
Abstract
Introduction
In recent years, it has been realized that innate lymphocytes do not act in isolation but potentiate their efficiency by interacting with each other, resulting even in the regulation of adaptive immune response. One such cross-talk exists between dendritic cells (DCs) and natural killer (NK) cells. Here, we summarize recent studies on which subsets of these two innate immune components participate in this interaction, how it influences immune responses, and to which extent similar stimuli are integrated by DCs and NK cells during innate immunity.
Conclusion
We suggest that this cross-talk should be harnessed by activating both of these innate leucocyte populations with new adjuvant formulations for immunotherapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the immune system, the different leukocytes act not only by mediating their own protective functions, but also by interacting with each other to optimize the response against a pathogen. Recently, relevant liaisons, occurring between natural killer (NK) cells and dendritic cells (DCs), have been extensively investigated, and we will review NK cell activation by DC, editing of DC induced immune responses by NK cells, and an emerging similarity in target cell recognition between these two innate leukocyte subsets. Our discussion will primarily focus on human DC/NK cell interactions.
DCs are critical for initiating immune responses [1]. At an immature stage, they act as sentinels in peripheral tissues, continuously sampling the environment, sensing the presence of pathogens, and secreting chemokines and cytokines to amplify the immune response. Upon activation by danger signals, they up-regulate chemokine receptors and costimulatory molecules, which allow them to migrate into lymph nodes and to efficiently induce T-cell responses [1].
NK cells were identified originally on a functional basis, as this term was assigned to lymphoid cells capable of killing a number of tumor cell lines in the absence of previous stimulation in vivo and in vitro [2]. The molecular mechanisms, by which NK cells discriminate between normal and tumor cells were revealed in recent years. It has been shown that NK cells recognize MHC class I molecules through surface receptors delivering inhibitory signals. As a consequence, NK cells can lyse target cells that have lost (or express low amounts of) MHC class I molecules, a frequent event in tumors or in cells infected by certain cytopathic viruses, primarily herpes viruses. This principle of NK cell activation was coined “missing-self” recognition [3]. Human NK cells express different HLA class I-specific inhibitory receptors. Those named “killer immunoglobulin (Ig)-like receptors” (KIR) are specific for allelic determinants of HLA class I molecules. In contrast, the Ig-like transcript (ILT)2 (LIR1) receptor is characterized by a broad specificity for various HLA class I molecules, while CD94/NKG2A recognizes the nonclassical MHC class I molecule HLA-E [4–7].
In addition to detection of MHC class I loss, NK cells require also an activating signal for target cell lysis. Absence of activating ligands allows them to spare somatic cells with low MHC class I expression like neurons, and strong activating signaling leads to killing of MHC class I-positive tumor cells. The archetypical activating NK cell receptor is CD16 (FcγRIIIA) [8, 9]. This intermediate affinity Fc receptor allows NK cells to target cells opsonized with antibodies and to mediate antibody-dependent cellular cytotoxicity (ADCC). Triggering receptors specific for HLA class I molecules, displaying a high homology with the corresponding inhibitory receptors, have also been identified on subsets of NK cells [10–12]. However, these receptors cannot account for the NK cell-mediated lysis of HLA class I negative target cells. Indeed, the major receptors responsible for NK cell triggering in the process of natural cytotoxicity are not HLA class I-specific. Three receptors, termed NKp46, NKp30, and NKp44, which are referred to collectively as “natural cytotoxicity receptors” (NCR) were the first activating NK cell receptors to be identified and characterized molecularly [5]. Although a direct correlation has been established between the surface density of NCR on NK cells and the intensity of their cytolytic activity [13], only limited information is available regarding the cell surface ligands recognized by NCRs [14, 15]. An additional activating receptor, NKG2D, is expressed not only by NK cells but also by cytotoxic T lymphocytes. NKG2D recognizes the stress-inducible MICA/B [16] and ULBPs proteins [17]. More recently, it has been shown that DNAM-1, a triggering receptor expressed by virtually all NK cells and partially shared with T lymphocytes and monocytes, specifically recognizes PVR (CD155) and Nectin-2 (CD112) [18], two members of the nectin family. Other activating surface molecules include 2B4, NTBA, and NKp80 [5], which contribute to NK cell triggering during the process of natural cytotoxicity. However, it appears that they primarily play a role as coreceptors, i.e., they may amplify the NK cell triggering induced by NCR or NKG2D. Although activating NK cell signals are, therefore, mediated by many receptors, all the available data are compatible with the concept that the ligands for NK cell-activating receptors are mainly expressed by “stressed” cells (e.g., tumor and infected cells or proliferating cells), for which genomic instability might be one hallmark [19].
These NK cell receptors—both inhibitory and activating—allowed for the subdivision of human NK cells and for the analysis of the anatomical distribution of distinct NK cell subsets, which resulted in new insights into NK cell immunobiology.
NK Cell Subsets
For many years, NK cells were considered to be a homogeneous lymphocyte population with excellent cytotoxic capability. Nowadays, NK cells rather appear to comprise various subsets that differ in function, in phenotype, and in anatomical localization.
In human peripheral blood (PB), the majority of NK cells (≥95%) belongs to the CD56dimCD16 + cytolytic NK subset [20–22]. These cells carry homing markers for inflamed peripheral sites and perforin to rapidly mediate cytotoxicity. The minor NK cell subset in blood (≤5%) is CD56brightCD16− [20–22]. These NK cells lack perforin (or display low level of it) but secrete large amounts of interferon (IFN)-γ and tumor necrosis factor (TNF)-α upon activation and are superior to CD56dim CD16 + NK cells in this latter function [21, 22]. CD56brightCD16− NK cells proliferate more vigorously than their CD56dimCD16 + counterparts. Consistent with this feature, they uniquely express the high-affinity receptor for interleukin (IL)-2 (CD25), the alpha chain of IL-7 receptor and CD117, the receptor for the stem cell factor, also named c-kit. In addition, they display homing markers for secondary lymphoid organs, namely, CCR7 and CD62L [20]. Notably, the major histocompatibility complex (MHC) class I allele-specific KIRs are expressed on subsets of CD56dimCD16 + cytolytic NK cells, whereas the immunoregulatory CD56brightCD16− NK subset expresses uniformly CD94/NKG2A and lacks KIRs [21]. Therefore, two main functional NK cell subsets have been characterized in peripheral blood, and we will discuss their anatomical distribution next.
NK Cells in Inflamed and Malignant Tissues
During inflammation, activated myeloid DCs (mDCs) and plasmacytoid DCs (pDCs) can secrete different chemokines such as MIP-1β/CCL4, RANTES/CCL5, fractalkine/CX3CL1, IL-8/CXCL8, and IP-10/CXCL10 [23–25]. The different NK cell subsets are able to migrate in response to these chemokines, according to the receptors they express. CD56lowCD16 + NK cells express CXCR1 and CX3CR1 [20, 26], receptors for IL-8/CXCL8 and fractalkine/CX3CL1, respectively. This last chemokine would mediate the firm adhesion of cells to the endothelium and subsequent migration toward IL-8 [27]. The CD56bright NK cells express CCR5, CXCR3, and CXCR4 which allow them to migrate in response to RANTES/CCL5, MIP-1β/CCL4, ITAC/CXCL11, and IP-10/CXCL10 [20]. Thus, DCs would attract the two types of NK cells in the site of inflammation, rendering their encounter possible. This hypothetical interaction is supported by in vivo observations, where NK cells have been found in close contact with DCs either in lesions of atopic eczema/dermatitis syndrome or in Gleevec-induced lichenoid dermatitis in gastrointestinal stromal tumor (GIST) bearing patients [28, 29]. However, in autoimmune inflamed lesions primarily the CD56brightCD16− NK cell subset was found to accumulate [30]. While NK cell infiltrates of inflamed tissues have been investigated to some extent, NK cells infiltrating malignant neoplasms have been poorly characterized, although it is clear that NK cells are capable of recognizing and killing tumor cells in vitro. In a recent study, nevertheless, NK cells have been isolated from non-small-cell lung cancers and analyzed in detail [31]. The CD56brightCD16− NK cell subset was consistently observed as being highly enriched in tumor infiltrate and displaying activation markers, including NKp44, CD69, and HLA-DR. Remarkably, lung cancers NK cells were mainly capable of producing cytokines rather than exerting direct cancer cell killing. A similar pattern of NK cell infiltration, characterized by abundant CD56brightCD16− NK cells with low cytolytic activity, has been also reported in renal cell carcinoma [32, 33]. It might be possible that a chemokine environment produced by stromal leukocytes might provide anchorage signals to tumor-infiltrating NK cells. For instance, it has been suggested that heterodimerization of the chemokine receptor CCR5, i.e., the receptor of MIP-1β, leads to an adhesive signal and to the arrest of leukocytes within tissue [34]. Consistent with this notion, only CD56brightCD16− PB NK cells express CCR5 [20], which might explain their accumulation in tumor tissues. Thus both CD56dimCD16+ and CD56brightCD16− NK cells might get attracted to inflamed tissues including tumor sites, but only the latter subset might be retained there.
NK Cells in Secondary Lymphoid Tissues
Recent reports have shown that a substantial amount of human NK cells home to secondary lymphoid organs (SLO). These account for around 5% of mononuclear cells in uninflamed lymph nodes (LN) and 0.4–1% in inflamed tonsils and LN [35, 36]. These NK cells constitute a remarkable pool of innate effector cells, since LN harbor 40% of all lymphocytes, whereas peripheral blood contains only 2% of all lymphocytes [37, 38]. Therefore, LN NK cells are under physiological conditions ten times more abundant than blood NK cells. Remarkably, SLOs might not only constitute substantial reservoirs of NK cells, but might even harbor NK cell subsets that are functionally distinct from their peripheral blood counterparts. For instance, NKp44+ cells of the mucosal tissue overlaying tonsils and Peyer’s patches might primarily secrete IL-22 upon activation [39–43]. This peculiar NK cell receptor carrying cellular subset expresses RORγt and seems to play a pivotal role in maintaining mucosal homeostasis. However, IL-22+ NK-like cells are rare cells with an exclusive localization in the mucosa surrounding the lymphoid follicles, which might be related to the putative protective role of IL-22, triggering bactericidal peptide production against microbial invasion at epithelial and mucosal surfaces. More in general, and as might be anticipated from their CCR7 and CD62L expression, SLO NK cells are mainly composed of the CD56brightCD16− NK cells subset [36]. Like CD56brightCD16− NK cells of peripheral blood, secondary lymphoid tissue NK cells are perforin low/negative and show extremely poor cytolytic activity. Distinguishing CD56brightCD16− SLO NK cells from their PB counterparts, however, is their around fivefold elevated ability to produce IFN-γ in response to activation [44, 45]. In addition, perforin and cytotoxicity can be promptly upregulated on SLO NK cells by cytokines, and at the same time, these cells acquire the expression of CD16, as well as KIRs [36]. Therefore, activation seems to convert secondary lymphoid organ NK cells into cytotoxic effector analogous to blood CD56dimCD16+ NK cells. Several studies have indeed recently investigated whether PB-CD56brightCD16− NK cells give rise in vitro and in vivo to cells akin to CD56dimCD16+ NK cells and whether SLO can be sites of NK cell maturation [46–48]. These investigations support the hypothesis that CD56dimCD16+ develop from CD56brightCD16− NK cells, and that this differentiation can take place both during immune activation in inflamed peripheral tissues, such as reactive LN, and in the steady state. Consistent with this hypothesis, CD56brightCD16− NK cells have longer telomeres than CD56dimCD16+ NK cells, and could therefore be their progeny [46, 47]. However, why do NK cells accumulate in SLOs. One possibility is that the resident population of NK cells in lymph node might be derived from blood CD56brightCD16− NK cells, which express CCR7 and CD62L, two molecules involved in lymphocyte trafficking to lymphoid tissues [20]. Alternatively, NK cells might also differentiate directly in SLOs from resident CD34dimCD45RA+ hematopoietic progenitor cells [49]. In addition, thymus-derived CD127+ mouse NK cells, which show characteristics reminiscent of human CD56brightCD16− NK cells, have been demonstrated to repopulate peripheral lymphoid organs [50]. Finally, long-lived NK cell populations might preferentially home to these sites [51]. Therefore, NK cells harbored in secondary lymphoid tissues could either home there from the blood after their generation in bone marrow or thymus and/or develop there as an independent NK-cell lineage.
NK Cell Activation by DCs
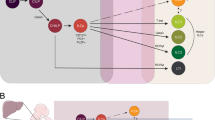
The realization that NK cells are not a homogenous population of innate lymphocytes, but can mount various types of innate immune responses that are either dominated by cytotoxicity or immunoregulatory cytokine secretion, suggests that the repertoire of NK cell responses might be differentially triggered by accessory cells. Dendritic cells (DCs) have been identified as one type of NK cell-activating cells, and preactivation of NK cells by DCs was even found to be required for efficient innate immune responses by these innate lymphocytes against a variety of pathogenic challenges in the mouse [52–54]. Previously, it has been realized that NK cells can be efficiently activated by DCs to elicit antitumor immune responses in mice [55] and to stimulate IFN-γ production, proliferation, and enhanced cytotoxicity of NK cells in humans [56–58]. Of the above-discussed NK cell subsets, the CD56brightCD16− NK cells, which are enriched in secondary lymphoid tissues [35, 36], were found to be especially responsive to activation by DCs [59, 60]. Myeloid DCs preferentially home to secondary lymphoid tissues upon encounter of inflammatory stimuli as well as pathogen constituents and were found to interact with NK cells in perifollicular T cell zones at these sites [59, 61, 62]. However, not only myeloid, but also plasmacytoid DCs can activate NK cells, and these two DC subsets were suggested to stimulate different NK cell functions via distinct signals [63] (Fig. 1). Plasmacytoid DCs have been identified as the main type I IFN-secreting cells upon pathogen encounter [64] and preferentially augment NK cell cytotoxicity via this cytokine [63]. Accordingly, protective NK cell cytotoxicity during infection with the murine cytomegalovirus (MCMV) was found to be type I IFN dependent [65], and these cytokines were preferentially produced by plasmacytoid DCs during this infection [66]. In contrast, myeloid DCs stimulate IFN-γ production by NK cells efficiently via IL-12 and IL-18 [45, 59, 63, 67]. In good agreement, IFN-γ production by NK cells during MCMV infection in vivo was also primarily IL-12 dependent [68]. IL-12 which is essential for resistance to MCMV, comes primarily from myeloid DCs for this innate immune response [66]. Therefore, plasmacytoid and myeloid DCs regulate different aspects of NK cell activation with type I IFN of plasmacytoid DCs increasing NK cell cytotoxicity and IL-12/IL-18 by myeloid DCs primarily triggering cytokine production by NK cells. In contrast to the fairly clear roles of type I IFNs and IL-12 in NK cell activation by DCs, multiple functions have been assigned to IL-15 in this interaction. IL-15 presented on DCs via binding to IL-15Rα has been suggested to stimulate NK cell proliferation [59], survival [69, 70], type I IFN production [71], and priming of protective NK cell responses [52]. These functions, in addition to its crucial role during NK cell development [72, 73], identify IL-15 as an essential cytokine for NK cell development and activity, which, depending on the amount produced by DCs under certain stimulation conditions, can probably trigger most NK cell functions. Along these lines, Langerhans cells, a DC subset originally described in skin, was found to produce more IL-15 than other myeloid DC subpopulations, thereby supporting NK cell survival efficiently [74]. Vice versa, maturation with polyinosinic-polycytidylic acid (polyI:C), a toll-like receptor (TLR) 3 agonist, elicited more IL-12 production by myeloid DCs than other maturation stimuli and the resulting DCs stimulated proliferation, cytokine secretion, and enhanced cytotoxicity induction primarily via this cytokine [45]. These studies suggest that NK cells require activation by DCs to reach their full functional potential, and this activation depends on the DC and NK cell subset involved as well as by which stimulus the DCs have been matured.

The cross-talk between dendritic cells (DCs) and NK cells and their shared receptors. Myeloid DCs (mDCs) primarily activate NK cells to secrete cytokines like IFN-γ and to proliferate/survive via production of IL-12/18 and IL-15, respectively. In contrast, NK cell cytotoxicity is primarily triggered via IFN-α/β, which is mainly produced by plasmacytoid DCs (pDCs). NK cells, mDCs and pDCs share pathogen-associated molecular pattern (PAMP) recognition via TLRs. In addition, both NK cells and mDCs get activated upon encounter of antibody opsonized targets via CD16/FcγRIIIA and inhibited via MHC class I recognition by ILT2/LIR1. Contrary to this synchronized activation or inhibition of mDCs and NK cells, pDCs get inhibited to secrete type I IFNs by the activating NK cell receptor NKp44, and vice versa activated to produce IFN-α/β by the inhibitory NK cell receptor IRp60 (CD300a)
NK Cell Assistance in T-Cell Priming by DCs
NK cells activated in this fashion can fulfill multiple functions. In addition to the above-discussed mechanisms for infected and transformed target cell recognition, they can also shape consecutive adaptive immune responses by causing further DC maturation and influencing the polarization of primary T-cell responses. In contrast to editing of immune responses by NK cells that will be discussed in the next section, these functions support protective immune responses against intracellular pathogens and tumors that are most efficiently targeted by Th1 polarized cell-mediated immunity [61, 75, 76]. NK cell assistance in T-cell priming by DCs is best achieved by CD56brightCD16− NK cells [44] and in secondary lymphoid tissues [77], where only low numbers of cytotoxic NK cells are present [58]. NK cell mediate DC differentiation and maturation primarily via TNF and IFN-γ [57, 58, 78]. TNF mainly mediates phenotypical DC maturation with the up-regulation of costimulatory receptors [57, 58], while IFN-γ is required for the differentiation of monocytes into tumor necrosis factor and inducible nitric oxide synthase producing DCs (TipDCs) during granuloma formation like after Listeria monocytogenes infection [78]. DC differentiation and maturation allows for the priming of protective adaptive T-cell responses, therefore offering with NK cell recognition and alternative mode of maturation apart from direct recognition of pathogen constituents by DCs [79–81]. Apart from this role in DC differentiation and maturation, IFN-γ produced by NK cells contributes to Th1 polarization by probably also directly acting on T cells during priming [44, 61, 75, 77]. Interestingly, cytokine secreting CD56brightCD16− NK cells are not only enriched in secondary lymphoid tissues, where DC-mediated T-cell priming mainly takes place, but these cells produce also around fivefold more IFN-γ than their peripheral blood counterparts and are, therefore, especially suited to assist in Th1 polarization of primary T-cell responses [44, 45]. These findings document that NK cells are not only activated by DCs, but also influence, in turn, adaptive immune responses that are initiated by these antigen-presenting cells (APCs).
NK Cell Editing of Myeloid APCs
In addition to their support of adaptive immune responses via DC maturation and T-cell polarization, NK cells have also the ability to edit myeloid APCs, including macrophages, DCs, and microglia, in order to possibly avoid immunopathology. This preferentially occurs at high numbers of activated NK cells [58]. Immature DCs can be killed by activated NK cells, and this killing is dependent on the activating NK cell receptors NKp30, NKp46, and DNAM-1 [56, 82, 83]. In contrast, mature DCs are protected from NK cell cytotoxicity via their up-regulation of MHC class I molecules [56]. Especially up-regulation of the nonclassical HLA class I molecule HLA-E and its mouse counterpart Qa1b mediates this protection against mainly CD94/NKG2A carrying NK cells [84, 85]. This cytolytic DC editing by NK cells has been suggested to reduce graft-versus-host disease in bone marrow transplantation [86] and graft rejection in solid organ transplantation [87, 88]. During ameliorated skin graft rejection via this mechanism NK cells homed to lymph nodes and killed there allogeneic DCs in a perforin-dependent manner [88]. Therefore, cytolytic DC editing can limit pathological immune responses. DCs are, however, not the only myeloid APCs that can be edited by activated NK cells. Macrophages can also be targeted by NK cell cytotoxicity. Contrary to DCs, however, they become more susceptible to NK cell lysis by activation with TLR agonists [89–91] or infection [92, 93]. Especially high doses of lipopolysaccharide (LPS), a TLR4 agonist, renders macrophages susceptible to NKG2D-dependent killing by NK cells [89, 90]. Thus, activated, but not resting macrophages are targeted by activated NK cells. A third APC population that can be edited by NK cell cytotoxicity are microglial cells, resident APCs of the central nervous system (CNS) [94, 95]. Similar to DCs, resting microglia is sensitive to NK cell cytotoxicity, whereas activation by TLR4 ligation with LPS protects these cells from NK cell lysis by up-regulation of MHC class I molecules [95]. Resting microglia recognition is mediated by the activating NK cell receptors NKG2D and NKp46 [95]. These studies suggest that resting DCs, activated macrophages, and resting microglial cells can be targeted by activated NK cells, and cytotoxic editing of these myeloid APCs attenuates immunopathology, for example, in transplantation settings.
NK Cell and DC Recognition of Similar Stimuli
Consistent with their concomitant role during the early phase of immune responses, NK cells and DCs are often able to sense similar stimuli in parallel. It has been reported that stimuli acting on TLRs not only activate immature dendritic cells but also render NK cells more prone to receive triggering signals from pathogen-associated molecules, thus exerting a regulatory control on the early steps of innate immune responses against infectious agents [96]. While TLR engagement results similarly for both DCs and NK cells in activation, recognition of self-MHC class I molecules, the main mechanism to restrain NK cell activity, can also inhibit other leukocyte populations. For intance, myelomonocytic cells, including myeloid DCs (mDCs), express ILT2, an inhibitory receptor expressed on subsets of NK cells. This receptor binds MHC class I molecules and delivers a negative signal that inhibits killing by NK cells. In addition, ILT2 engagement also dampens Ca2+ mobilization in mDCs triggered through human histocompatibility leukocyte antigens (HLA)-DR [97, 98]. Most recently, plasmacytoid DCs (pDCs), also known as natural interferon-producing cells, have been shown to express receptors that are typically employed for NK cell recognition. One of these receptor, NKp44, is present on a subset of pDCs in SLOs and is inducible on PB pDCs after in vitro culture with interleukin 3. Crosslinking of NKp44 on NK cells is associated with triggering of NK cell-mediated cytotoxicity. Paradoxically, crosslinking of NKp44 on pDCs does not trigger their functions but significantly inhibits interferon (IFN)-α production in response to cytosine-phosphate-guanosine (CpG) oligonucleotides [99]. Since IFN-α is a potent inducer of NK cell cytotoxicity, the recognition of the same ligand by receptors mediating divergent outcomes might appear contradictory. Nevertheless, pDCs in tonsils are in close contact with CD8+ T cells that are able to produce IL-3 [99]. In this context, expression of NKp44 on pDCs and the inhibition of IFN-α by putative NKp44 ligands would occur when an adaptive response is already taking place, and further, NK cell activation might no longer be required. In addition, IRp60 (CD300a) is another inhibitory receptor expressed by NK cells, as well as by many other leukocytes and also displayed an unpredicted role when cross-linked on pDCs. IRp60 triggering reduced, as expected, TNF-α but increased IFN-α secretion by pDCs [100]. Because exogenous TNF-α, a cytokine abundantly released by activated NK cells, inhibits IFN-α secretion by pDCs [101], these new findings support the notion that there is a balance between IFN-α and TNF-α levels produced by pDC and NK cells. However, pDCs express also CD300c, which shares with CD300a 80% amino acid sequence similarity in their Ig domains. CD300a contains three immunoreceptor tyrosine-based inhibitory motifs (ITIMs), of which at least one is functional in NK cells [102]. CD300c has a short intracellular domain with charged amino acid in the transmembrane domain, which may be associated with other signaling molecules. Thus, although these CD300 receptors may share a common ligand, they almost certainly have different intracellular pDC signaling capabilities. Defining the natural ligands of NKp44 and CD300a/c will shed further light on the complex cross-talk between DCs and NK cells upon recognition of similar stimuli early during immune responses, setting eventually the stage for establishing adaptive immune responses that confer long-term protection.
Conclusions
The cross-talk between NK cells and DCs suggests a critical role for NK cells in the initiation and regulation of immune responses. The considerable knowledge on the molecular basis of these cellular interactions offers opportunities for clinical intervention exploiting DC/NK cell cooperation. Indeed, NK cell activation by DCs is particularly efficient since DC promote both effector functions and survival or proliferation of NK cells. In addition, recent publications in the field of semi-allogeneic bone marrow transplantation have highlighted a beneficial role of NK cells in mediated graft-versus-leukemia effects, and NK cell-based immunotherapies are, therefore, currently being reconsidered [103]. Along these lines, the ability of NK cells to kill tumor cells may facilitate the generation of tumor-derived antigenic material that can be efficiently presented by DCs, further accelerating the induction of tumor-specific immunity [81].
In addition to their traditional role as major innate cytotoxic cells, NK cells have now been shown to regulate immune responses. On one hand, they provide immunoregulatory “helper” functions, being able to activate DCs for pro-inflammatory cytokine production and for efficient Th1 and cytotoxic T lymphocyte (CTL) stimulation. As a matter of fact, DCs activated by NK cells are better inducers of antitumor CTL response, at least in vitro, when compared with the standard mature DCs currently employed in DC-based clinical trials [104]. On the other hand, they can edit myeloid APC populations to attenuate and direct immune responses [105]
All these considerations provide a strong rationale for a combined targeting of NK cells and DCs in novel immunotherapeutic strategies, harnessing this cellular cross-talk in the treatment of patients with cancer and chronic infections that are resistant to conventional therapies.
References
Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998;392:245–52.
Trinchieri G. Biology of natural killer cells. Adv Immunol 1989;47:187–376.
Ljunggren HG, Karre K. Host resistance directed selectively against H-2-deficient lymphoma variants. Analysis of the mechanism. J Exp Med 1985;162:1745–59.
Yokoyama WM, Kim S, French AR. The dynamic life of natural killer cells. Annu Rev Immunol 2004;22:405–29.
Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol 2001;19:197–223.
Lanier LL. NK cell recognition. Annu Rev Immunol 2005;23:225–74.
Long EO. Negative signaling by inhibitory receptors: the NK cell paradigm. Immunol Rev 2008;224:70–84.
Perussia B, Starr S, Abraham S, Fanning V, Trinchieri G. Human natural killer cells analyzed by B73.1, a monoclonal antibody blocking Fc receptor functions. I. Characterization of the lymphocyte subset reactive with B73.1. J Immunol 1983;130:2133–41.
Perussia B, Acuto O, Terhorst C, Faust J, Lazarus R, Fanning V, et al. Human natural killer cells analyzed by B73.1, a monoclonal antibody blocking Fc receptor functions. II. Studies of B73.1 antibody-antigen interaction on the lymphocyte membrane. J Immunol 1983;130:2142–8.
Moretta A, Sivori S, Vitale M, Pende D, Morelli L, Augugliaro R, et al. Existence of both inhibitory (p58) and activatory (p50) receptors for HLA-C molecules in human natural killer cells. J Exp Med 1995;182:875–84.
Biassoni R, Cantoni C, Falco M, Verdiani S, Bottino C, Vitale M, et al. The human leukocyte antigen (HLA)-C-specific “activatory” or “inhibitory” natural killer cell receptors display highly homologous extracellular domains but differ in their transmembrane and intracytoplasmic portions. J Exp Med 1996;183:645–50.
Bottino C, Sivori S, Vitale M, Cantoni C, Falco M, Pende D, et al. A novel surface molecule homologous to the p58/p50 family of receptors is selectively expressed on a subset of human natural killer cells and induces both triggering of cell functions and proliferation. Eur J Immunol 1996;26:1816–24.
Sivori S, Pende D, Bottino C, Marcenaro E, Pessino A, Biassoni R, et al. NKp46 is the major triggering receptor involved in the natural cytotoxicity of fresh or cultured human NK cells. Correlation between surface density of NKp46 and natural cytotoxicity against autologous, allogeneic or xenogeneic target cells. Eur J Immunol 1999;29:1656–66.
Pogge von Strandmann E, Simhadri VR, von Tresckow B, Sasse S, Reiners KS, Hansen HP, et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity 2007;27:965–74.
Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, et al. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 2001;409:1055–60.
Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999;285:727–9.
Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001;14:123–33.
Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med 2003;198:557–67.
Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005;436:1186–90.
Campbell JJ, Qin S, Unutmaz D, Soler D, Murphy KE, Hodge MR, et al. Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J Immunol 2001;166:6477–82.
Jacobs R, Hintzen G, Kemper A, Beul K, Kempf S, Behrens G, et al. CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur J Immunol 2001;31:3121–7.
Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56bright subset. Blood 2001;97:3146–51.
Penna G, Sozzani S, Adorini L. Cutting edge: selective usage of chemokine receptors by plasmacytoid dendritic cells. J Immunol 2001;167:1862–6.
Papadopoulos EJ, Sassetti C, Saeki H, Yamada N, Kawamura T, Fitzhugh DJ, et al. Fractalkine, a CX3C chemokine, is expressed by dendritic cells and is up-regulated upon dendritic cell maturation. Eur J Immunol 1999;29:2551–9.
Megjugorac NJ, Young HA, Amrute SB, Olshalsky SL, Fitzgerald-Bocarsly P. Virally stimulated plasmacytoid dendritic cells produce chemokines and induce migration of T and NK cells. J Leukoc Biol 2004;75:504–14.
Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature 1997;385:640–4.
Nishimura M, Umehara H, Nakayama T, Yoneda O, Hieshima K, Kakizaki M, et al. Dual functions of fractalkine/CX3C ligand 1 in trafficking of perforin +/granzyme B + cytotoxic effector lymphocytes that are defined by CX3CR1 expression. J Immunol 2002;168:6173–80.
Buentke E, Heffler LC, Wilson JL, Wallin RP, Lofman C, Chambers BJ, et al. Natural killer and dendritic cell contact in lesional atopic dermatitis skin–Malassezia-influenced cell interaction. J Invest Dermatol 2002;119:850–7.
Borg C, Terme M, Taieb J, Menard C, Flament C, Robert C, et al. Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J Clin Invest 2004;114:379–88.
Dalbeth N, Gundle R, Davies RJ, Lee YC, McMichael AJ, Callan MF. CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation. J Immunol 2004;173:6418–26.
Carrega P, Morandi B, Costa R, Frumento G, Forte G, Altavilla G, et al. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56brightCD16- cells and display an impaired capability to kill tumor cells. Cancer 2008;112:863–75.
Schleypen JS, Baur N, Kammerer R, Nelson PJ, Rohrmann K, Grone EF, et al. Cytotoxic markers and frequency predict functional capacity of natural killer cells infiltrating renal cell carcinoma. Clin Cancer Res 2006;12:718–25.
Schleypen JS, Von Geldern M, Weiss EH, Kotzias N, Rohrmann K, Schendel DJ, et al. Renal cell carcinoma-infiltrating natural killer cells express differential repertoires of activating and inhibitory receptors and are inhibited by specific HLA class I allotypes. Int J Cancer 2003;106:905–12.
Buckley CD, Simmons DL. Sticky moments with sticky molecules. Immunol Today 2000;21:601–3.
Fehniger TA, Cooper MA, Nuovo GJ, Cella M, Facchetti F, Colonna M, et al. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell derived IL-2: a potential new link between adaptive and innate immunity. Blood 2003;102:3052–57.
Ferlazzo G, Thomas D, Lin SL, Goodman K, Morandi B, Muller WA, et al. The abundant NK cells in human lymphoid tissues require activation to express killer cell Ig-like receptors and become cytolytic. Journal of Immunology 2004;172:1455–62.
Westermann J, Pabst R. Distribution of lymphocyte subsets and natural killer cells in the human body. Clin Investig 1992;70:539–44.
Trepel F. Number and distribution of lymphocytes in man. A critical analysis. Klin Wochenschr 1974;52:511–5.
Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009;457:722–5.
Luci C, Reynders A, Ivanov II, Cognet C, Chiche L, Chasson L, et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol 2009;10:75–82.
Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46 + cells. Nat Immunol 2009;10:83–91.
Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 2008;29:958–70.
Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 2008;29:947–57.
Morandi B, Bougras G, Muller WA, Ferlazzo G, Münz C. NK cells of human secondary lymphoid tissues enhance T cell polarization via IFN-gamma secretion. Eur J Immunol 2006;36:2394–400.
Strowig T, Brilot F, Arrey F, Bougras G, Thomas D, Muller WA, et al. Tonsilar natural killer cells restrict B cell transformation by the Epstein Barr virus via IFN-γ. PLoS Pathog 2008;4:e27.
Romagnani C, Juelke K, Falco M, Morandi B, D’Agostino A, Costa R, et al. CD56brightCD16- killer Ig-like receptor- NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J Immunol 2007;178:4947–55.
Chan A, Hong DL, Atzberger A, Kollnberger S, Filer AD, Buckley CD, et al. CD56bright human NK cells differentiate into CD56dim cells: role of contact with peripheral fibroblasts. J Immunol 2007;179:89–94.
Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med 2009;206:25–34.
Freud AG, Becknell B, Roychowdhury S, Mao HC, Ferketich AK, Nuovo GJ, et al. A human CD34 + subset resides in lymph nodes and differentiates into CD56bright natural killer cells. Immunity 2005;22:295–304.
Vosshenrich CA, Garcia-Ojeda ME, Samson-Villeger SI, Pasqualetto V, Enault L, Richard-Le Goff O, et al. A thymic pathway of mouse natural killer cell development characterized by expression of GATA-3 and CD127. Nat Immunol 2006;7:1217–24.
Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature 2009;457:557–61.
Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007;26:503–17.
Kassim SH, Rajasagi NK, Zhao X, Chervenak R, Jennings SR. In vivo ablation of CD11c-positive dendritic cells increases susceptibility to herpes simplex virus type 1 infection and diminishes NK and T-cell responses. J Virol 2006;80:3985–93.
Andrews DM, Scalzo AA, Yokoyama WM, Smyth MJ, Degli-Esposti MA. Functional interactions between dendritic cells and NK cells during viral infection. Nat Immunol 2003;4:175–81.
Fernandez NC, Lozier A, Flament C, Ricciardi-Castagnoli P, Bellet D, Suter M, et al. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat Med 1999;5:405–11.
Ferlazzo G, Tsang ML, Moretta L, Melioli G, Steinman RM, Münz C. Human dendritic cells activate resting NK cells and are recognized via the NKp30 receptor by activated NK cells. J Exp Med 2002;195:343–51.
Gerosa F, Baldani-Guerra B, Nisii C, Marchesini V, Carra G, Trinchieri G. Reciprocal activating interaction between natural killer cells and dendritic cells. J Exp Med 2002;195:327–33.
Piccioli D, Sbrana S, Melandri E, Valiante NM. Contact-dependent stimulation and inhibition of dendritic cells by natural killer cells. J Exp Med 2002;195:335–41.
Ferlazzo G, Thomas D, Pack M, Paludan C, Schmid D, Strowig T, et al. Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. Proc Natl Acad Sci USA 2004;101:16606–11.
Vitale M, Della Chiesa M, Carlomagno S, Romagnani C, Thiel A, Moretta L, et al. The small subset of CD56brightCD16- natural killer cells is selectively responsible for both cell proliferation and interferon-gamma production upon interaction with dendritic cells. Eur J Immunol 2004;34:1715–22.
Bajenoff M, Breart B, Huang AY, Qi H, Cazareth J, Braud VM, et al. Natural killer cell behavior in lymph nodes revealed by static and real-time imaging. J Exp Med 2006;203:619–31.
Garrod KR, Wei SH, Parker I, Cahalan MD. Natural killer cells actively patrol peripheral lymph nodes forming stable conjugates to eliminate MHC-mismatched targets. Proc Natl Acad Sci USA 2007;104:12081–6.
Gerosa F, Gobbi A, Zorzi P, Burg S, Briere F, Carra G, et al. The reciprocal interaction of NK cells with plasmacytoid or myeloid dendritic cells profoundly affects innate resistance functions. J Immunol 2005;174:727–34.
Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol 2008;8:594–606.
Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei XQ, Liew FY, et al. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J Immunol 2002;169:4279–87.
Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, et al. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med 2002;195:517–28.
Andoniou CE, van Dommelen SL, Voigt V, Andrews DM, Brizard G, Asselin-Paturel C, et al. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol 2005;6:1011–19.
Orange JS, Biron CA. An absolute and restricted requirement for IL-12 in natural killer cell IFN-gamma production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J Immunol 1996;156:1138–42.
Brilot F, Strowig T, Roberts SM, Arrey F, Münz C. NK cell survival mediated through the regulatory synapse with human dendritic cells requires IL-15Ralpha. J Clin Invest 2007;117:3316–29.
Huntington ND, Puthalakath H, Gunn P, Naik E, Michalak EM, Smyth MJ, et al. Interleukin 15-mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat Immunol 2007;8:856–63.
Jinushi M, Takehara T, Tatsumi T, Kanto T, Groh V, Spies T, et al. Autocrine/paracrine IL-15 that is required for type I IFN-mediated dendritic cell expression of MHC class I-related chain A and B is impaired in hepatitis C virus infection. J Immunol 2003;171:5423–9.
Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med 2000;191:771–80.
Koka R, Burkett PR, Chien M, Chai S, Chan F, Lodolce JP, et al. Interleukin (IL)-15Ralpha-deficient natural killer cells survive in normal but not IL-15Ralpha-deficient mice. J Exp Med 2003;197:977–84.
Münz C, Dao T, Ferlazzo G, De Cos MA, Goodman K, Young JW. Mature myeloid dendritic cell subsets have distinct roles for activation and viability of circulating human natural killer cells. Blood 2005;105:266–73.
Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol 2005;6:600–7.
Ing R, Stevenson MM. Dendritic cell and NK cell reciprocal cross talk promotes gamma interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect Immun 2009;77:770–82.
Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol 2004;5:1260–5.
Kang SJ, Liang HE, Reizis B, Locksley RM. Regulation of hierarchical clustering and activation of innate immune cells by dendritic cells. Immunity 2008;29:819–33.
Mocikat R, Braumuller H, Gumy A, Egeter O, Ziegler H, Reusch U, et al. Natural killer cells activated by MHC class Ilow targets prime dendritic cells to induce protective CD8 T cell responses. Immunity 2003;19:561–9.
Adam C, King S, Allgeier T, Braumuller H, Luking C, Mysliwietz J, et al. DC-NK cell cross talk as a novel CD4 + T-cell-independent pathway for antitumor CTL induction. Blood 2005;106:338–44.
Münz C, Steinman RM, Fujii S. Dendritic cell maturation by innate lymphocytes: coordinated stimulation of innate and adaptive immunity. J Exp Med 2005;202:203–7.
Spaggiari GM, Carosio R, Pende D, Marcenaro S, Rivera P, Zocchi MR, et al. NK cell-mediated lysis of autologous antigen-presenting cells is triggered by the engagement of the phosphatidylinositol 3-kinase upon ligation of the natural cytotoxicity receptors NKp30 and NKp46. Eur J Immunol 2001;31:1656–65.
Pende D, Castriconi R, Romagnani P, Spaggiari GM, Marcenaro S, Dondero A, et al. Expression of the DNAM-1 ligands, Nectin-2 (CD112) and poliovirus receptor (CD155), on dendritic cells: relevance for natural killer-dendritic cell interaction. Blood 2006;107:2030–6.
Della Chiesa M, Vitale M, Carlomagno S, Ferlazzo G, Moretta L, Moretta A. The natural killer cell-mediated killing of autologous dendritic cells is confined to a cell subset expressing CD94/NKG2A, but lacking inhibitory killer Ig-like receptors. Eur J Immunol 2003;33:1657–66.
Persson CM, Assarsson E, Vahlne G, Brodin P, Chambers BJ. Critical role of Qa1b in the protection of mature dendritic cells from NK cell-mediated killing. Scand J Immunol 2008;67:30–6.
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002;295:2097–100.
Yu G, Xu X, Vu MD, Kilpatrick ED, Li XC. NK cells promote transplant tolerance by killing donor antigen-presenting cells. J Exp Med 2006;203:1851–8.
Laffont S, Seillet C, Ortaldo J, Coudert JD, Guery JC. Natural killer cells recruited into lymph nodes inhibit alloreactive T-cell activation through perforin-mediated killing of donor allogeneic dendritic cells. Blood 2008;112:661–71.
Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol 2006;177:2051–5.
Nedvetzki S, Sowinski S, Eagle RA, Harris J, Vely F, Pende D, et al. Reciprocal regulation of human natural killer cells and macrophages associated with distinct immune synapses. Blood 2007;109:3776–85.
Basu S, Eriksson M, Pioli PA, Conejo-Garcia J, Mselle TF, Yamamoto S, et al. Human uterine NK cells interact with uterine macrophages via NKG2D upon stimulation with PAMPs. Am J Reprod Immunol 2009;61:52–61.
Vankayalapati R, Garg A, Porgador A, Griffith DE, Klucar P, Safi H, et al. Role of NK cell-activating receptors and their ligands in the lysis of mononuclear phagocytes infected with an intracellular bacterium. J Immunol 2005;175:4611–7.
Siren J, Sareneva T, Pirhonen J, Strengell M, Veckman V, Julkunen I, et al. Cytokine and contact-dependent activation of natural killer cells by influenza A or Sendai virus-infected macrophages. J Gen Virol 2004;85:2357–64.
Saikali P, Antel JP, Newcombe J, Chen Z, Freedman M, Blain M, et al. NKG2D-mediated cytotoxicity toward oligodendrocytes suggests a mechanism for tissue injury in multiple sclerosis. J Neurosci 2007;27:1220–8.
Lünemann A, Lünemann JD, Roberts S, Messmer B, Barreira da Silva R, Raine CS, et al. Human NK cells kill resting but not activated microglia via NKG2D- and NKp46-mediated recognition. J Immunol 2008;181:6170–7.
Sivori S, Falco M, Della Chiesa M, Carlomagno S, Vitale M, Moretta L, et al. CpG and double-stranded RNA trigger human NK cells by Toll-like receptors: induction of cytokine release and cytotoxicity against tumors and dendritic cells. Proc Natl Acad Sci USA 2004;101:10116–21.
Colonna M, Navarro F, Bellon T, Llano M, Garcia P, Samaridis J, et al. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J Exp Med 1997;186:1809–18.
Allan DS, McMichael AJ, Braud VM. The ILT family of leukocyte receptors. Immunobiology 2000;202:34–41.
Fuchs A, Cella M, Kondo T, Colonna M. Paradoxic inhibition of human natural interferon-producing cells by the activating receptor NKp44. Blood 2005;106:2076–82.
Ju X, Zenke M, Hart DN, Clark GJ. CD300a/c regulate type I interferon and TNF-alpha secretion by human plasmacytoid dendritic cells stimulated with TLR7 and TLR9 ligands. Blood 2008;112:1184–94.
Palucka AK, Blanck JP, Bennett L, Pascual V, Banchereau J. Cross-regulation of TNF and IFN-alpha in autoimmune diseases. Proc Natl Acad Sci USA 2005;102:3372–7.
Cantoni C, Bottino C, Augugliaro R, Morelli L, Marcenaro E, Castriconi R, et al. Molecular and functional characterization of IRp60, a member of the immunoglobulin superfamily that functions as an inhibitory receptor in human NK cells. Eur J Immunol 1999;29:3148–59.
Terme M, Ullrich E, Delahaye NF, Chaput N, Zitvogel L. Natural killer cell-directed therapies: moving from unexpected results to successful strategies. Nat Immunol 2008;9:486–94.
Kalinski P, Nakamura Y, Watchmaker P, Giermasz A, Muthuswamy R, Mailliard RB. Helper roles of NK and CD8+ T cells in the induction of tumor immunity. Polarized dendritic cells as cancer vaccines. Immunol Res 2006;36:137–46.
Moretta A. Natural killer cells and dendritic cells: rendezvous in abused tissues. Nat Rev Immunol 2002;2:957–64.
Acknowledgements
Research in our laboratories is supported by the Burroughs Wellcome Fund, the Starr Foundation, the National Cancer Institute (R01CA108609 and R01CA101741), the National Institute of Allergy and Infectious Diseases (RFP-NIH-NIAID-DAIDS-BAA-06-19), and the Foundation for the National Institutes of Health (Grand Challenges in Global Health) to C.M., and by Associazione Italiana Ricerca sul Cancro (AIRC), Ministero Italiano della Salute—Programma Straordinario Ricerca Oncologica, Fondazione Banco di Sicilia, and Regione Sicilia—Ricerca Sanitaria Regionale 2007, to G.F.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ferlazzo, G., Münz, C. Dendritic Cell Interactions with NK Cells from Different Tissues. J Clin Immunol 29, 265–273 (2009). https://doi.org/10.1007/s10875-009-9283-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-009-9283-y