Gly NEMO abrogated IL-6 production for all ligands studied (p < 0.0001 versus WT NEMO). IL-6 production by NEMO−/− fibroblasts transduced with p.Asp113Asn NEMO was comparable to IL-6 production by NEMO−/− fibroblasts transduced with WT NEMO (p > 0.05 for all ligands studied). NEMO−/− fibroblasts transduced with p.Gly57Lys NEMO produced significantly less IL-6 after stimulation with IL-1β and TNF-α compared to NEMO−/− fibroblasts transduced with WT NEMO (p = 0.003 and p = 0.0423, respectively). Nevertheless, IL-6 production was higher in p.Gly57Lys NEMO-transduced fibroblasts than in p.Val146Gly NEMO-transduced fibroblasts (p < 0.01 for all ligands studied). Compared to IL-6 production in NEMO−/− fibroblasts transduced with WT NEMO, NEMO−/− fibroblasts transduced with p.Cys417Arg NEMO produced significantly lower IL-6 after stimulation with IL-1β (p < 0.0001) but not after stimulation with Poly I:C or TNF-α (p > 0.05). These findings are in line with pathogenicity predicted by several in silico tools, which predict that p.Glu57Lys, p.Cys417Arg, and p.Val146Gly missense variants are more pathogenic than p.Asp113Asn (Table S2).

SV40-transformed NEMO−/− fibroblasts were transduced with wild-type NEMO, p.Cys417Arg NEMO, p.Gly57Lys NEMO, p.Asp113Asn NEMO, p. Val146Gly NEMO, or GFP. a Western blot results displaying NEMO re-expressed versus loading control pan-14-3-3 in stably transduced SV40-transformed NEMO−/− fibroblasts. b IL-6 production was evaluated 24 h after stimulation with IL-1β, Poly I:C, and TNF-α. The results shown are mean ± SD from three independent experiments, each performed in duplicate
Due to the increasing amount of case reports on NEMO-ID patients (not including XR-MSMD patients), we compared the phenotypes of our patient with those of other NEMO-ID patients (Fig. 1 and Table 2). At present, there are five published NEMO-ID variants [7, 13,14,15,16,17,18,19]. All of these patients suffered from recurrent bacterial infections mostly localized in the lower respiratory tract. Sepsis and bacteremia were present in 80% of patients while sinusitis and/or otitis were only present in 20%. SPAD was observed in all patients, which is in line with the clinical presentation of EDA-ID causing hypomorphic NEMO mutations [7]. Defects in immunoglobulin production likely depend on the location of the mutation in the NEMO protein. Mutations affecting the N-terminus of NEMO and located in the first α-helical domain lead to decreased production of immunoglobulins, with the p.Met38fs*48 mutation leading to hyper-IgM and the p.Glu57Lys mutation to hypogammaglobulinemia [13, 14, and this study]. Mutations in the first coiled-coil domain, Δ271–276, or the absence of exon 9 was associated with normal immunoglobulin production [15,16,17,18,19]. Deletion of exon 9 was associated with a hyper-IgA phenotype [17,18,19]. NK cell cytotoxicity has been evaluated in two of the published NEMO-ID patients and was impaired in both cases. EDA-ID patients with IKBKG mutations also display impaired NK cell cytotoxicity [7, 16,17,18,19]. NK cell cytotoxicity was not evaluated in our patient. In patients with NEMO-ID, the TNF-R- and IL1R- signaling pathways are most likely to be affected, both in PBMCs and in skin-derived fibroblasts.
To investigate the occurrence of other potentially pathogenic genetic variants, WES was performed. The joint genotyping on a sample from the mother and the son generated 119.150 variants. After extensive filtration and prioritization of data, mainly based on pathogenicity and allele frequency [21], we obtained two potential pathogenic mutations in addition to p.Glu57Lys NEMO variant (Table 3); a de novo heterozygous variant in C7 (rs121964920) and a heterozygous inherited variant in CCDC40 (rs762938274). Both variants are considered to be of unknown significance due to non-corresponding inheritance pattern (autosomal recessive C7 deficiency [29] and primary ciliary dyskinesia, respectively [30]) and allele frequency in the Caucasian population (0.0028 and 0.0044, respectively [31]). In addition, a variant in TNFRSF13B (p.Cys104Arg, rs34557412, associated with common variable immunodeficiency [32, 33]) was identified in the mother but was not inherited by her son.
Discussion
We report a male patient with a disease-causing variant in IKBKG. As an infant, he suffered from an episode of otitis media that evolved into a bilateral mastoiditis. The immunological work up was performed because of the severity of the mastoiditis and the uncommon cause (Pseudomonas species).
Specific (polysaccharide) antibody deficiency and low switched memory B cell subsets were noticed. Although immunoglobulin levels appeared normal at the age of 8 months, an important decrease was observed a few months later for which immunoglobulin substitution therapy was initiated. The initial normal levels of immunoglobulins could have been due to residual maternal IgG. After initiation of IVIG therapy at the age of 2 years, the child has been doing well and remains infection-free. This mild clinical course is probably related to the initiation of appropriate therapy at a very young age. We hypothesize that the infection occurred as a result of hypogammaglobulinemia and a defective polysaccharide antibody response.
This is the second description of a patient with a hypomorphic p.Glu57Lys NEMO missense variant. The first patient was diagnosed with EDA-ID, caused by this IKBKG mutation and a coincident p.Arg156Cys missense mutation in the EDA gene (ectodysplasin A) [27]. EDA is the membrane-bound signaling molecule of the TNF superfamily that interacts with the EDA-receptor [34]. Based on the association of both mutations in one patient, clear conclusions about the phenotype caused by p.Glu57Lys could not be made. Interestingly, this patient also had low IgG and IgA levels and impaired pneumococcal antibody responses [27]. The p.Glu57Lys variant has also been associated with a mild form of IP [4]. The reported female IP patient exhibited only dermatological defects limited to the upper and lower limbs. In addition, she exhibited non-random but incomplete X-inactivation skewing at the time of diagnosis. Her mother also presented mild dermatological signs, compatible with familial IP [4]. Our patient’s mother was heterozygous for the variant and suffered from recurrent throat infections without signs of IP. There was no skewed X-inactivation, in agreement with the “mild” nature of the variant.
The p.Glu57Lys substitution affects the N-terminal sequence of the NEMO protein. Only 3% of hypomorphic IKBKG mutations are situated within the region coding for amino acid 50 to 120, containing the α-helical coiled-coil domain [7]. Deletion of this domain leads to reduced NEMO dimerization and NEMO-IKK-α/β interaction [35, 36]. Stabilization of the NEMO dimer is mediated by hydrophobic interactions at the N- and C-terminus [36]. The highly conserved Glu57 residue is one of the amino acids that mediate this interaction at the N-terminus [36]. Inside the domain, the region containing amino acids 57–69 is important for interaction with TRAF6 [37]. TRAF6 is a E3 ligase responsible for NEMO polyubiquitination and participates in several signaling pathways controlling immunity, osteoclastogenesis, skin development, and brain functions. Experimental introduction of the p.Glu57Lys variant in mouse embryonic fibroblasts resulted in impaired TRAF6 binding through defective direct interaction [37]. This led to non-functional IL-1β signaling in mouse embryonic fibroblasts but did not affect activation of the NF-κB pathway after TNF-α stimulation [37]. It has also been reported that NF-κB luciferase activity after LPS stimulation in Nemo−/− murine pre-B lymphocytes supplemented with p.Glu57Lys NEMO is similar to NF-κB luciferase activity in Nemo−/− murine pre-B lymphocytes supplemented with wild-type NEMO, indicating that TLR4 signaling was not influenced by the mutation [4]. Although there was no specific signaling defect in skin-derived fibroblasts in our patient, transduction of p.Glu57Lys NEMO in SV40-transformed NEMO−/− fibroblasts revealed a slight but significant decrease of IL6 induction upon stimulation with IL-1β and TNF-α. Taken together, the in vitro findings and the clinical presentation of the patient suggest that NEMO residue 57 is not required for EDA-receptor- and TNF-R-mediated function but is important for full IL-1R signaling and immunoglobulin and specific antibody production.
Nevertheless, linking genotype to phenotype (especially for single nucleotide polymorphisms) is challenging in the absence of large family or population studies. The allele frequencies of p.Asp113Asn (rs179363896), p.Val146Gly, and p.Cys417Arg are not known in public databases due to interference with the IKBKG pseudogene [31]. Still, different in silico pathogenicity prediction scores indicate that p.Asp113Asn is less likely to cause disease in comparison to the other 3 variants, which is in agreement with results obtained in our functional in vitro studies. The allele frequency of p.Glu57Lys (rs148695964) is 0,001 (ExAC database) in the European population [31]. Given this high allele frequency, we hypothesize that this allele has an incomplete or reduced penetrance. Thus, the allele can have an immunological phenotype without clinical expressivity. An alternative hypothesis is the polygenic hypothesis. It can be that the variant is a mild modifier and not solely responsible for the phenotype. In order to test this, we performed WES. WES revealed no other known pathogenic PID variants. However, it remains possible that additional modifier genes (incl. genetic variations not identified through WES), environmental factors, or epigenetic mechanisms underlie the clinical phenotype observed in our patient [38]. Evaluation of additional patients with the same IKBKG variant might provide more information.
Conclusion
The combination of increased susceptibility to bacterial infections (particularly pneumonia), specific antibody deficiency, impaired NF-κB signaling (mostly after IL-1R or TNF-R activation), and impaired NK function should trigger physicians to investigate alterations in the IKBKG gene even without the presence of EDA. In this study, we investigated the pathogenic impact of the p.Glu57Lys IKBKG variant in a patient suspected to suffer from NEMO-ID. We demonstrated that p.Glu57Lys leads to specific immunological defects in vitro. No other pathogenic PID variants were identified through WES. As rare polymorphisms have been described in IKBKG (incl. p.Asp113Asn) and polygenic inheritance remains an option in our presented case, our study emphasizes the need for thorough functional and genetic evaluation when encountering and interpreting suspected disease-causing NEMO-ID variants.
References
Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol. 2000;18:621–63.
Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. 2008;132:344–62.
Scheidereit C. IκB kinase complexes: gateways to NF-κB activation and transcription. Oncogene. 2006;25:6685–705.
Fusco F, Bardaro T, Fimiani G, Mercadante V, Miano MG, Falco G, et al. Molecular analysis of the genetic defect in a large cohort of IP patients and identification of novel NEMO mutations interfering with NF-kappaB activation. Hum Mol Genet. 2004;13:1763–73.
Uzel G. The range of defects associated with nuclear factor kappaB essential modulator. Curr Opin Allergy Clin Immunol. 2005;5:513–8.
Courtois G, Gilmore TD. Mutations in the NF-κB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–43.
Hanson EP, Monaco-Shawver L, Solt LA, Madge LA, Banerjee PP, May MJ, et al. Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allergy Clin Immunol. 2008;122:1169–77.
Fusco F, Pescatore A, Conte MI, Mirabelli P, Paciolla M, Esposito E, et al. EDA-ID and IP, two faces of the same coin: how the same IKBKG/NEMO mutation affecting the NF-κB pathway can cause immunodeficiency and/or inflammation. Int Rev Immunol. 2015;34:445–59.
Boisson B, Puel A, Picard C, Casanova JL. Human IκBα gain of function: a severe and syndromic immunodeficiency. J Clin Immunol. 2017;37:397–412.
Frans G, Meyts I, Picard C, Puel A, Zhang SY, Moens L, et al. Addressing diagnostic challenges in primary immunodeficiencies: laboratory evaluation of Toll-like receptor- and NF-κB-mediated immune responses. Crit Rev Clin Lab Sci. 2014;51:112–23.
Rosenzweig SD, Holland SM. Defects in the interferon-gamma and interleukin-12 pathways. Immunol Rev. 2005;203:38–47.
Filipe-Santos O, Bustamante J, Haverkamp MH, Vinolo E, Ku CL, Puel A, et al. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med. 2006;203:1745–59.
Puel A, Reichenbach J, Bustamante J, Ku CL, Feinberg J, Döffinger R, et al. The NEMO mutation creating the most-upstream premature stop codon is hypomorphic because of a reinitation of translation. Am J Hum Genet. 2006;78:691–701.
Niehues T, Reichenbach J, Neubert J, Gudowius S, Puel A, Horneff G, et al. Nuclear factor kappaB essential modulator-deficient child with immunodeficiency yet without anhidrotic ectodermal dysplasia. J Allergy Clin Immunol. 2004;114:1456–62.
Ku CL, Dupuis-Girod S, Dittrich AM, Bustamante J, Santos OF, Schulze I, et al. NEMO mutations in 2 unrelated boys with severe infections and conical teeth. Pediatrics. 2005;115:615–9.
Salt BH, Niemela JE, Pandey R, Hanson EP, Deering RP, Quinones R, et al. IKBKG (nuclear factor-κB essential modulator) mutation can be associated with opportunistic infection without impairing Toll-like receptor function. J Allergy Clin Immunol. 2008;121:976–82.
Orange JS, Levy O, Brodeur SR, Krzewski K, Roy RM, Niemela JE, et al. Human nuclear factor kappa B essential modulator mutation can result in immunodeficiency without ectodermal dysplasia. J Allergy Clin Immunol. 2004;114:650–6.
Orange JS, Jain A, Ballas ZK, Schneider LC, Geha RS, Bonilla FA. The presentation and natural history of immunodeficiency caused by nuclear factor kappaB essential modulator mutation. J Allergy Clin Immunol. 2004;113:725–33.
Dai YS, Liang MG, Gellis SE, Bonilla FA, Schneider LC, Geha RS, et al. Characteristics of mycobacterial infection in patients with immunodeficiency and nuclear factor-kappaB essential modulator mutation, with or without ectodermal dysplasia. J Am Acad Dermatol. 2004;51:718–22.
Tuerlinckx D, Vermeulen F, Pékus V, de Bilderling G, Glupczynski Y, Collet S, et al. Optimal assessment of the ability of children with recurrent respiratory tract infections to produce anti-polysaccharide antibodies. Clin Exp Immunol. 2007;149:295–302.
Shirkani A, Shahrooei M, Azizi G, Rokni-Zadeh H, Abolhassani H, Farrokhi S, et al. Novel mutation of ZAP-70-related combined immunodeficiency: first case from the National Iranian Registry and Review of the Literature. Immunol Investig. 2017;46:70–9.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–9.
Smahi A, Courtois G, Rabia SH, Doffinger R, Bodemer C, Munnich A, et al. The NF-kappaB signalling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet. 2002;11:2371–5.
Ibrahimi A, Vande Velde G, Reumers V, Toelen J, Thiry I, Vandeputte C, et al. Highly efficient multicistronic lentiviral vectors with peptide 2A sequences. Hum Gene Ther. 2009;20:845–60.
Devora GA, Sun L, Chen Z, van Oers NS, Hanson EP, Orange JS, et al. A novel missense mutation in the nuclear factor-κB essential modulator (NEMO) gene resulting in impaired activation of the NF-κB pathway and a unique clinical phenotype presenting as MRSA subdural empyema. J Clin Immunol. 2010;30:881–5.
Keller MD, Petersen M, Ong P, Church J, Risma K, Burham J, et al. Hypohidrotic ectodermal dysplasia and immunodeficiency with coincident NEMO and EDA mutations. Front Immunol. 2011;2:61.
Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535–43.
Rameix-Welti MA, Régnier CH, Bienaimé F, Blouin J, Schifferli J, Fridman WH, et al. Hereditary complement C7 deficiency in nine families: subtotal C7 deficiency revisited. Eur J Immunol. 2007;37:1377–85.
Becker-Heck A, Zohn IE, Okabe N, Pollock A, Lenhart KB, Sullivan-Brown J, et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat Genet. 2011;43:79–84.
Exome Aggregation Consortium (ExAC), Cambridge, MA, USA. Website: https://ExAC.broadinstitute.org. Accessed on: 5 Aug 2016.
Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37:829–34.
Freiberger T, Ravčuková B, Grodecká L, Pikulová Z, Stikarovská D, Pešák S, et al. Sequence variants of the TNFRSF13B gene in Czech CVID and IgAD patients in the context of other populations. Hum Immunol. 2012;73:1147–54.
Cui CY, Schlessinger D. EDA signaling and skin appendage development. Cell Cycle. 2006;5:2477–83.
Marienfeld RB, Palkowitsch L, Ghosh S. Dimerization of the I kappa B kinase-binding domain of NEMO is required for tumor necrosis factor alpha-induced NF-kappa B activity. Mol Cell Biol. 2006;26:9209–19.
Rushe M, Silvian L, Bixler S, Chen LL, Cheung A, Bowes S, et al. Structure of a NEMO/IKK-associating domain reveals architecture of the interaction site. Structure. 2008;16:798–808.
Gautheron J, Pescatore A, Fusco F, Esposito E, Yamaoka S, Agou F, et al. Identification of a new NEMO/TRAF6 interface affected in incontinentia pigmenti pathology. Hum Mol Genet. 2010;19:3138–49.
Bogaert DJ, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet. 2016;53:575–90.
Acknowledgments
We thank Jean-Laurent Casanova for providing NEMO-deficient SV40 transformed fibroblasts. We are indebted to the patient and his parents.
Funding
This work was supported by a GOA grant from the Research Council of the KU Leuven, Belgium. IM is supported by a KOF mandate of the KU Leuven, Belgium.
Author information
Authors and Affiliations
Contributions
GF drafted the manuscript and conducted experiments. JVDWTB characterized the immune deficiency and coordinates the clinical care of the patient. LM, RG, and GW conducted experiments. HRZ, MC-A, and MS performed whole exome sequencing and bioinformatics analysis. XB supervised the routine laboratory immunology work up and TLR testing. IM and XB designed the study and finalized the manuscript. Each author has critically revised the final version of the manuscript and has read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflicts of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
The study was performed in accordance with the 1964 Helsinki declaration and its later amendments. Informed consent was obtained for genetic analysis and report of the case. The study was approved by the Ethics Committee of UZ Leuven.
Electronic supplementary material
ESM 1
(DOCX 20 kb)
Rights and permissions
About this article

Cite this article
Frans, G., van der Werff Ten Bosch, J., Moens, L. et al. Functional Evaluation of an IKBKG Variant Suspected to Cause Immunodeficiency Without Ectodermal Dysplasia. J Clin Immunol 37, 801–810 (2017). https://doi.org/10.1007/s10875-017-0448-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-017-0448-9