
Abstract
A novel charge transfer complex (CTC), C14H10N2OS·C7H6N2S was synthesized by the reaction of 2-aminobenzothiazole (ABT) with its derivative Schiff base (2-((benzo[d]thiazol-2-ylimino) methyl) phenol is abbreviated as BYMBP) in THF. This complex was characterized by 1HNMR, 13CNMR, IR and UV–vis. Crystal structure of the complex has been determined by X-ray diffraction. The crystal is triclinic, space group P-1 with the unit cell parameters a = 9.7732(5), b = 9.9936(4), c = 11.0193(7) Å and α = 71.901(9), β = 78.686(10), γ = 70.397(9)°, Z = 2, R = 0.0499. wR 2 = 0.1190. The structure of the title complex was fully optimized at the RB3LYP/6-311++g(d,p) level of theory and CTC’s π–π stacking interaction, bond lengths (Å) and angles (°) were calculated.
Graphical Abstract
A novel 1:1 complex was synthesized because of π–π stacking , C–H⋯π and C–H⋯N interactions in its molecule.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The arrangements of the molecules forming the packed arrays are controlled by the interplay of several different weak intermolecular interactions: π–π stacking, C–H···π and C–H···N interactions, and these interactions bring about charge transfer. Charge transfer compounds are important components of optical material and have gradually been an active research area. Among these, D-A compounds exhibit particular optical properties because of their unique molecular structure, so they have been extensively investigated and widely used in luminescent, photoconductive, nonlinear optical material and so on [1–14]. Based on these features, more and more groups focus their research on charge transfer compounds, of which the ligands containing the conjugate systems of sulphur and nitrogen are widely studied currently. Amanym had studied charge transfer complexes of Schiff bases derived from 2-aminobenzothiazole with some acidic and nonacidic acceptors [15]. Dumur et al. has reported Q-TTF-Q(1) and Q-TTF-Q(2), these complexes by themselves contain electron donor–acceptor (EDA) which leads to charge transfer [16]. Liu et al. have shown charge transfer complex TTF–TCNAQ [17]. These phenomena are originated from interaction between electron donor and acceptor molecules in biomolecular equilibrium or in model compounds with intramolecular interaction, which has been an important topic of research in physical chemistry and biochemistry for many decades [1–14]. So, syntheses of these complexes are always an attractive theme in the area of physical chemistry and biochemistry.
In this paper, synthesis and crystal structure of a new charge transfer complex was reported. In addition, the structural details of the complex have been investigated using RB3LYP/6-311++g(d,p) calculations [18]. In the gas phase, π–π stacking interaction, bond lengths (Å) and angles (o) were calculated, and charge transfer (CT) can be seen in its molecule.
Experimental
General
Melting points were determined in sealed argon-filled capillaries and uncorrected. Carbon, hydrogen, and nitrogen analyse were performed by direct combustion on a Carlo Erba-1110 instrument; quoted data are the average of at least two independent determinations. The IR spectra were recorded on a Nicolet-550 FTIR spectrometer as KBr pellets. The single crystal X-ray diffraction data were recorded on a Rigaku Mercury CCD X-ray diffractometer. 1H NMR and 13C NMR spectra were recorded on a Unity Inova-300 spectrometer. UV–vis spectra were recorded on a Shimadzu UV-3150 spectrometer. All calculations were carried out with the RB3LYP/6-311++g(d,p) [18].
Synthesis
Synthesis and Character of Schiff Bases (BYMBP)
2-Aminobenzthiazole (ABT): The aniline (0.1 mol) and ammonium thiocyanate (0.2 mol) in 150 mL of glacial acetic acid were cooled in an ice bath and stirred mechanically. To the solution, bromine (0.2 mol) in 25 mL of glacial acetic acid was added dropwise at such a rate to keep the temperature below 10 °C throughout the addition. Stirring was continued for additional thirty minutes after the bromine addition. The precipitate of the benzthiazole hydrobromide was collected, dissolved in hot water and basified with a saturated sodium carbonate solution. The free substituted benzthiazol was collected, washed with water and dried under vacuum. Recrystallization from the appropriate solvent gave 2-aminobenzthiazol in good yield (88.5%). m.p.: 129–130 °C. 1HNMR (CDCl3, 300 MHz) (δ, ppm): 7.57–7.52 (m, 2 H, ArH), 7.29–7.12 (m, 2 H, ArH), 5.85 (brs, 2 H, –NH2); 13CNMR (CDC13, 75 MHz) (δ, ppm): 166.4, 152.0, 131.5, 126.0, 122.2, 120.9, 119.0. IR (KBr, cm−1): 3399(s), 3274(m), 3067(s), 2929(m), 1645(s), 1530(s), 1448(s), 1316(m), 1310(m), 1285(m), 1123(m), 1017(m), 920(m), 889(m), 741(s), 720(sh), 706(s), 687(m), 484(m).
Schiff bases (BTYMP): To a 10 mL EtOH solution of salicylaldehyde and a catalytic amount of formic acid was added a 5 mL EtOH solution of the appropriate amine (0.9 eq.). The mixture was refluxed for 8 h and the solution stored at 5 °C overnight. The resulting precipitate was collected by suction filtration and washed with Et2O (3 × 10 mL). Yield: 85% of an orange solid, m.p.: 150–151 °C. 1HNMR (CDC13, 300 MHz) (δ, ppm): 12.24 (s, 1 H, OH), 9.26 (s, 1 H, CH=N), 7.98 (d, J = 8.2 Hz, 1 H, Ar), 7.84 (d, J = 7.8 Hz, 1 H, Ar), 7.52–7.45 (ov m, 3 H, Ar), 7.40–7.35 (m, 1 H, Ar), 7.06 (d, J = 8.4 Hz, 1 H, Ar), 7.01–6.97 (m, 1 H, Ar); 13CNMR (CDC13, 75 MHz) (δ, ppm): 169.2, 167.6, 162.2, 151.7, 135.6, 134.8, 134.3, 126.9, 125.5, 123.2, 122.0, 120.0, 118.5, 117.9. IR(KBr, cm−1): 3300 (bs), 3055(s), 2949(s), 2873(s), 1606(v), 1570(vs), 1502(m), 1452(m), 1377(s), 1284(m), 1174(m), 1149(m), 1063(m), 901(s), 800(s), 754(s), 723(m), 679(m), 617(m).
Synthesis and Character of Charge Transfer Complex (CTC)
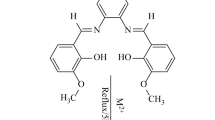
The CTC was prepared by mixing the equimolar amounts of BYMBP and ABT in THF at 60 °C for 8 h, during the time the pale yellow color changed gradually to yellow. The precipitate was removed from the reaction mixture by centrifugation, and THF was completely evaporated in vacuum, and ethanol was added to extract the product. Insoluble CTC was purified by being heated with ethanol several times to remove any contaminants, and the complex was filtered off, dried in vacuum. The crystal was grown from an ethanol-toluene mixture (1:10). Yellow microcrystals were obtained at room temperature in a few days, m.p.: 176–178 °C (dec). Anal. Calc. for C21H16N4OS2: C, 62.35; H, 3.99; N, 13.85; S, 15.85%. Found: C, 62.35; H, 3.99; N, 13.85; S, 15.85%. 1HNMR (CDC13, 300 MHz) (δ, ppm): 12.24 (s, 1 H, OH), 9.29 (s, 1 H, CH=N), 7.99 (d, J = 8.2 Hz, 1 H, Ar), 7.86 (d, J = 7.8 Hz, 1 H, Ar), 7.61–7.46 (ovm, 5 H, Ar), 7.38 (t, J = 7.7 Hz, 1 H, Ar), 7.32 (t, J = 7.7 Hz, 1 H, Ar), 7.14 (t, J = 7.8 Hz, 1 H, Ar), 7.07 (d, J = 8.4 Hz, 1 H, Ar), 7.01 (t, J = 7.6 Hz, 1 H, Ar), 5.25 (brs, 2 H, –NH2); 13CNMR (CDC13, 75 MHz) (δ, ppm): 170.4, 167.6, 135.7, 134.3, 127.0, 126.2, 125.5, 123.2, 122.6, 122.0, 121.1, 120.1, 119.5, 118.6, 118.5, 117.9. IR(KBr, cm−1): 3340(s,b), 3275(s,b), 3072(s), 2764(m), 2650(m), 1661(s), 1606(s), 1571(vs), 1541(s), 1501(s), 1478(s), 1448(s), 1316(m), 1288(s), 1255(m), 1239(m), 1182(s), 1149(s), 1123(m), 1064(m), 1017(m), 968(w), 931(w), 893(m), 889(s), 849(m), 788(m), 754(s), 724(s), 692(s).
Results and Discussion
Synthesis and Molecular Structure of the 1:1 Complex of ABT with BYMBP
BYMBP was prepared as described previously [15], and treated with 1 equiv of ABT in THF at 60 °C in order to prepare the corresponding 1:1 complex. The reaction went smoothly and after workup, the desired 1:1 complex was isolated in good yield as yellow crystals upon crystallization from toluene. Scheme 1 shows the reaction sequence.

Synthesis of ABT:BYMBP complex
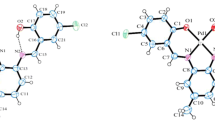
The IR spectra of this complex exhibited the strong absorptions near 1571 and 1541 cm−1 indicating partial C=N double bond character [19]. This complex was further characterized by X-ray structure analysis. Figure 1 and 2 show the molecular structure and the packing of the title compound, respectively. The value of calculation and test of selected bond lengths and bond angles are given in Table 1. Single-crystal X-ray structure determination reveals that one unit cell of the title compound contains two independent molecules BYMBP and ABT in its crystals. From Fig. 2 we see a very interesting property of ABT:BYMBP: along the b axis, the nearby units connecting with π–π stacking (ca.3.43 Å) of thiazole ring formed a 2D necklace-like supramolecular chain. These chains formed by N(3)–H(3B)···N(2) and N(3)–H(3A)···N(4) lying paralleled to each other, resulting in a two-dimensional hydrogen-bonded layer along the ab and ac plane, respectively (Fig. 2). The layers are further extended into a three-dimensional (3D) supramolecular array through hydrogen-bonded interactions, which are offset π–π stacking interactions with the face-to-face distance of ca. 3.43 Å between thiazole rings. Each uncoordinated N3 of thiazole from one chain forms an N···H–O hydrogen bond with a hydroxyl group from another nearby chain. The packing of the title compound can be described as parallel-sandwich geometry (Fig. 2). In addition, 2-aminobenzothiazole (ABT) acts as a donor and 2-((benzothiazol-2-ylimino)methyl)-4,6-di-tert-butylphenol (BYMBP) acts as a acceptor, four ABT occupy the four apical positions, formed the intermolecular hydrogen bond and π–π stacking of the complex when two ABT molecules were antiparallel, simultaneously, each ABT using the amino nitrogen atom and benzothiazole nitrogen atom of BYMBP, forms one kind of hydrogen bonds (N(3)–H(3B)···N(2), Table 1) [20, 21]. Each BYMBP making use of benzothiazole carbon atom and hydroxyl oxygen atom forms the other kind of hydrogen bonds (C(10)–H(10)···O(1), Table 1). Additionally, each ABT making use of thiazole nitrogen atom and amino nitrogen atom forms another kind of hydrogen bonds (N(3)–H(3A)···N(4), Table 1) in the same formation, and O(1)–H(1)···N(1) are the last hydrogen bonds which are the weakest in this 1:1 complex. It is very interestingly observed in this 2D array, and they are four types of hydrogen-bonded patterns A–D. All calculations were carried out with the RB3LYP/6-311++g(d,p) [18], and the result of the calculations were in accordance with the result of using X-Ray.

Crystal structure of ABT:BYMBP complex

Packing of the ABT:BYMBP complex
RB3LYP/6-311++g(d,p) Calculations
The structures of ABT:BYMBP optimized by the RB3LYP/6-311++g(d,p) [18] approach are shown in Fig. 3. The calculated bond lengths, bond and torsion angles are given in Table 2. Structure (a) shows a monomer of the 1:1 complex in which the hydroxyl group of BYMBP interacts with the amine group of ABT, through the N(3)–H(3B)···N(4) hydrogen bond of 2.121 Å, shorter by 0.001 Å than in the crystal (Table 2). In the optimized structure (b) the hydroxyl group of BYMBP is engaged in the N(3)–H(3A)···N(4) hydrogen bond, which is by 0.326 Å longer than in the crystal.

The optimized structures of ABT:BYMBP by the RB3LYP/6-311++g(d,p) method, a monomer connected by the N(3)–H(3B)···N(4) hydrogen bond, b monomer connected by N(3)–H(3A)···N(4) hydrogen bond
X-Ray Crystallography
Diffraction data were collected on a Rigaku Mercury CCD area detector. The structure was solved by direct methods and refined by full-matrix least-squares procedures based on |F|2. All non-hydrogen atoms were refined with anisotropic displacement coefficients. Hydrogen atoms were treated as idealized contributions. The structure was solved and refined using SHELXS-97 programs. Crystal and refinement data for complex are listed in Table 3.
Ultraviolet Spectral Studies
The electronic absorption spectra of the donor (ABT), acceptors (BYMBP) and the resulting complex in ethanol were recorded by UV–vis with a 1 cm quartz cell at room temperature. The ultraviolet spectra of these compounds are shown in Fig. 4. ABT:BYMBP compound shows a moderately intense absorption band at 376 nm, while such a band is absent in the parent ABT. However, at 326 nm for the parent BYMBP, the CT band with a red shift, ∆λ about 50 nm, indicates that BYMBP is an efficient electron acceptor, which makes the charge transfer (CT) easier. Thus, ABT:BYMBP compound is a good D-A compound.

Absorption spectra of ABT, BYMBP and ABT:BYMBP(10−6 mol L−1) in ethanol
Conclusion
The 1:1 complex of BYMBP with ABT was obtained. According to the structure analysis, three chemistry interactions can be seen in its structure, π–π stacking, hydrogen bond and charge transfer. Synthesizing more 1:1 complex remains an important task for further research.
Supplementary Data
Additional crystallographic data have been deposited at the Cambridge Crystallographic Data Centre, CCDC reference number is 746489 for the complex. The data can be obtained free of charge from 178 www.ccdc.cam.ac.uk/conts/retrieving.html (or from CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax:+44 1223 336 033; e-mail: deposit@ccdc.cam.ac.uk.)
References
Pickering AL, Seeber G, Long DL, Cronin L (2005) Cryst Eng Comm 7:504
Lawrence SD, Jiang T, Levitt M (1995) Chem Rev 95:2229
Sherrington DC, Taskinen KA (2001) Chem Soc Rev 30:83
Sigalov M, Vashchenko A, Khodorkovsky V (2005) J Org Chem 70:92
Michel O (2010) Photochem Photobiol Sci 9:637
Giese B, Biland A (2002) Chem Comm 38:667
Goes M, Verhoeven JW, Hofstraat H, Brunner K (2003) Chem Phys Chem 4:349
Al-Ahmary KM, Habeeb MM, Al-Solmy EA (2010) J Solution Chem 39:1264
Jakbiak R, Bao Z, Rothberg L (2000) Synth Met 114:61
Brueggermann K, Czernuszewicz RS, Kochi JK (1992) J Phys Chem 96:4405
Andrade SM, Costa SMB, Pansu R (2000) J Colloid Interface Sci 226:260
Gutmann F, Johnson C, Keyzer H, Molnár J (1997) Charge transfer complexes in biochemistry system. Marcel Dekker, Inc., New York
Dozal A, Keyzer H, Kin HK, Way WW (2000) Int J Antimicrob Agent 14:261
Bazzi HS, Mostafa A, AlQaradawi SY, Nour E (2007) J Mol Struct 842:1
Amanym AI (1993) Can J Chem 71:318
Frédéric D, Nicolac G, Yucel S (2004) J Org Chem 69:2164
Wu JC, Liu SX, Neels A, Derf FL, Salle M, Decurtins S (2007) Tetrahedron 63:11282
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven JT, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2003) Gaussian03. Gaussian Inc, Pittsburgh
Yao YM, Xue MQ, Luo YJ, Zhang ZQ, Jiao R, Zhang Y, Shen Q, Wong W (2003) J Organomet Chem 678:108
Desiraju GR, Steiner T (1999) The weak hydrogen bond in structural chemistry and biology. Oxford University Press, Oxford
Wang YT, Qin TX, Zhao C, Tang GM, Li TD, Cui YZ, Li JY (2009) J Mol Struct 938:291
Acknowledgments
We are grateful for the support from the Open Project Program (KJS1007) of Key Lab. of Organic Synthesis (Soochow University) of Jiangsu Province.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, H., Li, J., Chen, H. et al. Synthesis and Crystal Structure of Charge Transfer Complex (CTC) of 2-Aminobenzothiazole with Its Schiff Base. J Chem Crystallogr 41, 1844–1849 (2011). https://doi.org/10.1007/s10870-011-0185-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-011-0185-5