
Abstract
Purpose
Anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID) is caused by mutations in the NF-κB essential modulator (NEMO) or NF-κB inhibitor, alpha (IKBA) genes. A heterozygous NEMO mutation causes incontinentia pigmenti (IP) in females, while a hemizygous hypomorphic mutation of NEMO causes EDA-ID in males. In general, immunodeficiency is not shown in IP patients. Here, we investigated two female patients with IP and immunodeficiency.
Methods
The patients were initially suspected to have IRAK4 deficiency and Mendelian susceptibility to mycobacterial disease, respectively, because of recurrent pneumonia with delayed umbilical cord detachment or disseminated mycobacterial infectious disease. We measured tumor necrosis factor (TNF)-α production and performed mutation screening.
Results
The TNF-α production from lipopolysaccharide (LPS)-stimulated CD14-positive cells was partially defective in both female patients. A genetic analysis showed them to carry the heterozygous NEMO mutations c.1167_1168insC or c.1192C>T. Although NEMO mutations in IP patients are typically eliminated by X-inactivation skewing, an analysis of cDNA obtained from the somatic cells of the patients showed the persistence of these mutations in peripheral blood mononuclear cells and peripheral granulocytes. A NF-κB reporter gene analysis using NEMO-deficient HEK293 cells showed the loss of NF-κB activity in these NEMO mutants, while the NF-κB protein expression levels by the NEMO mutants were consistent with those of wild-type NEMO.
Conclusions
The delayed skewing of the mutant allele may be responsible for the observed innate immune defect in these patients. The detection of LPS unresponsiveness is suitable for identifying female IP patients with immunodeficiency.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Genetic defects in the signaling pathways of the Toll-like receptor (TLR) and interleukin (IL)-1 families are known to cause innate immune deficiency syndromes such as IL-1 receptor-associated kinase 4 (IRAK4) deficiency (MIM607676) and anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID; MIM300291 and 612132) [1]. EDA-ID is caused by mutations in the NF-κB essential modulator (NEMO) or NF-κB inhibitor, alpha (IKBA) genes. NEMO deficiency is an X-linked inheritance disease. A hemizygous hypomorphic mutation of NEMO causes EDA-ID in males, while a heterozygous NEMO mutation causes incontinentia pigmenti (IP; MIM308300) in females. NEMO is also one of the causative genes for Mendelian susceptibility to mycobacterial diseases (MSMD) [2, 3]. Several screening systems have been developed for the rapid diagnosis of IRAK4 deficiency, including using flow cytometry for the detection of unresponsiveness to lipopolysaccharide (LPS) as a representative TLR ligand. EDA-ID also shows partial LPS unresponsiveness [4,5,6].
IP, also known as Bloch–Sulzberger syndrome, is a progressive skin disorder that involves four characteristic sequential cutaneous disease stages: perinatal inflammatory vesicles, verrucous patches, a distinctive pattern of hyperpigmentation, and dermal scarring. IP patients also have abnormalities of the hair, nails, teeth, eyes, and central nervous system. Approximately 80–90% of the IP patients have a common heterozygous large deletion of NEMO, known as Δexon 4–10. Other IP patients carry nonsense, frameshift, or missense mutations of NEMO [7].
Typically, female IP patients do not show immunodeficiency. Here, we present two female IP patients with immunodeficiency (IP-ID) who we identified using our rapid screening for the detection of lipopolysaccharide (LPS) unresponsiveness.
Methods
Patients
P1.
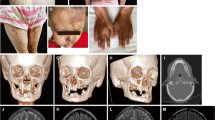
The patient is a 6-year-old female whose umbilical cord detachment was delayed (28 days after birth). She showed an atypical progress of IP with no skin rash during early infancy. At the age of 6 months, she developed a skin ulcer following the Bacillus Calmette–Guérin (BCG) vaccination, after which generalized pustules appeared. At the age of 1 year, her skin presentation gradually changed to linear and whorled nevoid hypermelanosis. The skin appearance in the patient at the age of 3 years is shown in Fig. 1a. Febrile episodes became frequent after the age of 4 months, and from 3 years of age, she had recurrent pneumonia. Nontypable Haemophilus influenzae was detected in her throat using a bacterial culture test, although her serum titer levels of antibodies against H. influenzae were sufficient owing to Hib vaccination. Her immunological parameters at the age of 3 years are shown in Table 1. After the start of prophylaxis with sulfamethoxazole/trimethoprim, the frequency of her infections decreased. Her family members have no symptoms suggestive of IP or immunodeficiency. She was initially suspected to have IRAK4 deficiency because of her recurrent infections, delay in umbilical cord detachment, and below-mentioned LPS unresponsiveness [4, 8].

Pedigrees of the families with heterozygous NEMO mutations and clinical manifestations in the affected patients. a Family tree of P1 and her skin appearance at the age of 3 years. b Family tree of P2, her facial appearance with continuous swelling around the lips at the age of 11 years, and the slight hyperpigmentation following the Blaschko line on her arm at the age of 14 years. T2 imaging and the corresponding histology, as well as dental panoramic radiography, are shown. This photography is included with permission from the patients’ parents
P2.
The patient is a 14-year-old female. No delay was seen in her umbilical cord detachment. At the age of 5 months, she received the BCG vaccination. At the age of 6 months, she developed a fever, a skin ulcer at the BCG inoculation site, an axillary lymphadenitis on the same side, and BCG dermatitis. After the administration of antibiotics for 10 days, only the axillary lymphadenopathy remained. At the age of 3 years, swelling appeared around her lips. This was diagnosed as granulomatous cheilitis, and disseminated BCG disease was suspected. At the age of 9 years, she received isonicotinic acid hydrazide (INH) for 5 months, after which her symptoms improved. However, after the completion of INH administration, her lip swelling relapsed. The patient’s facial appearance, with continuous swelling around the lips, at the age of 11 years is shown in Fig. 1b. T2 imaging showed a left axillary lymphadenopathy, and the corresponding histology showed necrotic tissue with a thin layer of epithelioid cells. Finally, after the extraction of her axillary lymph node and the administration of roxithromycin and tranilast for 3 and 5 months, respectively, she recovered. Mycobacterium tuberculosis was detected in her extracted axillary lymph node using a PCR analysis that could detect the M. tuberculosis complex, including Mycobacterium bovis. However, no additional analyses were conducted to identify the specific strain. Her immunological parameters at the age of 14 years are shown in Table 1. She was initially suspected to have MSMD. However, after genetic diagnosis, her history of skin disorders was disclosed, which led to an updated diagnosis of IP with immunodeficiency. At present, a slight hyperpigmentation following the Blaschko lines remains on her skin, and her left lower third molar tooth is defective. Her family members have no skin disorder symptoms suggestive of IP.
Cell Culture
Blood samples were provided from both patients and their parents, two 1-year-old typical IP patients (NEMO Δexon 4–10) diagnosed by a previously described method [9], an XL-EDA-ED patient (NEMO E391X), an IRAK4-deficient patient (IRAK4 c.118-123insA) [10], and age-matched healthy child controls. Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood with gradient centrifugation using Ficoll–Paque (GE Healthcare, Uppsala, Sweden). PBMCs were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum (FCS), l-glutamine (2 mmol/L), penicillin (100 U/mL), and streptomycin (100 μg/mL). PBMCs were seeded at a density of 106 cells/mL and cultured in the presence or absence of 100 ng/mL LPS from Escherichia coli 0127:B8 (Sigma-Aldrich, St. Louis, MO, USA) for 24 h in 24-well plates at 37 °C in a humidified atmosphere containing 5% CO2. Human embryonic kidney (HEK) 293 cells (purchased from the Japanese Collection of Research Bioresources, Osaka, Japan) were cultured in high glucose Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat-inactivated FCS (Sigma-Aldrich), penicillin (100 U/mL), and streptomycin (100 μg/mL).
Detection of TNF-α-Producing Cells Using Intracellular Cytokine Staining
The responsiveness to LPS was evaluated by the intracellular staining of CD14+ monocytes for the production of tumor necrosis factor (TNF)-α. Heparinized whole blood (0.5 mL) was added to 0.5 mL of RPMI-1640 medium and then incubated with or without 100 ng/mL ultra-pure LPS (InvivoGen, San Diego, CA, USA) in the presence of 10 μg/mL of brefeldin A (Sigma-Aldrich) for 4 h at 37 °C in a humidified atmosphere containing 5% CO2. Surface staining for CD14 using an IOTest fluorescein isothiocyanate-conjugated anti-CD14 antibody (Beckman Coulter, Brea, CA, USA) was performed at room temperature for 20 min. The blood samples were then fixed and permeabilized with IntraPrep™ Permeabilization Reagent (Beckman Coulter). Finally, the cells were incubated with IOTest TNFα-PE (Beckman Coulter) for 40 min and then analyzed with flow cytometry. The percentages of TNF-α-producing cells in the CD14+ cell population were evaluated. The proportion of TNF-α-positive cells stimulated with LPS was over 90% in the control subjects [4, 6, 11].
Analysis of Cytokine Levels with an Enzyme-Linked Immunosorbent Assay
Culture supernatants of PBMCs were centrifuged in test tubes to remove cells and stored at −80 °C until assayed. TNF-α and IL-12 p40/p70 concentrations were each measured using an ELISA Kit (Invitrogen) with a detection limit of 1.7 and 2.0 pg/mL, respectively. The statistical significance of differences between the patient and the age-matched control subjects was analyzed by a one-way ANOVA with Tukey’s multiple comparisons test using PRISM version 6.0 (GraphPad software, San Diego, CA, USA), and p values of <0.05 were considered statistically significant.
Mutation Analysis
Genomic DNA was extracted from peripheral blood using the QIAamp DNA Blood Mini Kit (Qiagen, Venlo, the Netherlands) according to the manufacturer’s instructions. DNA fragments of IRAK4, MYD88, TIRAP, NEMO, and IKBA were amplified using PCR and analyzed with the BigDye Terminator v3.1 Cycle Sequencing Kit and an Applied Biosystems 3730xl or 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Known pathogenic genes of MSMD (IFNGR1, IFNGR2, NEMO, IL12B, IL12RB1, STAT1, CYBB, IRF8, and ISG15) were analyzed using MiSeq (Illumina, San Diego, CA, USA), and the possible variants detected were confirmed using the Sanger sequencing method. DNA fragments of NEMO were amplified by long-range PCR using the forward primer 5′-GGTGAATTATCAGCATTCTG-3′ and the reverse primer 5′-AACAGCTGAAGCGTAAGGTG-3′, and then secondary PCR was performed for each exon because of the existence of a highly homologous pseudogene.
NEMO mRNA Analysis
The Oragene RNA collection kit (DNA Genotek Inc., Ottawa, Canada) was used to extract messenger RNA (mRNA) from saliva, and the QIAzol Lysis Reagent (Qiagen) was used to obtain mRNA from peripheral blood and urine. SuperScript III Reverse Transcriptase (Invitrogen) was then used to synthesize complementary DNA (cDNA) from the resulting mRNA. The regions of NEMO in which the mutations c.1167_1168insC and c.1192C>T are located were amplified by PCR, and the resulting DNA sequences were analyzed using a similar method to that used for the analysis of genomic DNA (described previously). The allele frequency of c.1192C>T in cDNA was estimated by real-time PCR analysis using a TaqMan SNP genotyping assay (Thermo Fisher Scientific, Waltham, MA, USA) according to the previously described method [12]. The allele frequency of c.1167_1168insC in cDNA was estimated by subcloning into pGEM-T (Promega, Fitchburg, WI, USA) and then performing a sequencing analysis, because this genotype was not thought to be suitable for analysis by TaqMan assay.
Vector Preparation
The cDNA encoding human NEMO tagged at the N-terminus with a c-myc-epitope (myc-NEMO) was cloned into plasmid pcDNA3.1+ (Invitrogen). NEMO mutants c.793insA (p.Q265TfsX19), c.1167_1168insC (p.E390RfsX5), c.1192C>T (p.P398S), and c.1217A>T (p.D406V) were generated using the KOD Plus Mutagenesis Kit (Toyobo Co., Osaka, Japan) according to the manufacturer’s instructions. Expression plasmids of TLR4 and its co-receptors (pUNO-hTLR4A-HA and pDUO2-hMD2/CD14) were purchased from InvivoGen. The NF-κB luciferase reporter vector (pGL4.32-luc2P/NF-kappaB-RE/Hygro) and the Renilla luciferase reporter vector (pGL4.74-hRluc/TK) were purchased from Promega. The IKKγ CRISPR/Cas9 KO plasmid (h) for disrupting NEMO was purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
NEMO-Deficient Cell Preparation
NEMO-deficient HEK293 cells were created by the CRISPR/Cas9 system. HEK293 cells were transfected with the IKKγ CRISPR/Cas9 KO plasmid (h) by Nucleofector II and the Cell Line Nucleofector kit V (Lonza, Basel, Switzerland) using the program Q-001. Single cell clones adjusted by the limited dilution method were then cultured. Successful NEMO knockout was verified by the detection of a DNA fragment of the target site and the direct sequencing of genomic DNA from candidate clones along with the detection of endogenous protein expression with an immunoblot. The below-mentioned NF-κB reporter gene activity assay was also performed after TNF-α, IL-18, or LPS stimulation (Fig. S1).
NF-κB Reporter Gene Activity
NEMO-deficient HEK293 cells were transfected with 50 ng per well of pcDNA3.1+ control vector or pcDNA3.1+ myc-NEMO (wild type or mutant) in 96-well plates using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. TLR4 and MD2-CD14 (for LPS stimulation) or IL-18Rβ (for IL-18 stimulation) expression plasmids, the NF-κB luciferase reporter, and the Renilla luciferase reporter vectors were co-transfected. After transfection, the cells were incubated for 24 h and then stimulated with 100 ng/mL LPS, 10 ng/mL IL-18, or 10 ng/mL TNF-α (R&D, Minneapolis, MN, USA) for 6 h. The mature form of human IL-18 was prepared using an E. coli expression system according to previously reported methods [13]. Luciferase reporter activity was analyzed using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. The statistical significance of differences in luciferase activity between wild-type and mutant gene activity in the NF-κB reporter assays was analyzed using a one-way ANOVA with Tukey’s multiple comparisons test, and p values of <0.05 were considered statistically significant.
Western Blot Analysis
To detect protein expression, NEMO-deficient HEK293 cells were seeded in six-well plates at a density of 2 × 105/mL and transfected with 1 μg of pcDNA3.1+ control vector or pcDNA3.1+ myc-NEMO (wild type or mutant) using Lipofectamine 2000 according to the manufacturer’s instructions. After 48 h of incubation, the cells were harvested and lysed using hypotonic lysis buffer (pH 7.5: 10 mM Tris-HCl, 10 mM NaCl, 0.5% Triton-X100, and 10 mM EDTA) containing a protease inhibitor mix (Roche Applied Science, Indianapolis, IN, USA). All extracts were adjusted to contain equal amounts of total cellular proteins, as determined using the Bradford method. The supernatants of cell lysates were separated by electrophoresis on sodium dodecyl sulfate polyacrylamide gels and transferred to nitrocellulose membranes using an iBlot Gel Transfer Device (Invitrogen). NEMO and β-actin proteins were detected with an anti-myc antibody (Invitrogen), purified mouse anti-IKKγ (BD Pharmingen, Franklin Lakes, NJ, USA), or anti-β-actin antibody (Sigma-Aldrich) followed by incubation with an anti-mouse IgG–horseradish peroxidase conjugate (Promega).
Results
The percentages of TNF-α-producing cells in the CD14+ population of LPS-stimulated cells in both Patient 1 (P1) (57.2%) and Patient 2 (P2) (70.8%) were partially decreased. The IRAK4-deficient patient was almost completely defective in TNF-α production (29.7%), and the XL-EDA-ID male patient showed a partial reduction in TNF-α production (62.6%), while the two typical IP patients showed a normal response (97.3 or 98.5%). A representative result from this experiment is shown in Fig. 2. The TNF-α and IL-12 p40/p70 production levels from LPS-stimulated PBMCs of the IRAK4-deficient patient, the XL-EDA-ID patients, and the two present cases were also significantly decreased compared with those of the healthy control subjects (Fig. S2a and S2b).

Evaluation of the response against LPS. TNF-α production of LPS-stimulated CD14-positive cells was measured using flow cytometry
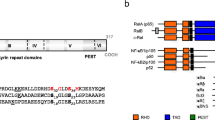
A genetic analysis revealed pathogenic NEMO mutations in the present cases. The heterozygous mutation c.1167_1168insC (E390RfsX5) was detected in the genomic DNA of P1, and the heterozygous mutation c.1192C>T (p.P398S) was detected in the genomic DNA of P2 (Fig. 3a). No mutations were detected in the genomic DNA of the patients’ parents. The peaks of these mutant alleles were detected at similar levels to those of wild alleles with the genomic DNA from the cells of whole blood, saliva, and urine. It should be noted that P398S is a novel mutation of NEMO.

Mutations identified in P1 and P2. a Heterozygous mutations c.1167_1168insC in P1 and c.1192C>T in P2 were detected in genomic DNA from peripheral blood. b Analyses of mutations in cDNA from somatic cells. cDNA samples were obtained from P1 at the age of 6 years and P2 at the age of 14 years
Although NEMO mutations in IP patients are typically eliminated by X-inactivation skewing, an analysis of the cDNA from P1 obtained at 6 years of age revealed the presence of the c.1167_1168insC mutation in her PBMCs and peripheral granulocytes. Additionally, a small mutation peak was also seen in the cDNA from her urine but not in the cDNA samples obtained from her saliva. Furthermore, an analysis of the cDNA from P2 obtained at 14 years of age revealed that the expression of the mutated allele (T allele) was clearly dominant in her PBMCs, peripheral granulocytes, and saliva (Fig. 3b). The mutant allele frequencies in the cDNA of PBMCs, peripheral granulocytes, urine, and saliva from P1 were 70 (21/30), 45.2 (14/31), 51.7 (15/29), and 0% (0/34), respectively. Those of P2 were 76.1 ± 1.04, 86.8 ± 2.20, 63.9 ± 1.02, and 88.8 ± 0.64%, respectively.
An NF-κB reporter gene analysis using NEMO-deficient HEK293 cells created by the CRISPR/Cas9 system showed a loss of activity of the NEMO mutants Q265TfsX19, E390RfsX5, P398S, and D406V compared with the wild type. The three mutants E390RfsX5, P398S, and D406V showed significantly weaker reporter activity than the wild type, and Q265TfsX19 showed a complete lack of response against LPS (Fig. 4a). The data shown are the mean ± SD of triplicate wells and are representative of three independent experiments with similar results.

In vitro functional analysis of NEMO variants. a NF-κB reporter gene analysis measuring the activity of NEMO mutants using NEMO-deficient HEK293 cells. The data shown are the mean ± SD of triplicate wells and are representative of three independent experiments with similar results. b Amounts of protein expression of the mutants as measured by a Western blot
The amounts of protein expression for myc-tagged NEMO mutants E390RfsX5, P398S, and D406V were similar to the wild-type expression levels, but that of Q265TfsX19 was decreased (Fig. 4b).
Discussion
Because P1 had recurrent infections and a delay in her umbilical cord detachment and P2 had no immunodeficiency symptoms other than disseminated mycobacterial infectious disease, these cases were initially suspected to have IRAK4 deficiency and MSMD, respectively. Although IP patients typically do not show immunodeficiency, heterozygous pathogenic NEMO mutations were identified in these two patients. Both P1 and P2 showed partial LPS unresponsiveness, but neither had any obvious reduction in T cell proliferation or in the production of immunoglobulins (Table 1).
Previously, only two suspected cases of female patients with IP and immunodeficiency (IP-ID) have been described. Kosaki et al. reported the first suspected case of IP-ID [14]. This individual had a heterozygous c.1167_1168insC NEMO mutation, which is also carried by the present case P1, and showed recurrent infections, including respiratory tract infections, cervical lymphadenitis, otitis media, cellulitis, and soft tissue abscesses at 4 years of age. She died at 11 years of age after cardiac catheterization for the evaluation of her pulmonary hypertension derived from secondary bronchiectasis. The patient reported by Martinez-Pomar et al. was the second possible case of IP-ID, with a heterozygous c.793insA NEMO mutation [15]. She suffered transient immunodeficiency with a low T cell proliferation and CD40L expression. Her immunodeficiency symptoms disappeared at the age of 4 years after X-inactivation skewing was completed. Furthermore, one familial case with IP presenting as intestinal Behçet’s disease with a heterozygous NEMO missense mutation, D406V, has also been reported [11]. Interestingly, although a mild partial unresponsiveness against LPS was determined in the proband of this family as well as in the present study, the clinical presentation was Behçet’s disease rather than immunodeficiency. Of note, the mother of the proband of this family has not yet undergone remission. Meanwhile, a recent report summarizing the clinical data on a quite large cohort of IP patients provided clear evidence of the disease heterogeneity, and it indicated that a genotype–phenotype correlation is not always observed in IP. Intriguingly, 11.7% of the IP patients showed recurrent infections [16]. Thus, immunodeficiency in IP seems to be a rare but possible symptom of this condition.
Although the present two patients are 6 and 14 years old, their partial unresponsiveness against LPS has been retained. In general, mutated alleles of NEMO would be expected to be eliminated by X-inactivation skewing at an early stage [11]. Indeed, the two typical IP patients with NEMO Δexon 4–10, which is a common heterozygous large deletion of NEMO (null allele), that were included in this study showed a completely normal response against LPS and had no symptoms of immunodeficiency at 1 year of age. However, in the cases presented here, the expression of the mutated alleles was retained in all of the analyzed cells from P1, except for the cells derived from saliva, and in the all of the analyzed cells from P2 (Fig. 3). The abovementioned IP patient with Behçet’s disease as well as her mother also showed a delay in X-inactivation skewing [11]. Thus, we consider that such a delay within immune cells might be the cause of LPS unresponsiveness. In fact, our collaborators previously showed the expression of mutant NEMO in various blood cell lineages of the mothers of XL-EDA-ID patients, and their monocytes with mutant NEMO expression were also retained at various proportions [17]. The reason for this is unknown, but we explored one of the possible explanations in our in vitro assay of the NEMO-deficient cell line, the results of which demonstrated that NF-κB activity and protein expression were retained by the NEMO mutants. Because NF-κB is associated with cell survival, null alleles can therefore cause more rapid skewing than hypomorphic mutations, which are less strongly selected against and persist for longer. However, the cells with hypomorphic mutant expression might be responsible for the delay in skewing. Additionally, mutated alleles in immune cells might be dominantly inclined by an initially random lyonization. The existence of several symptomatic female patients of X-linked recessive inherited diseases can be explained by this theory [18]. Here, P2 had a dominantly mutated allele expressed by blood cells and cells from saliva. Therefore, we conclude that patients with a part of hypomorphic mutations such as E390RfsX5, P398S, or D406V might be expected to occasionally show IP-ID.
In this study, we revealed that the novel disease concept IP-ID appears to be caused by a hypomorphic mutation of NEMO and a delay in X-inactivation skewing in immune cells. These rare cases can be detected using an evaluation of LPS unresponsiveness.
References
Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014;5:162.
Doffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277–85.
Filipe-Santos O, Bustamante J, Haverkamp MH, Vinolo E, Ku CL, Puel A, et al. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med. 2006;203:1745–59.
Takada H, Yoshikawa H, Imaizumi M, Kitamura T, Takeyama J, Kumaki S, et al. Delayed separation of the umbilical cord in two siblings with interleukin-1 receptor-associated kinase 4 deficiency: rapid screening by flow cytometer. J Pediatr. 2006;148:546–8.
Mizukami T, Obara M, Nishikomori R, Kawai T, Tahara Y, Sameshima N, et al. Successful treatment with infliximab for inflammatory colitis in a patient with X-linked anhidrotic ectodermal dysplasia with immunodeficiency. J Clin Immunol. 2012;32:39–49.
Ohnishi H, Miyata R, Suzuki T, Nose T, Kubota K, Kato Z, et al. A rapid screening method to detect autosomal-dominant ectodermal dysplasia with immune deficiency syndrome. J Allergy Clin Immunol. 2012;129:578–80.
Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature. 2000;405:466–72.
Takada H, Ishimura M, Takimoto T, Kohagura T, Yoshikawa H, Imaizumi M, et al. Invasive bacterial infection in patients with interleukin-1 receptor-associated kinase 4 deficiency: case report. Medicine (Baltimore). 2016;95:e2437.
Hsiao PF, Lin SP, Chiang SS, Wu YH, Chen HC, Lin YC. NEMO gene mutations in Chinese patients with incontinentia pigmenti. J Formos Med Assoc. 2010;109:192–200.
Yoshikawa H, Watanabe S, Imaizumi M. Successful prevention of severe infection in Japanese siblings with interleukin-1 receptor-associated kinase 4 deficiency. J Pediatr. 2010;156:168.
Takada H, Nomura A, Ishimura M, Ichiyama M, Ohga S, Hara T. NEMO mutation as a cause of familial occurrence of Behçet’s disease in female patients. Clin Genet. 2010;78:575–9.
Lo HS, Wang Z, Hu Y, Yang HH, Gere S, Buetow KH, et al. Allelic variation in gene expression is common in the human genome. Genome Res. 2003;13:1855–62.
Kato Z, Jee J, Shikano H, Mishima M, Ohki I, Ohnishi H, et al. The structure and binding mode of interleukin-18. Nat Struct Biol. 2003;10:966–71.
Kosaki K, Shimasaki N, Fukushima H, Hara M, Ogata T, Matsuo N. Female patient showing hypohidrotic ectodermal dysplasia and immunodeficiency (HED-ID). Am J Hum Genet. 2001;69:664–6.
Martinez-Pomar N, Munoz-Saa I, Heine-Suner D, Martin A, Smahi A, Matamoros N. A new mutation in exon 7 of NEMO gene: late skewed X-chromosome inactivation in an incontinentia pigmenti female patient with immunodeficiency. Hum Genet. 2005;118:458–65.
Fusco F, Paciolla M, Conte MI, Pescatore A, Esposito E, Mirabelli P, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014;9:93.
Kawai T, Nishikomori R, Izawa K, Murata Y, Tanaka N, Sakai H, et al. Frequent somatic mosaicism of NEMO in T cells of patients with X-linked anhidrotic ectodermal dysplasia with immunodeficiency. Blood. 2012;119:5458–66.
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51:1229–39.
Acknowledgments
Patient samples were kindly provided by Dr. Togashi (Miyagi Children’s Hospital) and Dr. Yamamoto (Teikyo University Hospital). Hib antibody titers were evaluated by Dr. Akeda (Osaka University). We also thank M. Yamamoto, H. Kiriyama, and K. Kasahara (Gifu University) for their technical help. This work was supported by the Health and Labor Science Research Grants for Research on Intractable Diseases from the Ministry of Health, Labour and Welfare of Japan [grant numbers 17933688 and 17933299].
Author information
Authors and Affiliations
Contributions
H.O. designed the study, collected the data, contributed to data interpretation, wrote the manuscript, and approved the final manuscript. Y.K., T.T., Mi.N., Ma.N., O.O., and K.O. collected the data, contributed to data interpretation, and approved the final manuscript. N.K., T.K., and T.F. contributed to data interpretation and approved the final manuscript.
Corresponding author
Ethics declarations
Informed consent was obtained from the patients and their families in accord with the Declaration of Helsinki and based on the protocol approved by the Internal Review Board of Gifu University.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Electronic supplementary material
ESM 1
(DOCX 1142 kb).
Rights and permissions
About this article

Cite this article
Ohnishi, H., Kishimoto, Y., Taguchi, T. et al. Immunodeficiency in Two Female Patients with Incontinentia Pigmenti with Heterozygous NEMO Mutation Diagnosed by LPS Unresponsiveness. J Clin Immunol 37, 529–538 (2017). https://doi.org/10.1007/s10875-017-0417-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-017-0417-3