
Abstract
Purpose
Chronic granulomatous disease (CGD) is an inherited phagocytic disorder characterized by recurrent infections with usually catalase-positive organisms. Infections in CGD from developing countries are expected to be different from those in the Western countries. We report the profile of infections in children diagnosed with CGD from a tertiary care center in North India.
Methodology
Case records of children diagnosed with CGD at Pediatric Immunodeficiency Clinic, Advanced Pediatrics Centre, Postgraduate Institute of Medical Education and Research, Chandigarh, India, from August 1993 to April 2016 (23 years) were analyzed.
Results
Thirty-eight children were diagnosed to have CGD. Median follow-up of patients was 2 years (interquartile range 0.75, 6.0). Staphylococcus aureus and Pseudomonas spp. were the two most common causative bacteria isolated. Aspergillus was the most common fungus isolated. The most common organ involved was the lung (94.7%). Liver abscesses were identified in 5 patients (13.2%), and 20 (52.6%) patients had lymphadenitis. Infections with Pseudomonas spp. were high in our cohort (15.7%) compared to the other studies. Infections with some unusual organisms (e.g., Fusarium dimerium and Chryseobacterium gleum) were also seen in our cohort. Children with X-linked CGD presented earlier and also had a greater number of infections as compared to autosomal recessive CGD.
Conclusions
Various socioeconomic factors coupled with the lack of awareness and paucity of readily available diagnostic facilities for primary immunodeficiencies accounted for a late clinical presentation with severe infections and increased mortality (28.9%) in our cohort. However, mortality was similar in X-linked and autosomal recessive CGD as was the number of fungal infections. The incidence of infections and mortality was significantly lower after initiation of antibacterial and antifungal prophylaxis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic granulomatous disease (CGD) is an inherited primary immunodeficiency disorder that results in defective phagocyte oxidative function [1]. Five different types of CGD have been described based on defects in one of the five proteins constituting the NADPH oxidase system. A defect in CYBB gene which encodes for gp91phox results in X-linked CGD. Defects in CYBA, NCF-1, NCF-2, and NCF-4 result in autosomal recessive CGD due to deficiency of gp21phox, p47phox, p67phox, and p40phox proteins, respectively [1, 2]. X-linked CGD (XL-CGD) is the most common type of CGD reported in the Western countries, whereas autosomal recessive CGD (AR-CGD) is the most common subtype in countries with the high rates of endogamous marriages.
The clinical phenotype is characterized by recurrent infections due to catalase-positive organisms such as Staphylococcus aureus and Aspergillus spp. [1]. Common clinical presentations include suppurative adenitis, deep-seated abscesses, pneumonia, and osteomyelitis. The use of prophylactic antimicrobials, interferon gamma (IFN), and bone marrow transplantation has resulted in a significant reduction in morbidity and mortality in CGD [3, 4]. Data on infection pattern and causative microorganisms provide added information to the CGD infection spectrum and help in early identification and management of CGD. Infections in CGD are likely to be persistent and usually require a prolonged duration of antimicrobial or antifungal therapy to clear the organisms [5]. However, the pattern, frequency, and severity of infections may vary among different subtypes [1, 5]. Identification of deleterious infections is of paramount importance to guide the empirical choice of antibiotics or antifungal therapy until confirmatory microbiological investigations are available.
Data regarding infections in CGD have predominantly emanated from developed countries [3, 5, 7–12], and literature from developing countries is scarce [13–18]. Infection profile from developing countries is expected to be different to that reported from the Western countries due to the difference in environmental and socioeconomic factors. Hence, the guidelines for the management of infectious complications in CGD cannot be extrapolated to developing nations. We previously reported 17 cases of CGD from our center in 2013 [18]. We have now diagnosed 21 new cases of CGD in the last 3 years and analyzed the infection pattern and mortality in all the patients.
Methodology
The study was conducted in the Pediatric Allergy Immunology Unit, Advanced Pediatrics Centre, Postgraduate Institute of Medical Education and Research, Chandigarh, India. In the last 23 years (August 1993–April 2016), we diagnosed 38 children with CGD. Data of all 38 patients were retrieved from the case records and files of the Pediatric Immunodeficiency Clinic. The study was approved by the Departmental Review Board. Diagnosis of CGD was based on the nitroblue tetrazolium test (NBT) alone in 10 cases, dihydrorhodamine (DHR) testing alone in 2 cases, and by both in 26 cases. Heparinized blood was used for the DHR assay by flow cytometry. Stimulation of the neutrophils was done by phorbol 12-myristate 13-acetate (PMA). DHR was also performed on the parents and siblings wherever necessary. b558 (gp91phox/p22phox) staining was carried out by flow cytometry in selected cases where suspicion of XL-CGD was based on clinical grounds. p47phox and p67phox staining by flow cytometry was performed in cases where autosomal recessive CGD was suspected. Genetic diagnosis of CGD was established in 25 out of the 38 children. Mutational analysis was performed at the National Defense Medical College, Saitama, Japan, and at the Department of Pediatrics and Adolescent Medicine, The University of Hong Kong, Hong Kong, after obtaining written informed consent from the parents.
Age at onset of infection, infection and microbiological pattern before and after diagnosis of CGD, duration of follow-up, and mortality data were analyzed in all 38 patients. Complete blood counts, immunoglobulin profile, blood cultures, chest radiograph, and ultrasound abdomen were performed in all cases. Computerized tomography (CT), fine needle aspiration or biopsy from tissue specimens, body fluid cultures, and fungal blood serology were performed wherein considered necessary on clinical grounds. Bacterial cultures were carried out by conventional methods until 2010, and matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS, Bruker Daltonics, Germany) analysis was used for identification of bacteria from 2010 onwards. Diagnosis of fungal infection was based on fungal cultures or histopathological evidence. Fungal serology was considered positive for antigen, when the galactomannan index (Aspergillus) was >1.0, whereas for antibody (Candida, Aspergillus fumigatus, Aspergillus flavus, Aspergillus niger), it was considered significant when the clear precipitin band was seen on immunodiffusion plate.
Data were analyzed by using SPSS version 21 software (SPSS, Inc., Chicago, IL).
Calculation of Stimulation Index by Flow Cytometry-Based Dihydrorhodamine Testing
Fluorescence of DHR was measured on the FL1 histogram. Percentage positivity and median fluorescence intensity (MFI) were taken in consideration for reporting. MFI of stimulated tube (C) was divided by MFI of the unstimulated tube (B) to calculate neutrophil oxidative index (NOI) or stimulation index (SI). A healthy control sample was simultaneously tested during each run for comparison.
Results
A total of 38 cases of CGD were diagnosed in the last 23 years. We had diagnosed only 17 cases of CGD until 2013 [18], and 21 additional new cases of CGD (55.3%) have been diagnosed in the last 3 years after that period. Median age of onset of infections was 5 months with interquartile range (IQR) between 2 and 21 months. Median age at diagnosis of CGD was 2 years (IQR 0.7, 4.5). Median delay in the diagnosis of CGD was 1.17 years (IQR 0.15, 2.34). Sixteen children (42.1%) had XL-CGD—13 of them were found to have a mutation in the CYBB gene, and 3 have been identified as X-linked based on b558 staining pattern on flow cytometry. Autosomal recessive form of CGD (AR-CGD) was identified in 18 children (47.4%)—8 were found to have a mutation in the NCF1 gene, 3 in the NCF2 gene, 1 of the patients was a sibling of a child with mutation in NCF1 gene, 2 children had an abnormal DHR, but no mutation has been identified in the CYBB gene, and 4 girls had an abnormal DHR who were presumed to have an AR-CGD (Suppl. Table S1). The type of CGD could not be identified in four patients (10.5%). One patient with NCF-2 mutation was lost to follow-up. Median follow-up of patients who survived the first infection at the time of diagnosis of CGD was 2 years (IQR 0.75, 6.0).
Microbiological Patterns
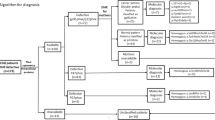
Bacteria were isolated during 34 infectious episodes, and majority of them were in XL-CGD group. Similarly, confirmatory evidence of fungal infection, either on histopathology or fungal culture positivity, was evident during 14 infectious episodes (Suppl. Table S2 and Fig. 1).

Organisms isolated in a north Indian cohort of 38 patients with chronic granulomatous disease (CGD). a Total microbiological isolates over a period of 23 years. b Microbiological isolates before the diagnosis of CGD and initiation of antibiotic prophylaxis. c Microbiological isolates after the diagnosis of CGD and initiation of antibiotic prophylaxis
After initiation of cotrimoxazole and itraconazole prophylaxis, there was a marked reduction in the incidence of culture-proven bacterial and fungal infections (Fig. 1). Except for an episode of Staphylococcus hemolyticus septicemia, there was no documented Staphylococcus infection after the initiation of prophylaxis. Glucocorticoids were used in four patients—two with colitis and one each with intestinal obstruction and hemophagocytic lymphohistiocytosis. Except for one patient (S.No. 28) who developed an Aspergillus pneumonia during oral prednisolone therapy, none of the others developed significant infections while on glucocorticoid therapy.
Mixed Infections
A patient was said to have mixed infection when more than one organism was isolated during an infectious episode. Mixed infections were seen in 10 patients (26.3%). Five patients were in the XL-CGD group—S.No. 3 (pneumonia due to Mucor spp. and Mycobacterium tuberculosis), S.No. 5 (Candida albicans in urine culture and Burkholderia cepacia septicemia), S.No. 9 (Enterobacter spp. septicemia and a probable fungal pneumonia), S.No. 12 (fungal pneumonia, rib, and vertebral osteomyelitis and Acinetobacter spp. paravertebral soft tissue infection), and S.No. 14 (Klebsiella pneumonia from retropharyngeal abscess and pneumonia due to Pseudomonas aeruginosa).
Four children with AR-CGD had mixed infections—S.No. 17 (S. aureus and C. albicans in lung aspirate cultures), S.No. 27 (Nocardia spp. lung abscess and S. hemolyticus septicemia), S.No. 29 (pneumonia due to A. fumigatus and P. aeruginosa), and S.No. 30 (septicemia due to Pseudomonas stutzeri and Acinetobacter spp.). A patient (S.No. 36) with unclassified CGD had an episode of pneumonia with P. aeruginosa and septicemia with S. aureus.
Localization of Infections
Predominant infections were pneumonia, followed by skin and soft tissue infections, lymphadenitis, septicemia, and deep organ infections (Suppl. Table S2 and Table 1).
Lung
Eighty-four episodes of pneumonia were noted in 36 patients (94.7%). Two patients with XL-CGD developed contiguous bone involvement, and a patient with NCF-2 mutation developed a lung abscess. Pneumonia was complicated by septicemia in eight patients, and four of them died due to severe sepsis and respiratory failure. Twelve (31.6%) patients had received empirical antitubercular therapy before the diagnosis of CGD was made. Confirmatory evidence for the causative pathogen by culture from respiratory secretions or histopathology of lung aspirate could be identified during 16 episodes (Suppl. Table S2).
Galactomannan levels in broncheoalveolar lavage (BAL) specimens were assessed in two children (S.Nos. 28, 31) with suspected fungal pneumonia, and the BAL galactomannan optical density (OD) index was 0.98 and 2.9, respectively, in them. Both the OD index values are ≥0.8, our laboratory cutoff to identify invasive pulmonary aspergillosis (sensitivity 86.4%, specificity 90.7%) [19]. A. fumigatus grew from the BAL fungal cultures in one patient (S.No. 28). Although no growth was detected from the BAL fluid in the other patient (S.No. 31), she showed a clinical response to empirical intravenous amphotericin therapy.
Liver
Six episodes of liver abscess were documented in five patients (13.2%) (three XL-CGD, one AR-CGD, and one unclassified CGD). Four patients had multifocal liver abscesses (S.Nos 5, 7, 20, and 38), and one patient had a single loci of collection at presentation. S. aureus was isolated from liver pus in three patients (S.Nos 7, 20, and 38). Liver abscess in a patient (S.No. 38) ruptured into the peritoneal space and a laparoscopic drainage was performed for its resolution. Glucocorticoids were not used for the management of any of these liver abscesses.
Brain
One patient with unclassified CGD developed multifocal brain abscesses and endophthalmitis due to A. fumigatus. An episode of meningoencephalitis occurred in a patient with NCF-1 mutation. However, no organism was identified.
Skin and Soft Tissue
Forty episodes of skin and soft tissue infections were noted among 15 children (39.5%). Surgical drainage of the abscesses was required in 10 episodes. Microbiological evidence could be identified in only two patients as most of the episodes resolved with empirical antistaphylococcal antimicrobials such as cloxacillin or amoxicillin-clavulanate. Except for an episode of cellulitis due to P. aeruginosa, all other episodes had occurred before the initiation of cotrimoxazole prophylaxis.
Lymph Node
Twenty children (52.6%) were noted to have lymphadenitis in our cohort. Most of these were suppurative adenitis that occurred before the initiation of cotrimoxazole prophylaxis, and resolution could be achieved in nine cases only after surgical drainage. S. aureus was isolated in two patients before the initiation of cotrimoxazole prophylaxis. Lymphadenitis due to Klebsiella pneumoniae (n = 3) and B. cepacia (n = 1) was seen after the initiation of cotrimoxazole prophylaxis. Bacillus Calmette-Guérin (BCG) adenitis was noted in three patients—two with XL-CGD and one with unclassified CGD. All three patients responded to antitubercular therapy.
Bone
Four episodes of bone infection were documented. Two patients with XL-CGD had contiguous rib involvement along with pneumonia. Spread of infection to vertebra leading to osteomyelitis and gibbus was seen in one of the patients [20]. Both of the patients had fungal pneumonia at the time of bone involvement (Mucor spp. and a probable Aspergillus spp.). Osteomyelitis of the elbow joint was noted in one patient (S.No. 21) before the diagnosis of CGD was made.
Other Infections
Blood stream infections were identified in nine patients (three XL-CGD, four AR-CGD, and two unclassified CGD) proved to be fatal in five of them. Organisms isolated during the infectious episodes included B. cepacia, Enterobacter spp., S. aureus, S. hemolyticus, P. aeruginosa, P. stutzeri, Acinetobacter spp., non-typhoidal Salmonella spp., K. pneumoniae, and Candida tropicalis. An episode each of onychomycosis, esophageal candidiasis, and urinary tract infection with Candida spp. were noted in the XL-CGD group. Oral thrush was also seen in two patients with an NCF-1 mutation.
Comparison of Infections Among Types of CGD
Age at onset of infections was earlier in XL-CGD when compared to AR-CGD (Table 2). Pneumonia (56.3%), skin and soft tissue infection (25%), lymphadenitis (12.5%), and liver abscess (6.3%) were the presenting infectious manifestations in the XL-CGD group. Lymphadenitis (52.9%) followed by pneumonia (41.2%) and skin and soft tissue infections (5.9%) were the initial manifestations in the AR-CGD group. The number of infections was also higher in the XL-CGD group (Table 1). However, the mortality, the number of patients with fungal infections, and the mixed infections were comparable among both the groups (Table 2). Comparison of SI values between different types of CGD are shown in Fig. 2a.

Comparison of PMA stimulation index (SI) values in DHR oxidation assay and survival between different types of CGD. a Comparison of PMA SI values in DHR assay between different types of CGD. b Survival and follow-up: XL-CGD vs AR-CGD. c Survival and follow-up: SI ≥ 3 vs SI ≤ 1.5
Mortality
There were 11 deaths (28.9%) in our cohort of 38 patients. Median age at death was 8 months (IQR 7, 13) (Table 3). All patients died due to infections and its complications. Mortality was higher in patients with lower age at onset of symptoms and diagnosis. Ten deaths occurred at the initial detection of CGD, and a girl with AR-CGD died 5 years after the diagnosis of CGD due to pneumonia. Pneumonia was identified in nine patients (81.8%) at the time of death. Bloodstream infections were detected in five patients (45.5%).
Discussion
Prevalence of XL-CGD and AR-CGD was almost equal among in our cohort of CGD. This is in contrast to the increased prevalence of XL-CGD that has been reported from Western countries [3, 6–8, 12]. AR-CGD outnumbered XL-CGD in series reported from Iran, Turkey, and Israel, where the rate of consanguineous marriages is high [11, 13, 15]. Median age at diagnosis was 2 years which is similar to the reports from Western countries [21]. High rates of endogamous marriages within close-knit caste and community-based groups in India could have accounted for higher numbers of AR-CGD in our cohort. An early age at onset and an increased frequency of infections were noted in children with XL-CGD when compared to AR-CGD. The incidence of deep abscesses was also higher in the XL-CGD group. However, incidence of fungal infections, blood stream infections, and mortality was similar among both the groups, which is contrast to the reports from other countries (Suppl. Table S3). A report from Turkey showed that early age of onset and severity of infections were dependent on the residual phox activity and not on the mode of inheritance. All patients with gp91phox defects had SI ≤ 1.5, and all patients with p47phox defects had SI ≥ 3 [11]. However, in our series, the median SI values among gp91phox and p47phox-deficient patients were comparable (Fig. 2). A recent study by Kulkarni et al. [22] from India also noted SI values <1.5 in 14 out of 21 patients with p47phox defect. Survival also did not seem to depend upon the SI values in our cohort (Fig. 2 and Suppl. Table S4). However, the follow-up period in the majority of patients was very short (<3 years), and a longer follow-up period would provide convincing data on survival patterns. The higher mortality in AR-CGD observed in our cohort is probably due to the preferential identification of patients with severe manifestations and unrecognition of many patients with milder forms of disease [23].
Pneumonia is the most common infection in our cohort which is similar to previous reports from other countries. The incidence of lymphadenitis is also comparable to previous studies. However, incidence of osteomyelitis and liver abscess was lower in our cohort when compared to other reports. We could not use IFN prophylactic therapy in our patients due to difficulty in procurement and financial constraints. However, the number of infections and mortality was significantly reduced after initiation of cotrimoxazole and itraconazole prophylaxis in our cohort. This is in contrast to the reports from UK, where in spite of prophylactic cotrimoxazole and itraconazole, infections were frequent [6]. Reasons could be due to difference in compliance of patients with the drugs and severity of underlying defects which may vary among different ethnicities. Though we could not objectively document the compliance of antiinfective prophylactic therapy, all patients except one were on regular prophylaxis.
Staphylococcus followed by Aspergillus infections were commonest in our cohort which is similar to other reports. Overcrowding and a lower socioeconomic status may have played an additional role in the development of staphylococcal infections. We also noted a significant drop in staphylococcal infections after initiation of cotrimoxazole prophylaxis (Fig. 1). Pseudomonas spp. which are also catalase-positive organisms have not been commonly reported in CGD (Suppl. Table S3). We noted that Pseudomonas spp. was the second most common bacterial isolate in our cohort. No patients with documented Pseudomonas infections in our cohort were on concomitant immunosuppressive medications. A combination of acquired risk factors such as prolonged stay in the intensive care unit, placement of the endotracheal tube in situ, and the underlying CGD would have predisposed to fulminant Pseudomonas infections in patient nos. 30 and 36 [24]. Community-acquired Pseudomonas infections are reported mainly in patients with underlying risk factors, and in India, community-acquired pneumonia in children due to Pseudomonas spp. are not frequently encountered [24, 25]. Patient 6 had a community-acquired cellulitis, and patient nos. 23 and 29 had community-acquired pneumonia due to P. aeruginosa. We believe that underlying CGD status had played a major role in the acquisition of pseudomonas infections. A similarly high rate of Pseudomonas infections (13%) was also documented in a CGD cohort from Israel [14]. However, a similar rate of Pseudomonas infections has not been documented from Iran [13]. Hot and humid weather that is experienced in the subtropical and tropical countries may have contributed to the development of infection with Pseudomonas spp. Pseudomonas spp. have also been reported to grow well in warm conditions such as hot tubs [26].
Infections due to Burkholderia pseudomallei are more frequent in the tropical regions with heavy rainfall and coastal areas [27, 28]. B. pseudomallei infections in CGD have been reported from Singapore, Puerto Rico, and Guadeloupe, regions in close proximity to the sea [29–31]. The northern part of India neither experiences a heavy rainfall nor is it very close to the sea. This might be the reason for the absence of B. pseudomallei infections in our cohort. Infections due to Salmonella spp. were documented in 5.4 and 5.6% of patients with CGD in a cohort from Iran and Turkey, respectively [11, 13]. A low prevalence of Salmonella spp. in our cohort could be due to the widespread use of empirical antibiotics such as cephalosporins in the community for all kinds of infections which could have lowered the culture yield of Salmonella spp. [32].
Chryseobacterium gleum, a catalase-positive organism, was isolated from respiratory secretions of an infant with XL-CGD and pneumonia (Suppl. Table S1). Chryseobacterium spp. is known to colonize in natural habitats such as tap water and soil. Clinical isolates of C. gleum were initially reported in neonatal infections, and isolates were mainly from the sputum and urine [33–35]. Chryseobacterium isolates have been found to be susceptible to minocycline and cotrimoxazole, but resistant to other antibiotics [34]. The C. gleum isolate in our patient was sensitive to minocycline, cotrimoxazole, and piperacillin tazobactum. The child improved with 4 weeks of IV piperacillin tazobactum and oral cotrimoxazole therapy. Infection with C. gleum has previously not been documented in CGD.
Mycobacterial infections are also commonly reported in CGD, especially from countries where tuberculosis is endemic and BCG vaccination in neonatal period is a prevalent practice [11, 13, 14, 36, 37]. We documented BCG adenitis in four patients and M. tuberculosis pneumonia in two patients. The incidence of mycobacterial infections was low in our cohort despite India being an endemic nation for tuberculosis and a mandatory neonatal BCG vaccination as per the National Immunization Schedule of India. High rates of empirical antitubercular therapy (12/38) before the diagnosis of CGD could have accounted for a low yield of mycobacterium. Other reason could be due to the difference in virulence potential of BCG strain used in different countries. The BCG strains used in India are the Pasteur and Copenhagen strains [38].
Fungal infections were predominantly due to Aspergillus spp. which is similar to the previous reports. Confirmatory evidence of Aspergillus was documented in seven patients. Six patients were suspected to have Aspergillus pneumonia because they had a persistent pneumonia not responding to antibiotics, a positive fungal serology for Aspergillus, and a good clinical response to antifungal therapy such as amphotericin B or voriconazole. Two children with suspected fungal pneumonia had an elevated galactomannan OD index values in BAL and a good clinical response to antifungal therapy. The utility of BAL galactomannan assay in the diagnosis of Aspergillus pneumonia in CGD has not been explored in large cohorts. In an epidemiological study done in the community comprising two provinces of Northern India, Aspergillus accounted for ∼20% of all viable spores in the air with higher counts during the winters coinciding with the wheat-harvesting period in these areas [39]. In another study done at our institute, high spore counts of Aspergillus in the air were found [40]. These reports clearly indicate that Aspergillus is quite common in our region. Pneumonia due to Fusarium spp. was previously documented in three patients with CGD in the US cohort, and we noted the presence of Fusarium dimerum in one of the patients with p47phox defect [3].
Mortality in our cohort is high when compared to other cohorts, and this could be speculated to be due to non-availability of IFN and difficulties in arranging hematopoietic stem cell transplantation (HSCT). However, prophylactic therapy with IFN is mainly used in USA, and many parts of Europe still do not use IFN routinely in CGD. The survival rates between transplanted (90%) and non-transplanted children (90%) with CGD were also reported to be similar in a series by Cole et al.; however, the duration of follow-up was short (median post-transplantation follow-up 3.84 years) [41]. Ten out of 11 patients died due to severe infections at the time of diagnosis of CGD in our cohort, and only one patient succumbed to pneumonia 5 years after diagnosis and initiation of antiinfective prophylaxis. A higher incidence of septicemia in patients who had mortality was probably due to a late clinical presentation, dissemination of infection, and acquired sepsis as a result of prolonged hospital stay. A late clinical presentation of patients was probably due to a lack of awareness of primary immunodeficiency diseases (PIDs) among physicians. Moreover, diagnostic tests for PID are not readily available in many parts of the country, and the patients have to be referred to a tertiary care center for diagnosis and management of PID [23].
Though many infections before the diagnosis of CGD improved with antimicrobials, documentation of microorganisms could not be obtained in many of them. However, this is perhaps one of the largest series from a developing country that has focused on the infection profile in CGD. High rates of Pseudomonas infections (15.7%) were seen in our cohort. Moreover, we have documented the presence of unusual infections such as F. dimerium and C. gleum. Various factors such as lower socioeconomic status, overcrowding, and environmental factors such as tropical weather play an important role in the development of infections. A lack of awareness of PID and non-availability of diagnostic facilities for PID in the community could have accounted for a late clinical diagnosis and increased mortality in CGD in our cohort. We also recognize the lack of easy access to HSCT and IFN therapy. Though the survival and mortality were similar in XL-CGD and AR-CGD in our cohort, a longer follow-up would be needed to draw definitive conclusions.
Abbreviations
- XL:
-
X-linked
- AR:
-
Autosomal recessive
- MALDI-TOF:
-
Matrix-assisted laser desorption ionization time-of-flight mass spectrometry
- SI:
-
Stimulation index
References
Roos D. Chronic granulomatous disease. Br Med Bull. 2016;118(1):50–63.
de Boer M, Hilarius-Stokman PM, Hossle JP, Verhoeven AJ, Graf N, Kenney RT, et al. Autosomal recessive chronic granulomatous disease with absence of the 67-kD cytosolic NADPH oxidase component: identification of mutation and detection of carriers. Blood. 1994;83(2):531–6.
Winkelstein JA, Marino MC, Johnston RB, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore). 2000;79(3):155–69.
Chiriaco M, Salfa I, Di Matteo G, Rossi P, Finocchi A. Chronic granulomatous disease: clinical, molecular, and therapeutic aspects. Pediatr Allergy Immunol. 2016;27(3):242–53.
Rawat A, Bhattad S, Singh S. Chronic granulomatous disease. Indian J Pediatr. 2016;83(4):345–53.
Jones LBKR, McGrogan P, Flood TJ, Gennery AR, Morton L, Thrasher A, et al. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol. 2008;152(2):211–8.
Martire B, Rondelli R, Soresina A, Pignata C, Broccoletti T, Finocchi A, et al. Clinical features, long-term follow-up and outcome of a large cohort of patients with chronic granulomatous disease: an Italian multicenter study. Clin Immunol. 2008;126(2):155–64.
Kobayashi S, Murayama S, Takanashi S, Takahashi K, Miyatsuka S, Fujita T, et al. Clinical features and prognoses of 23 patients with chronic granulomatous disease followed for 21 years by a single hospital in Japan. Eur J Pediatr. 2008;167(12):1389–94.
Ahlin A, De Boer M, Roos D, Leusen J, Smith CI, Sundin U, et al. Prevalence, genetics and clinical presentation of chronic granulomatous disease in Sweden. Acta Paediatr. 1995;84(12):1386–94.
Soler-Palacín P, Margareto C, Llobet P, Asensio O, Hernández M, Caragol I, et al. Chronic granulomatous disease in pediatric patients: 25 years of experience. Allergol Immunopathol (Madr). 2007;35(3):83–9.
Köker MY, Camcıoğlu Y, van Leeuwen K, Kılıç SŞ, Barlan I, Yılmaz M, et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allergy Clin Immunol. 2013;132(5):1156–1163.e5.
Marciano BE, Spalding C, Fitzgerald A, Mann D, Brown T, Osgood S, et al. Common severe infections in chronic granulomatous disease. Clin Infect Dis. 2015;60(8):1176–83.
Fattahi F, Badalzadeh M, Sedighipour L, Movahedi M, Fazlollahi MR, Mansouri SD, et al. Inheritance pattern and clinical aspects of 93 Iranian patients with chronic granulomatous disease. J Clin Immunol. 2011;31(5):792–801.
Xu H, Tian W, Li S-J, Zhang L-Y, Liu W, Zhao Y, et al. Clinical and molecular features of 38 children with chronic granulomatous disease in mainland China. J Clin Immunol. 2014;34(6):633–41.
Wolach B, Gavrieli R, de Boer M, Gottesman G, Ben-Ari J, Rottem M, et al. Chronic granulomatous disease in Israel: clinical, functional and molecular studies of 38 patients. Clin Immunol. 2008;129(1):103–14.
de Oliveira-Junior EB, Zurro NB, Prando C, Cabral-Marques O, Pereira PVS, Schimke L-F, et al. Clinical and genotypic spectrum of chronic granulomatous disease in 71 Latin American patients: first report from the LASID registry. Pediatr Blood Cancer. 2015;62(12):2101–7.
Meshaal S, El Hawary R, Abd Elaziz D, Alkady R, Galal N, Boutros J, et al. Chronic granulomatous disease: review of a cohort of Egyptian patients. Allergol Immunopathol (Madr). 2015;43(3):279–85.
Rawat A, Singh S, Suri D, Gupta A, Saikia B, Minz RW, et al. Chronic granulomatous disease: two decades of experience from a tertiary care centre in North West India. J Clin Immunol. 2014;34(1):58–67.
D’Haese J, Theunissen K, Vermeulen E, Schoemans H, De Vlieger G, Lammertijn L, et al. Detection of galactomannan in bronchoalveolar lavage fluid samples of patients at risk for invasive pulmonary aspergillosis: analytical and clinical validity. J Clin Microbiol. 2012;50(4):1258–63.
Vignesh P, Bhattad S, Shandilya J-K, Vyas S, Garg R, Rawat A. Vertebral osteomyelitis and Acinetobacter spp. paravertebral soft tissue infection in a 4-year-old boy with X-linked chronic granulomatous disease. Pediatr Infect Dis J. 2016;35(9):1043–5.
Leiding JW, Holland SM. Chronic granulomatous disease. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, et al., editors. GeneReviews(®) [Internet]. Seattle (WA): University of Washington, Seattle; 1993. [cited 2017 Feb 2]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK99496/.
Kulkarni M, Desai M, Gupta M, Dalvi A, Taur P, Terrance A, et al. Clinical, immunological, and molecular findings of patients with p47(phox) defect chronic granulomatous disease (CGD) in Indian families. J Clin Immunol. 2016;36:774–84.
Gupta S, Madkaikar M, Singh S, Sehgal S. Primary immunodeficiencies in India: a perspective. Ann N Y Acad Sci. 2012;1250:73–9.
Gellatly SL, Hancock REW. Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog Dis. 2013;67(3):159–73.
Mathew JL, Singhi S, Ray P, Hagel E, Saghafian-Hedengren S, Bansal A, et al. Etiology of community acquired pneumonia among children in India: prospective, cohort study. J Glob Health. 2015;5(2):050418.
Centers for Disease Control and Prevention (CDC). Pseudomonas dermatitis/folliculitis associated with pools and hot tubs—Colorado and Maine, 1999-2000. MMWR Morb Mortal Wkly Rep. 2000;49(48):1087–91.
Leelarasamee A. Recent development in melioidosis. Curr Opin Infect Dis. 2004;17(2):131–6.
Lee PP-W, Lau Y-L. Improving care, education, and research: the Asian primary immunodeficiency network. Ann N Y Acad Sci. 2011;1238:33–41.
Renella R, Perez J-M, Chollet-Martin S, Sarnacki S, Fischer A, Blanche S, et al. Burkholderia pseudomallei infection in chronic granulomatous disease. Eur J Pediatr. 2006;165(3):175–7.
Dorman SE, Gill VJ, Gallin JI, Holland SM. Burkholderia pseudomallei infection in a Puerto Rican patient with chronic granulomatous disease: case report and review of occurrences in the Americas. Clin Infect Dis. 1998;26(4):889–94.
Tarlow MJ, Lloyd J. Melioidosis and chronic granulomatous disease. Proc R Soc Med. 1971;64(1):19–20.
Yewale VN. Mission: avoid antibiotic abuse. Indian Pediatr. 2014;51(10):773.
Virok DP, Ábrók M, Szél B, Tajti Z, Mader K, Urbán E, et al. Chryseobacterium gleum—a novel bacterium species detected in neonatal respiratory tract infections. J Matern-Fetal Neonatal Med. 2014;27(18):1926–9.
Lo H-H, Chang S-M. Identification, characterization, and biofilm formation of clinical Chryseobacterium gleum isolates. Diagn Microbiol Infect Dis. 2014;79(3):298–302.
Kirby JT, Sader HS, Walsh TR, Jones RN. Antimicrobial susceptibility and epidemiology of a worldwide collection of Chryseobacterium spp: report from the SENTRY Antimicrobial Surveillance Program (1997-2001). J Clin Microbiol. 2004;42(1):445–8.
Conti F, Lugo-Reyes SO, Blancas Galicia L, He J, Aksu G, Borges de Oliveira E, et al. Mycobacterial disease in patients with chronic granulomatous disease: a retrospective analysis of 71 cases. J Allergy Clin Immunol. 2016;138(1):241–248.e3.
Deffert C, Cachat J, Krause K-H. Phagocyte NADPH oxidase, chronic granulomatous disease and mycobacterial infections. Cell Microbiol. 2014;16(8):1168–78.
IAPCOI.com [Internet]. [cited 2017 Jan 5]. Available from: http://www.iapcoi.com/hp/iap_guidebook.php.
Chakrabarti A, Rudramurthy SM, Panda N, Das A, Singh A. Epidemiology of chronic fungal rhinosinusitis in rural India. Mycoses. 2015;58(5):294–302.
Rudramurthy SM, Singh G, Hallur V, Verma S, Chakrabarti A. High fungal spore burden with predominance of Aspergillus in hospital air of a tertiary care hospital in Chandigarh. Indian J Med Microbiol. 2016;34(4):529–32.
Cole T, Pearce MS, Cant AJ, Cale CM, Goldblatt D, Gennery AR. Clinical outcome in children with chronic granulomatous disease managed conservatively or with hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2013;132(5):1150–5.
Acknowledgements
Authors thankfully acknowledge India Council of Medical Research, New Delhi, India, and Department of Health Research, Ministry of Health and Family Welfare, Government of India, New Delhi, India, for funding vide Grant No. GIA/48/2014-DHR and the Foundation for Primary Immunodeficiencies (FPID), USA. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Grant No. GIA/48/2014-DHR to one of co-authors (SS) from Indian Council of Medical Research, New Delhi, and Department of Health Research, Ministry of Health and Family Welfare, Government of India, New Delhi.
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Rawat, A., Vignesh, P., Sharma, A. et al. Infection Profile in Chronic Granulomatous Disease: a 23-Year Experience from a Tertiary Care Center in North India. J Clin Immunol 37, 319–328 (2017). https://doi.org/10.1007/s10875-017-0382-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-017-0382-x