
Abstract
Purpose
A20 gene functions in negative immunoregulation and its SNP is related to SLE disease. But its expression level in immune cells from SLE patients is still unclear. The aim of this study is to investigate whether the expression of A20 is associated with pathogenesis of SLE.
Methods
Real-time transcription-polymerase chain reaction analysis (RT-PCR) was used to determine expression of A20 mRNA in peripheral blood mononuclear cells (PBMC) from 37 patients with SLE and 32 healthy controls.
Results
A20 expression was decreased in SLE patients compared with healthy controls (p = 0.0133). The expression level of A20 gene negatively correlated with the SLE disease activity index (SLEDAI) (r =−0.4661, p = 0.0036) and erythrocyte sedimentation rate (ESR) (r =−0.5222, p = 0.0009). Furthermore, SLE patients with nephritis had a lower expression of A20 than those without nephritis (p = 0.0188).
Conclusions
Our results suggest that the insufficient expression of A20 gene in PBMC may take part in the pathogenesis of SLE disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by chronic inflammation and consequent tissue damage. The chronic inflammation is attributed to breakdown of immune homostasis, which results in activation of immune cells and elevation of pro-inflammatory cytokines. The immune activation leads to deposition of autoimmune complexes and infiltration of tissue destructive T cells into susceptible organs while the pro-inflammatory cytokines is responsible for maintainance of the immune activation [1].
Dysfunction of negative immunoregulatory genes is one of important reasons for the failure of immune homostasis. For instance, TGF-β is an immunosuppressive cytokine and its expression decreases in SLE patients [2]. CTLA-4 is a negative regulator of T cell activation and is considered as a susceptibility gene for SLE. The serum level of soluble CTLA-4 elevates in SLE patients [3]. SOCS-1 can negatively regulate both antigen receptor and TLR signaling and its dysfunction is considered as a pathogenic factor of SLE [4]. FoxP3 gene is a master conrol of development and function of CD4 + CD25+ regulatory T cells and its expression is decreased in PBMC from SLE patients [5].
A20 gene, also known as tumor necrosis factor α induced protein-3 (TNFAIP3) is an important negative immunoregulatory gene. Constitutive expression of A20 is restricted in lymphoid tissues, like thymus and spleen. In A20 knockout mice, its deficiency leads to death shortly after birth by severe inflammation and tissue damage in multiple organs [6, 7]. In immune cells, overexpression of A20 can terminate NF-κB signaling transduced from TNF receptors, toll-like receptors, nucleotide-binding oligomerization domain containing 2 (NOD2) receptors or T cell receptors [8, 9]. ubiquitin-editing domains, C-terminal zinc-finger ubiquitin-binding domain and N-terminal deubiquitinase domain. The C-terminal domain is involved in K48-polyubiquitination, while the N-terminal domain is thought to promote de-ubiquitination of K63-polyubiquitin chains. Activation of NF-κB is controlled by both K48- and K63- polyubiquitination of upstream signaling proteins, respectively triggering proteasome-mediated degradation or interaction with other signaling proteins. Thus, A20 turns off activation of NF-KB by modulating both types of ubiquitination [10].
Although there is a relationship between SNP of A20 gene and SLE disease [11, 21, 22], expression level of A20 in immune cells from SLE patients is still a puzzle. Therefore, we compared the expression of A20 mRNA in PBMC between SLE patients and healthy controls and analyzed the relationship between A20 expression level and disease activity in order to elucidate the role of A20 expression in pathogenesis of SLE.
Materials and Methods
Human Subjects
Thirty-seven patients with SLE diagnosed according to the criteria of the American College of Rheumatology were enrolled. At the same time, thirty-two healthy controls were recruited, who were sex- and age-matched with the patients and did not have any rheumatologic conditions. Individual disease activity was quantified using the SLE disease activity index (SLEDAI) score. Peripheral bloods were sampled from all the patients before they took any immunosuppressive drug to exclude the influence of immunosuppressive drug on A20 expression. All the blood samples from the patients and healthy controls were used with informed consent and approval from the Ethics Committee of Shandong University, China. The characteristics of the patients and healthy subjects are showed in Table I.
Laboratory Measurement
For all the SLE patients, serum levels of C3, C4, IgG, IgA and IgM, as well as autoantibodies such as ANA, anti-dsDNA and anti-Sm antibodies, were detected using qualitative enzyme linked immunoabsorbent assay (ELISA) according to the instruction of the manufacturer (Sino-American biotechnology company, Luoyang, China). For all the subjects including the patients and healthy controls, erythrocyte sedimentation rates (ESR) were determined by Westergren test.
Preparation of PBMC and Extraction of RNA
PBMC was separated by density gradient centrifugation from the peripheral blood anticoagulated with sodium citrate. Total RNA was extracted from 5 × 105 PBMC using TRIzol according to the instructions of the manufacturer (Invitrogen, California, USA), then quantified by photometrical measurement.
Quantitative Real-Time RT-PCR
One microgram of RNA was reversely transcribed to cDNA using reverse transcription system kit (Promega, WI, USA) for each sample. The expression of A20 mRNA was evaluated by quantitative real-time PCR in triplicate and the level of β-actin mRNA was also detected as an internal control. Real-time PCR was performed using SYBR Green I real-time PCR kit (TAKARA, Dalian, China) on an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Forster city, CA, USA). The first step of PCR protocol is 95 °C for 10s, followed by 40 cycles of 95 °Cfor 5 s and 60 °C for 40s as the second step. Primers used were as follows: A20 forward: 5′-CGTCCAGGTTCCAGAACACCATTC-3′, and reverse: 5′-TGCGCTGGCTCGATCTCAGTTG-3′; β-actin forward: 5′GACTACCTCATGAAGATCCTCACC3′, and reverse: 5′TCTCCTTAATGTCACGCACGATT3′.
A melting-curve analysis was performed to ensure specificity of the PCR products and all the PCR products were subjected to electrophoresis in an agarose gel to confined to a single band of the expected size. The expression of A20 gene was normalized to β-actin and determined using the comparative (2−⊿⊿Ct) method.
Statistical Analysis
The relative expression of A20 mRNA fore each sample was presented as mean ± standard deviation. The difference in A20 mRNA level between subject groups was analyzed by Mann-Whitney test. Correlations analysis was carried out using Spearman’s rank test. A probability level of 0.05 was accepted to indicate significant difference. All analyses were performed with the GraphPad Prism, software, version 5.0.
Results
Laboratory Measurements of the Patients with SLE
The Demographic characteristics, clinical manifestation and laboratory measurements in the SLE patients are presented in Table I. Lupus nephritis (LN) were found in 23 of the 37 SLE patients. The arthritis, serositis and central nervous system (CNS) disease were found in 21, 12 and 3 patients, respectively. The mean value of SLEDAI was 16.66 with the rang from 2 to 39. The ANA, anti- dsDNA and anti-Sm autoantibodies were detected in 37, 18 and 13 patients. The mean value of ESR for the patients was 67 mm/h with the rang from 11 to 139 mm/h.
Quantification of A20 mRNA Expression in PBMC from SLE Patients and Healthy Controls by Real-Time RT-PCR
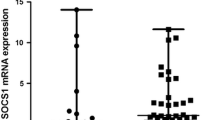
We examined the expression of A20 mRNA in PBMC from 37 SLE patients and 32 sex- and age-matched healthy controls using quantitative real-time RT-PCR. The mean of A20 mRNA expression in PBMC from SLE patients (26.66) was significantly decreased compared to healthy controls (43.62) (p = 0.0133) (Fig. 1). Besides, the expression of A20 mRNA in SLE patients with LN (21.09) was significantly lower than in those without LN (35.82)(p = 0.0188) (Fig. 2).

A20 mRNA expression levels in PBMC from patients with SLE and healthy controls Real time RT-PCR was performed to quantify the expression of A20 gene in SLE patients (n = 37) and healthy controls (n = 32). Horizontal lines indicate medians (26.66 in the patient group, 43.62 in the control group). There was a significant decrease in A20 expression in SLE patients compared with healthy controls (p = 0.0133)

A20 mRNA expression levels in PBMC from SLE patients without LN and those with LN Real time RT-PCR was performed to quantify the expression of A20 gene in SLE patients without LN (n = 14) and those with LN (n = 23). Horizontal lines indicate medians (35.82 in the patients without LN, 21.09 in the patients with LN). There was a significant decrease in A20 expression in SLE patients with LN compared with those without LN (p = 0.0188)
Analysis of Relationships Between A20 mRNA Expression and Characteristics or Laboratory Parameters in the Patients with SLE
Association of A20 with demographic characteristics, clinical manifestations and laboratory parameters were analyzed. The results showed that the expression level of A20 mRNA was negatively correlated with SLEDAI (r =−0.4661, P = 0.0036) (Fig. 3) and ESR (r =−0.5222, P = 0.0036) (Fig. 4) in 37 SLE patients. No statistically significant relationships were found between A20 mRNA expression levels and other characteristics, clinical manifestations or laboratory parameters in the patients with SLE.

Negative correlation between A20 mRNA expression levels and SLEDAI in all the SLE patients (n = 37)

Negative correlation between A20 mRNA expression levels and ESR in all the SLE patients (n = 37)
Discussion
SLE is a chronic inflammatory disease associated with dysfunction of multiple immunoregulatory genes. A20 gene plays a critical role in negative regulation of immunity. In this study, we first reported down-regulation of A20 expression in SLE patients and the negative correlation between A20 expression and disease activity. The results suggest that the decreased expression of A20 may be involved in the pathogenesis of SLE
The deficiency of A20 expression may contribute to the pathogenesis of SLE through several mechanisms. One of possible mechanisms is that the insufficient expression of A20 may cause hyperactivation of autoreactive T cell, which is one of hallmarks of SLE. Since A20 gene has a function in limiting activation of T cell by deubiquitinating Malt1 to disrupt T cell receptor signaling to NF-κB [9], its decreased expression may induce hyperactivation of T cell and the subsequent tissue damage occurring in susceptible organs of SLE patients. Another possible mechanism is that the decreased expression of A20 may enhance the deposition of autoimmune complexes, which is also a key marker of SLE. A20 restricts B cell survival and prevents dendritic cell activation [23, 24]. By contrast, A20 deficiency in B cells enhances B-cell proliferation and results in the development of autoantibodies [12]. Therefore, the insufficient expression of A20 may lead to hyperactivation of B cells and accumulation of autoantibodies. Besides, the decreased expression of A20 in SLE patients may also contribute to high serum level of proinflammatory cytokines including IL-4, IL-6, IL-12, TNF-αand IFN-γ [1, 13], which is another characteristic of SLE. The major source of the proinflammatory cytokines is monocytes and macrophages. Myeloid-A20-deficient mice have high levels of inflammatory cytokines in their serum, consistent with a sustained NF-κB activation and higher TNF-α production by the macrophages [14]. Thus, the elevated production of proinflammatory in SLE may be partially ascribed to the insufficient expression of A20. Collectively, the down-regulated expression of A20 may give rise to SLE disease since its normal expression controls the activation of multiple immune cells.
The inverse correlation between A20 expression and SLE disease activity indicated by SLEDAI and ESR suggest that the insufficient expression of A20 may contribute to severity of the disease. SLEDAI is a global score reflecting all aspects of disease activity and a reliable instrument for the assessment of disease activity in SLE. Higher scores indicate more severe disease activity [15, 16]. Hence, the negative correlation between the A20 mRNA expression and SLEDAI in the SLE patients means that the lower expression of A20, the more severity of the disease. Moreover, ESR is also an indication of the degree of inflammation and used to monitor disease activity. The level of A20 expression may be an index of disease activity since the A20 mRNA expression was negatively correlated with the ESR in all the SLE patients. In addition, there was a significant difference in A20 expression between SLE patients without LN and those with LN. LN represents one of the most serious manifestations of systemic lupus erythematosus (SLE). The association of A20 expression with LN in SLE patients also indicates reduction of A20 expression was involved in severity of SLE diseases. Taken together, the analysis of relationship between A20 expression and SLEDAI, ESR and LN further suggests a potential role of decreased A20 expression in pathogenesis of SLE.
As for causes of the low expression of A20 in SLE, there are two explanations at least. One reason may be polymorphism of A20 gene [21, 22]. Some SNPs can influence expression of A20 gene, such as two intronic SNPs (rs610604 and rs5029930) associated independently with lower gene expression and increased risk of coronary artery disease [17]. What is more, a haplotype of A20 gene with a TT > A polymorphic dinucleotide (deletion T followed by a T to A) resulting in reduced TNFAIP3 mRNA and A20 protein expression is associated with SLE [18]. Therefore, the deficiency of A20 expression may be partially attributed to polymorphism of A20 gene. The other possible reason is promoter methylation of A20 gene. A20 is targeted by promoter methylation in some hematological malignancies [19, 20]. Thus, the low expression of A20 gene in SLE may be due to in part its aberrant methylation. This remains to be further investigated.
This study is limited by sole real-time RT-PCR method in measurement of A20 expression and lacks protein data and mechanistic data. Quantification of A20 protein by flow cytometry and Western blot methods, as well as elucidation of the mechanism, will be an emphasis in our future research work.
In summary, we report that A20 mRNA expression was down-regulated and inversely correlated with disease activity in SLE patients. The results suggest that reduction of A20 expression may contribute to pathogenesis of SLE disease. These data support that A20 gene may be a target for therapy of SLE disease.
References
Apostolidis SA, Lieberman LA, Kis-Toth K, et al. The dysregulation of cytokine networks in systemic lupus erythematosus. J Interferon Cytokine Res. 2011;31:769–79.
Hrycek A, Kusmierz D, Dybała T, et al. Expression of messenger RNA for transforming growth factor-beta1 and for transforming growth factor-beta receptors in peripheral blood of systemic lupus erythematosus patients treated with low doses of quinagolide. Autoimmunity. 2007;40:23–30.
Wong CK, Lit LC, Tam LS, et al. Aberrant production of soluble costimulatory molecules CTLA-4, CD28, CD80 and CD86 in patients with systemic lupus erythematosus. Rheumatology (Oxford). 2005;44:989–94.
Fujimoto M, Tsutsui H, Xinshou O, et al. Inadequate induction of suppressor of cytokine signaling-1 causes systemic autoimmune diseases. Int Immunol. 2004;16:303–14.
Zhang B, Zhang X, Tang F, et al. Reduction of forkhead box P3 levels in CD4 + CD25high T cells in patients with new-onset systemic lupus erythematosus. Clin Exp Immunol. 2008;153:182–7.
Vereecke L, Beyaert R, van Loo G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009;30:383–91.
Coornaert B, Carpentier I, Beyaert R. A20: central gatekeeper in inflammation and immunity. J Biol Chem. 2009;284:8217–21.
Kingeter LM, Paul S, Maynard SK, et al. Cutting edge: TCR ligation triggers digital activation of NF-kappaB. J Immunol. 2010;185:4520–4.
Düwel M, Welteke V, Oeckinghaus A, et al. A20 negatively regulates T cell receptor signaling to NF-kappaB by cleaving Malt1 ubiquitin chains. J Immunol. 2009;182:7718–28.
Verstrepen L, Verhelst K, van Loo G, et al. Expression, biological activities and mechanisms of action of A20 (TNFAIP3). Biochem Pharmacol. 2010;80:2009–20.
Vereecke L, Beyaert R, van Loo G. Genetic relationships between A20/TNFAIP3, chronic inflammation and autoimmune disease. Biochem Soc Trans. 2011;39:1086–91.
Hövelmeyer N, Reissig S, Xuan NT, et al. A20 deficiency in B cells enhances B-cell proliferation and results in the development of autoantibodies. Eur J Immunol. 2011;41:595–601.
Ohl K, Tenbrock K. Inflammatory cytokines in systemic lupus erythematosus. J Biomed Biotechnol. 2011;2011:432595.
Matmati M, Jacques P, Maelfait J, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet. 2011;43:908–12.
Bombardier C, Gladman DD, Hurwitz MB, et al. Derivation of the SLEDAI: a disease activity index for lupus patients. The committee on prognosis studies in SLE. Arthritis Rheum. 1992;35:630–40.
Hawker G, Gabriel S, Bombardier C, et al. A reliability study of SLEDAI: a disease activity index for systemic lupus erythematosus. J Rheumatol. 1993;20:657–60.
Boonyasrisawat W, Eberle D, Bacci S, et al. Tag polymorphisms at the A20 (TNFAIP3) locus are associated with lower gene expression and increased risk of coronary artery disease in type 2 diabetes. Diabetes. 2007;56:499–505.
Adrianto I, Wen F, Templeton A, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet. 2011;43:253–8.
Frenzel LP, Claus R, Plume N, et al. Sustained NF-kappaB activity in chronic lymphocytic leukemia is independent of genetic and epigenetic alterations in the TNFAIP3 (A20) locus. Int J Cancer. 2011;128:2495–500.
Chanudet E, Huang Y, Ichimura K, et al. A20 is targeted by promoter methylation, deletion and inactivating mutation in MALT lymphoma. Leukemia. 2010;24:483–7.
Graham RR, Cotsapas C, Davies L, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:1059–61.
Musone SL, Taylor KE, Lu TT, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. 2008;40:1062–4.
Tavares RM, Turer EE, Liu CL, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. 2010;33:181–91.
Kool M, van Loo G, Waelput W, et al. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity. 2011;35:82–96.
Acknowledgments
This work was supported by grants from the National Nature Science Foundation of China (No. 81071705 and No. 30500591), the Award Funds for Excellent Young and Middle-aged Scientists of Shandong Province (BS2009YY002) and the National “973” Program of China (No. 2011CB503906).
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Di Li and Lei Wang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Li, D., Wang, L., Fan, Y. et al. Down-Regulation of A20 mRNA Expression in Peripheral Blood Mononuclear Cells from Patients with Systemic Lupus Erythematosus. J Clin Immunol 32, 1287–1291 (2012). https://doi.org/10.1007/s10875-012-9764-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-012-9764-2