
Abstract
The synthesis of a novel ligand bis(3-methoxysalicylidene)-4-methylbenzene-1,2-diamine (H2L) has been carried out by the condensation of 4-methylbenzene-1,2-diamine with 3-methoxysalicylaldehyde. The Ni(II) and Pd(II) Schiff base complexes have been synthesized from the reaction of Ni(OAc)2·4H2O and Pd(OAc)2 with the Schiff base ligand (H2L) in methanol and acetonitrile, respectively. The ligand and its complexes have been characterized by elemental analyses, FT-IR, 1H-NMR and UV–Vis spectroscopy. Moreover, the crystal structure of the ligand has been determined by single crystal X-ray crystallography. To have better understanding of electronic transitions of the ligand and its complexes, density functional theory (DFT) calculations have been performed. The geometries and electronic properties of the compounds have been obtained with the M062X level at Def2-TZVP basis set. Additionally, NBO analysis has been performed to interpret electronic transitions by determining their included occupied and virtual orbitals. The theoretical parameters confirmed the suggested structure of complexes. The spectroscopic data obtained from DFT calculations show acceptable agreement with the experimental results.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Schiff bases, characterized by the azomethine group (–CH=N–), form a significant class of compounds in medicinal and pharmaceutical chemistry [1] are known to have biological applications such as, anti-inflammatory [2], antifungal [3], antiproliferative [4], antibacterial [5], antitubercular [6], anticonvulsant [7] and antiviral [8]. The transition metal complexes containing these bases have extensive applications like biological activities [9,10,11], anticancer [12], antibacterial [13], in chemical analysis [14], catalytic properties [15,16,17], and potential applications in designing the analytical systems like optical sensors, bulk liquid membrane transport, solid phase extraction and in magneto-structural correlations in molecular systems and materials [18,19,20,21,22]. The tetradentate N2O2 ligands and their transition metal complexes have been of research interest for their applications, particularly on the development of agrochemical and pharmaceutical industries, as catalysts and in materials chemistry [23, 24]. The complexes of Schiff bases containing various central metals such as Ni and Pd have been studied in great detail for their crystallographic features, enzymatic reactions, steric effects, structure-redox property relationships, mesogenic characteristics, biological and catalytic activities and in the design and construction of new magnetic materials. The study of these compounds is of great importance in different fields of chemistry [25,26,27]. Moreover, palladium(II) complexes as catalyst in Suzuki–Miyaura reaction have been used widely for the synthesis of natural products, pharmaceutical intermediates, conducting polymers, pesticides and liquid crystals [15, 28, 29]. In recent years, quantum chemical calculation and spectroscopic characterization of many compounds have found considerable amount of interest [30,31,32]. The increasing use of DFT, as one of the most extensively used quantum mechanical method, can be attributed to its accuracy and computational speed [33]. DFT studies have been shown to possess great accuracy in molecular structural properties, FT-IR frequencies, spectroscopic parameters, etc [34,35,36].
In this article, we report the synthesis and characterization of a new tetradentate Schiff base ligand and its complexes with nickel(II) and palladium(II) (Fig. 1). The compounds were characterized using FT-IR, UV–Vis, 1H-NMR, elemental analyses (CHN) and the crystal structure of the ligand has been determined by single crystal X-ray crystallography. Moreover, the geometries and electronic structure of the ligand and its nickel(II) and palladium(II) complexes have been calculated using density functional theory (DFT).

Synthetic procedure for the preparation of ligand and its complexes
Experimental
Materials and physical methods
All reagents were purchased from commercial sources and used without additional purification. Elemental analyses were performed on a Heraeus CHN-O-FLASH EA 1112 elemental analyzer. Infrared spectra were recorded on a FT-IR Prestige21 spectrophotometer using KBr pellets. The 1H-NMR (400 MHz) spectra were obtained at ambient temperature on a BRUKER AVANCE 400 MHz spectrometer. The chemical shift values (δ) are given in ppm. UV–Vis absorption spectra were recorded on a UV-1601PC. The single crystal X-ray diffraction structure was determined on a Bruker SMART APEX CCD X-ray diffractometer. Full spheres of reciprocal lattice were scanned by 0.3° steps in omega with a crystal-to-detector distance of 5 cm. Cell refinement and data reduction were performed with the help of the SAINT program [37]. Corrections for absorption were made with the multi-scan method and SADABS program [37]. The structures were solved with direct methods using SHELXS and structure refinement on F2 was carried out with the SHELXL-2014/7 program. All non-hydrogen atoms were refined using anisotropic displacement parameters [38]. All crystallographic calculations were done with PLATON [39].
Synthesis of the ligand, (H2L)
The ligand of bis(3-methoxysalicylidene)-4-methylbenzene-1,2-diamine (H2L) was synthesized by adding 3-methoxysalicylaldehyde (0.30 g, 2 mmol) to a solution of 4-methylbenzene-1,2-diamine (0.12 g, 1 mmol) in methanol (20 ml) under refluxing for 1 h. The orange crystals, suitable for X-ray data collection, were obtained by slow evaporation of the methanol solution after several days. Anal. Calcd. for C23H22N2O4 (%): C, 70.75; H, 5.68; N, 7.18. Found: C, 71.04; H, 5.59; N, 7.32. FT-IR (KBr, cm−1): 1616 (C=N), 1573, 1465 (C=C), 1249 (C–O). 1H-NMR (400 MHz, CDCl3, ppm): δ = 13.32 (s, 1 H, O–H), 13.25 (s, 1 H, O–H), 8.63 (s, 1 H, HiC=N), 8.61 (s, 1 H, Hi′C=N), 6.84–7.17 (m, 9 H, aromatic), 3.90 (s, 6 H, –OCH3), 2.44 (s, 3 H, –CH3). UV–Vis ((2×10−5 M) DMF) λmax(nm): 265, 307, 321, 335.
Synthesis of the nickel(II) complex, (NiL)
The nickel(II) complex was synthesized by reaction of Ni(OAc)2·4H2O (0.25 g, 1 mmol) and bis(3-methoxysalicylidene)-4-methylbenzene-1,2-diamine (0.39 g, 1 mmol) in methanol (20 ml) under reflux condition for 5 h. The product was obtained by slow evaporation of the methanol solution. Anal. Calcd. for [Ni(C23H20N2O4)] (%): C, 61.78; H, 4.51; N, 6.27. Found: C, 62.32; H, 4.67; N, 6.04. FT-IR (KBr, cm−1): 1612 (C=N), 1543, 1465 (C=C), 1384 (C–O), 590 (Ni–O), 505 (Ni–N). 1H-NMR (400 MHz, DMSO-d6, ppm): δ = 8.61 (s, 1 H, HiC=N), 8.77 (s, 1 H, Hi′C=N), 6.53-8.00 (m, 9 H, aromatic), 3.76 (s, 6 H, –OCH3), 2.37 (s, 3 H, –CH3). UV–Vis ((2×10−5 M) DMF) λmax(nm): 266, 299, 380, 488.
Synthesis of the palladium(II) complex, (PdL)
The palladium(II) complex was prepared by adding Schiff base ligand (0.39 g, 1 mmol) to a solution of Pd(OAc)2 (0.22 g, 1 mmol) in acetonitrile (20 mL) under reflux condition for 1 h. After filtering, an orange colored solid was obtained upon slow evaporation of the filtrate. Anal. Calcd. for [Pd(C23H20N2O4)] (%): C, 55.83; H, 4.07; N, 5.66. Found: C, 56.12; H, 4.26; N, 5.38. FT-IR (KBr, cm−1): 1604 (C=N), 1539, 1435 (C=C), 1330 (C–O), 570 (Pd–O), 501 (Pd–N). 1H-NMR (400 MHz, DMSO-d6, ppm): δ = 9.17 (s, 1 H, HiC=N), 9.13 (s, 1 H, Hi′C=N), 6.68–8.27 (m, 9 H, aromatic), 3.85 (s, 6 H, –OCH3), 2.43 (s, 3 H, –CH3). UV–Vis [(2×10−5 M) DMF] λmax(nm): 289, 336, 359, 376, 484.
Computational methods
The molecular structures of the H2L, NiL and PdL were optimized by a DFT method at M062X level [40] and Def2-TZVP basis set [41], using the Gaussian 09 package (Revision D.01) [42]. DFT calculations of electronic properties at the fully optimized geometry of the compounds were performed in the gas phase. NBO calculations and determining atomic orbitals composition were performed by NBO 6.0 and chemissian programs, respectively [43, 44].
Results and discussion
Synthesis
The Schiff base ligand was synthesized from 3-methoxysalicylaldehyde and 4-methylbenzene-1,2-diamine with the molar ratio 2:1 in methanol. Its Ni(II) and Pd(II) complexes have been prepared by the reaction of Schiff base ligand with Ni(OAc)2.4H2O and Pd(OAc)2 in 1:1 molar ratio, respectively.
FT-IR spectra
The IR spectra of the Schiff base ligand and its nickel(II) and palladium(II) complexes are shown in Fig. 2. The characteristic IR data of the compounds agree with their structures. IR spectral data of the Schiff base ligand and its complexes are listed in Table 1. In the IR spectra of all compounds, vibrational bands at 1400–1600 cm−1 range are assigned for C=C stretching vibration of the benzene rings (see Fig. 2; Table 1) [45].

FT-IR spectra of a H2L, b NiL and c PdL
The IR spectrum of ligand exhibit a υ(C=N) peak at 1616 cm−1 [46]. This peak was shifted to lower frequencies of 1612, 1604 cm−1 in the Ni(II) and Pd(II) complexes, respectively, upon coordination of the nitrogen atom of the azomethine group to Ni(II) and Pd(II) ions [47, 48]. These shifts are consistent with π-back bonding from the electron-rich metals to the imine group [47]. After the metal-ion complexation, the C–O stretching band of the free ligand shift from 1249 to 1384 and 1330 cm−1 in the NiL and PdL, respectively. Upon coordination of the free ligand with the central metals, the new weak bands at 500–600 cm−1 ranges are observed from the spectra of both Ni(II) and Pd(II) metal complexes can be attributed to M–O and M–N stretching bands [49, 50].
NMR spectra
The signals at 13.32 and 13.25 ppm in the 1H-NMR spectrum of the free ligand (Fig. 3a), corresponded to the phenolic OH resonances, are disappeared in the spectra of the NiL and PdL complexes (Fig. 3b, c), which clearly indicate that the Schiff base ligand is coordinated as an anionic ligand to the Ni(II) and Pd(II) ions. In the 1H-NMR spectra of the NiL and PdL complexes, the signals at δ = 8.81, 8.77 ppm and δ = 9.17, 9.13 ppm are assigned to the Hi–C=N– and Hi′–C=N– proton of azomethine, respectively. Appearance of two 1H-NMR peaks for existing azomethine protons in the complexes are indicative of the magnetically non-equivalence environment of these protons. The 1H-NMR spectra of the complexes show the downfield shift of the azomethine H, confirming coordination of the azomethine nitrogen to metal atoms. The aromatic protons of NiL and PdL complexes appear in the range of δ = 6.53–8.00 ppm and δ = 6.68–7.27 ppm, respectively. In the 1H-NMR spectra of NiL and PdL complexes, the –OCH3 and –CH3 are observed as a singlet at δ = 3.76, δ = 2.37 ppm and δ = 3.85, δ = 2.43 ppm, respectively.

1H-NMR spectra of the ligand (a) in CDCl3, and its NiII (b) and PdII (c) complexes in DMSO, 400 MHz, 293 K
UV–Vis spectra
The electronic absorption spectra of the H2L ligand and its Ni(II) and Pd(II) complexes were recorded in DMF between 260 and 600 nm (Fig. 4). The UV–Vis spectra of the compounds have a similar pattern. The UV–Vis absorption spectrum of the Schiff base ligand shows the bands at 265, 307, 321 and 335 nm. The band at 265 nm can be attributed to the π–π* transition of the benzene rings. The absorption bands at around 307 and 321 nm are due to a π–π* transition of the C=N groups. A low-intensity band in the lower energy region at 335 nm is assigned to the n–π* transition of non-bonding electrons on the nitrogen of azomethine group in the free ligand.

UV–Vis spectra of a H2L (solid line), b NiL (dashed line) and c PdL (dotted line) in DMF
Maximal wavelength (λmax) due to the π–π* transition of the benzene rings and π–π* transition of the C=N groups in the nickel(II) and palladium(II) complexes are increased (Fig. 4; Table 2). These red shifts in the UV–Vis spectra of the complexes are ascribed to the hydroxyl deprotonation of the Schiff base ligand during complexation, and formation of coordination bonds [48]. The electronic spectra of both Ni(II) and Pd(II) metal complexes show an additional band in the visible region (488 and 484 nm, respectively), which is attributed to charge transfer.
For better understanding of the electronic properties, DFT calculations and NBO analysis were performed for all compounds at the M062X level and Def2-TZVP basis set. The electronic spectroscopic data such as absorption wavelengths maxima, excitation energies and other electronic properties of ligand and its Ni(II) and Pd(II) complexes are collected in Table 2.
In the calculations of Ni(II) complex, two peaks at 403 and 381 nm with major contribution from H-1 → L with 64% and H → L with 72% are obtained. These transitions could be assigned to experimental results observed at 488 and 379 nm. The results show that these absorption bands have inter ligand charge-transfer (ILCT) character. DFT calculations of Ni(II) complex show that the all transitions have inter ligand charge-transfer (ILCT) character (Table 2). In the calculations of Pd(II) complex the major contributions are from H-1 → L (46%) and H → L (79%) for the transitions at 424 and 401 nm, respectively. These transitions would be compared to experimental data observed at 484 and 375 nm. For these transitions, the charge transfer characters are assigned to ligand-to-metal charge-transfer (LMCT) and inter ligand charge-transfer (ILCT) transitions, respectively. The theoretical calculations of Pd(II) complex, predict that the all transitions have a mixed ILCT and LMCT character (Table 2). The calculations show that the shifts in the main absorptions of the Pd(II) complex are larger than its Ni(II) complex. Some above calculated electronic transitions are higher than the experimental ones, and some others are smaller; however, these differences are small. Similar behavior has been seen in previous studies [51]. The obtained spectroscopic parameters are in consistent agreement with the experimental data. The minor discrepancies observed in the wavelengths maxima. The main reason for these discrepancies is that the calculated data were obtained in the gas phase, whereas the experimental parameters are solvent phase.
To obtain the orbital distributions of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy of ligand and both Ni(II) and Pd(II) metal complexes, calculations were performed by M062X level and Def2-TZVP basis set. The results are presented in Fig. 5. The energy gaps of H2L, NiL and PdL are 6.07, 5.11 and 4.98 eV, respectively. The energy gap of Ni(II) complex is greater than Pd(II) metal complex. It can be attributed to higher stability of Ni(II) complex. After complexation, λmax of the Ni(II) and Pd(II) complexes increases (Fig. 5; Table 2), indicating that the energy gap (Eg = ELUMO − EHOMO) decreases (Fig. 5) [47].

Calculated spatial distributions and energies of the HOMO and LUMO levels of H2L, NiL and PdL
Crystal structure
The solid state structure of H2L was determined by X-ray diffraction. The crystal data and refinement parameters are summarized in Table 3, and ORTEP plot of H2L ligand is shown in Fig. 6. The asymmetric unit of H2L ligand, comprises a potentially tetradentate Schiff base ligand. The compound crystallizes in monoclinic system with P21/c space group. The absolute structure parameter (Flack parameter) was determined successfully. In H2L ligand, N(1)···H(1)‒O(1) and N(2)···H(3)‒O(3) intramolecular hydrogen bonds between N atoms and the OH groups generate a S(6) motif that help to stabilize the planarity of the molecule. The intramolecular H-bond distances and angle are also in the expected range [O(1)‒H(1): 0.82 Å, H(1)···N(1): 1.88 Å, N(1)···O(1): 2.610 (4) Å, N(1)‒H(1)‒O(1): 146.9° and O(3)‒H(3): 0.82 Å, H(3)···N(2): 1.84 Å, N(2)···O(3): 2.569 (3) Å and N(2)‒H(3)‒O(3): 146.6°] (Table 4) [52]. According to the hydrogen-bonding classification [53] these intramolecular hydrogen bonding are moderate interactions.

The optimized and the X-ray crystal structures of H2L ligand
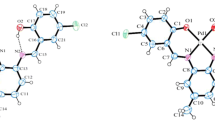
The calculations were undertaken at the M062X/Def2-TZVP level of theory to gain insight into the geometric parameters and structures of the ligand and its Ni(II) and Pd(II) complexes. The optimized and the X-ray crystal structures of H2L ligand and the optimized structures obtained by DFT for Ni(II) and Pd(II) complexes are given in Figs. 6 and 7. Both complexes have a similar structure. Some of the optimized geometrical parameters of compounds are collected in Table 5.

The optimized structures for NiL and PdL complexes at the M062X/Def2-TZVP method
Table 5 displays experimental bond lengths (Å) and bond angles (°) that are very close to those calculated for H2L. The theoretical calculations of Ni(II) and Pd(II) complexes, predict that the imine C=N bond length is longer in complexes than free ligand while the phenolic C–O and the C‒C bond lengths between the azomethine group and phenyl ring are shorter than the ligand [47, 50]. The imine C=N distance is longer in Ni(II) complex than in Pd(II) complex, also the N–M–O chelating angle is larger in Ni(II) complex when compared with Pd(II) complex (Table 5). Both Ni(II) and Pd(II) complexes have a distorted square planar geometry formed by two phenolic oxygen atoms and two imine nitrogen atoms. The bond lengths and angles involving this H2L ligand are similar in both complexes. On moving from Ni(II) complex to Pd(II) complex, the M–N and M–O bond lengths [(Ni–N) 1.919, 1.918 Å – (Pd-N) 1.986, 1.985 Å – (Ni–O) 1.869, 1.869 Å – (Pd–O) 2.016, 2.014 Å] increase (Table 5). Similar behavior has been seen in other researches [54,55,56]. For Ni(II) complex, the M–N distances are larger than the M–O distances while for Pd(II) complex, the M–O distances are larger than the M–N distances (Table 5).
Conclusion
A ligand bis(3-methoxysalicylidene)-4-methylbenzene-1,2-diamine and its Ni(II) and Pd(II) complexes were synthesized and characterized by elemental analyses, FT-IR, 1H-NMR and UV–Vis spectroscopy. Moreover, the crystal structure of the Schiff base ligand has been determined by X-ray crystallography. The geometries and electronic parameters of the compounds obtained by DFT calculations are in satisfactory agreement with experimental data. Calculation results of Ni(II) complex show that the all transitions have inter ligand charge-transfer (ILCT) character. The theoretical calculations of Pd(II) complex, show that the all transitions have a mixed ILCT and LMCT character. The DFT calculations predict that ligand acted as tetradentate Schiff base and coordinated to Ni(II) and Pd(II) via two N and two O atoms. We conclude that the Ni(II) and Pd(II) complexes have distorted square planar geometry.
Supplementary data
Crystallographic data has been deposited at the Cambridge Crystallographic Data Centre. CCDCs 1866059, and contain the supplementary crystallographic data for ligand. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving. html, or from the CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK; fax: (+ 44) 01223-336-033; e-mail: deposit@ ccdc.cam.ac.
References
G.O. Carlisle, A.S. Yamal, K.K. Ganguli, L.J. Theriot, J. Inorg. Nucl. Chem. 34, 2761 (1972)
S. Sharma, V.K. Srivastava, A. Kumar, Eur. J. Med. Chem. 37, 689 (2002)
A.S. El-Tabl, S.A. El-Enein, J. Coord. Chem. 57, 281 (2004)
P. Vicini, M. Incerti, I.A. Doytchinova, P.L. Colla, B. Busonera, R. Loddo, Eur. J. Med. Chem. 41, 624 (2006)
A. Adabi Ardakani, H. Kargar, N. Feizi, M.N. Tahir, J. Iran. Chem. Soc. 15, 1495 (2018)
M.C.S. Lourenco, M.V.N. Souza, A.C. Pinheiro, M.L. Ferreira, R.S.B. Goncalves, T.C.M. Nogueira, M.A. Peraltab, Arkivoc 15, 181 (2007)
J.V. Ragavendran, D. Sriram, S.K. Patel, I.V. Reddy, N. Bharathwajan, J. Stables, P. Yogeeswari, Eur. J. Med. Chem. 42, 146 (2007)
X. Qiao, Z.Y. Ma, C.Z. Xie, F. Xue, Y.W. Zhang, J.Y. Xu, Z.Y. Qiang, J.S. Lou, G.J. Chen, S.P. Yan, J. Inorg. Biochem. 105, 728 (2011)
H.G. Aslan, S. Akkoc, Z. Kokbudak, L. Aydin, J. Iran. Chem. Soc. 14, 2263 (2017)
V. Mishra, S.N. Pandeya, S. Anathan, Acta Pharm. Turc. 42, 139 (2000)
A. Sahraei, H. Kargar, M. Hakimi, M.N. Tahir, J. Mol. Struct. 1149, 576 (2017)
K. Neelima, S. Poonia, M. Siddiqui, D.A. Kumar, Spectrochim. Acta Part A 155, 146 (2016)
M. Galini, M. Salehi, M. Kubicki, A. Amiri, A. Khaleghian, Inorg. Chim. Acta 461, 167 (2017)
E.J. Baran, J. Inorg. Biochem. 80, 1 (2000)
H. Khojasteh, V. Mirkhani, M. Moghadam, S. Tangestaninejad, I. Mohammadpoor-Baltork, J. Iran. Chem. Soc. 14, 1139 (2017)
M. Hatefi Ardakani, M. Moghadam, S. Saeednia, Z. Pakdin-Parizi, J. Iran. Chem. Soc. 13, 631 (2016)
H. Kargar, Transit. Met. Chem. 39, 811 (2014)
M.K. Amini, J.H. Khorasani, S.S. Khaloo, S. Tangestaninejad, Anal. Biochem. 320, 32 (2003)
M.B. Gholivand, A. Yari, M. Joshaghani, Anal. Chim. Acta 538, 225 (2005)
M.B. Gholivand, F. Ahmadi, E. Rafiee, Sep. Sci. Technol. 41, 315 (2006)
M. B. Gholivand, F. Ahmadi, E. Rafiee, Sep. Sci. Technol. 42, 897 (2007)
A.D. Khalaji, H. Stoekli-Evans, Polyhedron 28, 3769 (2009)
M. Sedighipoor, A.H. Kianfar, W.A. Kamil Mahmood, M.H. Azarian, Inorg. Chim. Acta 457, 116 (2017)
R. K.Mahajan,K. V.Inderpreet,M.Sharma,M.Kumar,Sensors2,417(2002)
A.A. Khandar, K. Nejati, Polyhedron 19, 607 (2000)
Z. Beigi, A.H. Kianfar, H. Farrokhpour, M. Roushani, M.H. Azarian, W.A. Kamil, Mahmood, J. Mol. Liq. 249, 117 (2018)
W.A. Zordok, S.A. Sadeed, A.F. El-Faragy, J. Iran. Chem. Soc. 14, 2529 (2017)
M. Nikoorazm, A. Ghorbani-Choghamarani, A. Panani, B. Tahmasbi, N. Noori, J. Iran. Chem. Soc. 15, 181 (2018)
M. Sedighipoor, A.H. Kianfar, G. Mohammadnezhad, H. Gorls, W. Plass, Inorg. Chim. Acta 476, 20 (2018)
S. Thorwirth, M.C. McCarthy, J.B. Dudek, P. Thaddeus, J. Chem. Phys. 122, 184308 (2005)
M. Dostani, A.H. Kianfar, W.A. Kamil Mahmood, M. Dinari, H. Farrokhpour, M.R. Sabzalian, F. Abyar, M.H. Azarian, Spectrochim. Acta Part A 180, 144 (2017)
A.H. Kianfar, S. Tavanapour, K.E. Skandari, M.H. Azarian, W.K. Mahmood, M. Bagheri, J. Iran. Chem. Soc. 15, 369 (2018)
W. Koch, M.C. Holthausen, A Chemist’s Guide to Density Functional Theory (Wiley–VCH, Weinheim, 2003)
H. Kargar, V. Torabi, A. Akbari, R. Behjatmanesh-Ardakani, M. N. Tahir, J. Mol. Struct. 1179, 732 (2019)
A.H. Kianfar, H. Farrokhpour, P. Dehghani, H.R. Khavasi, Spectrochim. Acta, Part A 150, 220 (2015)
V. Torabi, H. Kargar, A. Akbari, R. Behjatmanesh-Ardakani, H. Amiri Rudbari, M.N. Tahir, J. Coord. Chem. (2018). https://doi.org/10.1080/00958972.2018
W. Bruker, AXS Programs: SMART, version 5.625; SAINT, version 6.45; SADABS, version 2.10; XPREP, version 6.14 (Bruker AXS Inc., Madison, 2003)
G.M. Sheldrick, Acta Crystallogr. A 64, 112 (2008)
S. Al, Acta Crystallogr. D 65, 148 (2009)
Y. Zhao, D.G. Truhlar, Theor. Chem. Acc. 120, 215 (2008)
F. Weigend, R. Ahlrichs, Phys. Chem. Chem. Phys. 7, 3297 (2005)
M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery Jr., J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, O. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, GAUSSIAN 09 (Revision D.01) (Gaussian, Inc., Wallingford, 2013)
E.D. Glendening, J.K. Badenhoop, A.E. Reed, J.E. Carpenter, J.A. Bohmann, C.M. Morales, C.R. Landis, F. Weinhold, (Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013). http://nbo6.chem.wisc.edu
S.E.H. Etaiw, D.M. Abd El-Aziz, E.H. Abd, E.A. El-Zaher, Ali, Spectrochim. Acta Part A 79, 1331 (2011)
F. Hiroki, Y. Kazuto, K. Yumiko, Y. Takakazu, Macromolecules 43, 10366 (2010)
O.A. Blackburn, B.J. Coe, J. Fielden, M. Helliwell, J.J.W. McDouall, M.G. Hutchings, Inorg. Chem. 49, 9136 (2010)
Y. Fan, W. You, W. Huang, J.L. Liu, Y.N. Wang, Polyhedron 29, 1149 (2010)
S. Ilhan, H. Temel, I. Yilmaz, M. Sekerci, Polyhedron 26, 2795 (2007)
A. Akbari, Z. Alinia, Turk. J. Chem. 37, 867 (2013)
G. Cosquer, F. Pointillart, B.L. Guennic, Y.L. Gal, S. Golhen, O. Cador, L. Ouahab, Inorg. Chem. 51, 8488 (2012)
A. Jamshidvand, M. Sahihi, V. Mirkhani, M. Moghadam, I. Mohammadpoor-Baltork, S. Tangestaninejad, H. Amiri, H. Rudbari, R. Kargar, S. Keshavarzi, Gharaghani, J. Mol. Liq. 253, 61 (2018)
T. Steiner, Angew. Chem. Int. Ed. 41, 48 (2002)
B. Castro, C. Freire, M.T. Duarte, M.F.M. Piedade, I.C. Santos, Acta Crystallogr. C 57, 370 (2001)
W. Sawodny, U. Thewalt, E. Potthoff, R. Ohl, Acta Crystallogr. C 55, 2060 (1999)
S. Yuichi, T. Yajima, F. Tani, S. Karasawa, K. Fukui, Y. Naruta, O. Yamauchi, J. Am. Chem. Soc. 129, 2559 (2007)
Acknowledgements
The support of this work by Payame Noor University and Ardakan University are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kargar, H., Torabi, V., Akbari, A. et al. Synthesis, crystal structure, experimental and theoretical studies of tetradentate N2O2 Schiff base ligand and its Ni(II) and Pd(II) complexes. J IRAN CHEM SOC 16, 1081–1090 (2019). https://doi.org/10.1007/s13738-018-01583-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-018-01583-1