
Abstract
The J-UNIO (JCSG protocol using the software UNIO) procedure for automated protein structure determination by NMR in solution is introduced. In the present implementation, J-UNIO makes use of APSY-NMR spectroscopy, 3D heteronuclear-resolved [1H,1H]-NOESY experiments, and the software UNIO. Applications with proteins from the JCSG target list with sizes up to 150 residues showed that the procedure is highly robust and efficient. In all instances the correct polypeptide fold was obtained in the first round of automated data analysis and structure calculation. After interactive validation of the data obtained from the automated routine, the quality of the final structures was comparable to results from interactive structure determination. Special advantages are that the NMR data have been recorded with 6–10 days of instrument time per protein, that there is only a single step of chemical shift adjustments to relate the backbone signals in the APSY-NMR spectra with the corresponding backbone signals in the NOESY spectra, and that the NOE-based amino acid side chain chemical shift assignments are automatically focused on those residues that are heavily weighted in the structure calculation. The individual working steps of J-UNIO are illustrated with the structure determination of the protein YP_926445.1 from Shewanella amazonensis, and the results obtained with 17 JCSG targets are critically evaluated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
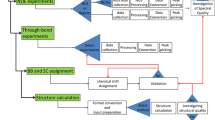
This paper is focused on automated NMR structure determination of soluble proteins in the size range up to about 150 residues. In recent papers we documented that a novel, extensively automated approach yields NMR structures that are in close agreement with the corresponding crystal structures, and on the basis of the NMR data sets used for the structure determination (Fig. 1) further provides qualitative information on function-related conformational equilibria and intramolecular rate processes (Jaudzems et al. 2010; Mohanty et al. 2010; Serrano et al. 2010; Wüthrich 2010). Here we describe the procedure that was used for obtaining this structural data, J-UNIO (protocol of the Joint Center for Structural Genomics (JCSG: www.jcsg.org) using the software UNIO).

J-UNIO protocol for automation of protein structure determination by NMR in solution. A NMR structure-quality protein solution, as characterized by the NMR-profile (see text) is used to obtain the seven NMR data sets listed on the left. These are then analyzed with the software listed on the right. Following the protocol from top to bottom, yellow boxes represent fully automated steps and white boxes represent interactive steps. The latter include one or multiple rounds of interactive interventions to check and complete the automatic chemical shift assignments, and to validate the resulting NMR structure
Our aim in pursuing the J-UNIO project was to establish a robust protocol for obtaining high-quality protein structures with minimal use of NMR spectrometer time and minimal workload for interactive spectral analysis. Following the example provided by the JCSG high-throughput crystal structure determination pipeline (Elsliger et al. 2010; Lesley et al. 2002), interactive intervention for expanding and validating the results of the automated steps was inserted at three critical points of the procedure (Fig. 1), emphasizing that the primary goal is to efficiently obtain high-quality structures rather than achieving full automation.
The manuscript starts with a survey of the J-UNIO protocol. The procedure is then illustrated with YP_926445.1, which is representative of a group of 17 JCSG target proteins for which metrics on the course of the structure determination and its results are presented in a third section of the paper. Two key elements of the J-UNIO protocol, i.e., the use of “NMR profiles” for characterization of “structure-quality” protein solutions and polypeptide backbone chemical shift assignments with APSY-NMR, are briefly summarized here and will be described in detail elsewhere (B. Pedrini et al., in preparation).
Survey of the J-UNIO protein structure determination protocol
The J-UNIO approach to automated NMR structure determination (Fig. 1) starts with characterization of “structure-quality” protein solutions by [15N,1H]-COSY-based “NMR-profiles” (here we used the [15N,1H]-HSQC experiment). The polypeptide backbone chemical shift assignment is then accomplished with a standard set of three APSY-NMR experiments, i.e., 4D APSY-HACANH, 5D APSY-CBCACONH and 5D APSY-HACACONH (Hiller et al. 2008). Individual analysis of each of these three APSY data sets with the software GAPRO (Hiller et al. 2005) yields a 4-dimensional and two 5-dimensional peak lists. These three peak lists are used as input for the software UNIO-MATCH (Volk et al. 2008). UNIO-MATCH first assembles the different backbone atom correlations of the three APSY peak lists into a single list of higher-dimensional generic spin systems, and then uses an evolutionary optimization scheme for placing these spin systems in unique locations along the sequence of the protein, which results in automated assignment of the chemical shifts for the atoms Hα, Cα, HN, N, C′ and Cβ (Fig. 2). It is of key importance at this point that the chemical shifts in the NOESY spectra are adapted to those in the APSY-NMR spectra and the UNIO-MATCH output is interactively validated. In addition, if applicable, the backbone assignments are extended with the use of the same 3D heteronuclear-resolved [1H,1H]-NOESY spectra that will subsequently be used to obtain the amino acid side chain assignments and to collect NOE distance constraints as input for the structure calculation. The validated backbone chemical shifts and the three 3D heteronuclear-resolved [1H,1H]-NOESY data sets (Fig. 1) then provide the input for automated chemical shift assignment of the amino acid side chains (Fig. 2) with the software UNIO-ATNOS/ASCAN (Fiorito et al. 2008). The polypeptide backbone chemical shift assignments and the automated amino acid side chain assignments obtained at this point represent the input for a first round of automated signal identification (“peak picking”) and NOE assignment in the three NOESY data sets (Fig. 1) with the software UNIO-ATNOS/CANDID (Herrmann et al. 2002a, b) and structure calculation with the simulated annealing routine of CYANA (Güntert et al. 1997). This step yields “Structure A” (“A” refers to “automated side chain assignments before interactive validation”). Using the Structure A as a reference, the three [1H,1H]-NOESY spectra are further interactively examined in order to validate and extend the automated side chain chemical shift assignments. This provides the input for the calculation of the “Structure V” (“V” stands for “validated interactively”), which is subjected to one or multiple rounds of further interactive refinement, using UNIO-ATNOS/CANDID and the simulated annealing routine of CYANA with the updated input, before validation with an in-house collection of tools and deposition to the Protein Data Bank (Fig. 1).

J-UNIO strategy for resonance assignment and structural interpretation of the NMR data, illustrated with the tripeptide segment –Ala–Thr–Phe–. Hydrogen, carbon, nitrogen and oxygen atoms are represented by white, black, blue and red spheres, respectively. The red line encloses the atoms considered in the APSY-based backbone assignment, and the blue frame surrounds all the atoms considered in the NOESY-based side chain chemical shift assignment. The overlap of APSY and NOESY data at the backbone atom and β-carbon positions ensures that the overall chemical shift assignment presents a robust platform for the automated structure determination, which is based on elucidating the NOE network among all hydrogen atoms in the protein
Methods: J-UNIO NMR structure determination of the Shewanella amazonensis protein YP_926445.1
Production of a structure-quality YP_926445.1 solution
The plasmid encoding YP_926445.1 was transformed into the E. coli strain BL21 (DE3) (Novagen). Expression of the uniformly 13C,15N-labeled 115-residue construct of YP_926445.1 (Fig. 3a) was carried out by growing the cells in M9 minimal medium containing 15NH4Cl (1 g/L) and [13C6]-d-glucose (4 g/L) as the sole nitrogen and carbon sources, respectively. Cell cultures were shaken at 37 °C to an OD600 nm of 0.6 before expression of YP_926445.1 was induced with 1 mM IPTG. The cells were then grown for 16 h at 18 °C, harvested by centrifugation, resuspended in extraction buffer [20 mM sodium phosphate at pH = 7.5, 200 mM NaCl, 10 mM imidazole, Complete EDTA-free protease inhibitor cocktail tablets (Roche)] and lysed by sonication. The cell debris was removed by centrifugation at 20,000g for 30 min and the supernatant loaded onto a Ni2+ affinity column (HisTrap HP column; GE Healthcare) equilibrated with buffer A (20 mM phosphate at pH 7.5, 200 mM sodium chloride, 10 mM imidazole). The imidazole concentration was stepwise increased, first to 30 mM in order to remove non-specifically bound proteins, and then to 500 mM to elute the target protein. After overnight cleavage of the expression tag with TEV protease at room temperature, the protein was loaded onto a desalting column (HiPrep™ 26/10, GE Healthcare) and eluted with buffer A. The protein fractions were then passed through a Ni+2 affinity column (HisTrap HP column, GE Healthcare) equilibrated with buffer A, in order to remove the TEV protease and the cleaved His-tag from the target protein. Fractions containing the target protein, as determined by SDS-PAGE, were pooled and loaded onto a size exclusion column (HiLoad™ 26/60 Superdex™ 75, GE Healthcare) equilibrated with NMR buffer (20 mM phosphate at pH 6.0, 50 mM sodium chloride) and eluted with the same buffer. The fractions containing the target protein were concentrated to a final volume of 550 μL for a final protein concentration of about 1.1 mM, using 3 kDa-cut-off centrifugal filter devices (Millipore). The NMR samples were supplemented with 5 % 2H2O (v/v) and 4.5 mM NaN3.

Protein YP_926445.1: amino acid sequence, characterization of a structure-quality NMR sample by recording of a [15N,1H]-HSQC spectrum and generation of an NMR-profile (see text), extent of the automated backbone chemical shift assignments, and locations of regular secondary structures in the NMR structure. a Amino acid sequence (the N-terminal glycine in position-1 is not part of the natural protein and its addition is a result of the cloning strategy used). For the underlined polypeptide segments the sequential connectivities were established by the automated UNIO-MATCH routine, whereby for each residue the chemical shifts of at least those atoms were automatically assigned which are needed to establish the sequential connectivities (see text). b 700 MHz microcoil 2D [15N,1H]-HSQC spectrum at 298 K. c NMR-profile obtained from the data in b, with the [15N,1H]-HSQC cross peaks arranged along the horizontal axis in the order of decreasing intensity. The vertical broken line indicates the number of backbone amide and tryptophane indole 15N–1H signals expected from the amino acid sequence. The horizontal broken line indicates an intensity cutoff established by the microcoil experiment. For the residues with [15N,1H]-HSQC cross peak intensities above this line, we expect to observe sequential connectivities by APSY-NMR (see text)
NMR spectroscopy
The three APSY-NMR spectra of YP_926445.1 indicated in Fig. 1 were recorded at 25 °C on a BRUKER AVANCE 600 MHz spectrometer equipped with a CPTCI HCN z-gradient cryogenic probehead, which has a sensitivity of 840:1 for observation of the DSS signal in a standard Bruker aqueous sucrose sample. Eight scans were accumulated, resulting in a total recording time of 27 h for the three experiments. For 4D APSY-HACANH, 27 projections were recorded with a resolution of 102 × 1,280 complex data points. For 5D APSY-CBCACONH and 5D APSY-HACACONH, 36 projections were recorded with 100 × 1,800 data points. Before Fourier transformation the spectra were multiplied in both dimensions with a 45°-shifted sine bell (DeMarco and Wüthrich 1976).
The three 3D heteronuclear-resolved [1H,1H]-NOESY spectra (Fig. 1) were acquired on an 800 MHz Bruker Avance spectrometer equipped with a 5 mm room temperature TXI probehead. The mixing time was 65 ms, and the following values for t1,max, t2,max and t3,max were used: 3D 15N-resolved NOESY, 11.7 ms, 20 ms, 96 ms; 3D 13Cali-resolved NOESY: 12 ms, 5.9 ms, 98 ms; 3D 13Caro-resolved NOESY: 9.0 ms, 7.6 ms, 98 ms. The 15N-, 13Cali- and 13Caro-resolved spectra were recorded with resolutions of 220 × 100 × 2,048, 240 × 100 × 2,300 and 200 × 80 × 2,200 complex data points, respectively. The total measurement time for the three data sets was 7 days. Prior to Fourier transformation the time domain data were multiplied with a sine-squared window.
NMR-profile monitors structure-quality YP_926445.1 solution
The protein solutions were initially assessed by the recording of a 700 MHz 2D [15N,1H]-COSY spectrum (Fig. 3b) with a microcoil probehead. A series of samples with different solution conditions could thus be screened with minimal expense of 15N-labeled protein. For selected samples a “NMR-profile” was then generated by arranging the cross peaks in the 2D [15N,1H]-COSY spectrum in the order of their intensities along a horizontal axis (Fig. 3c). The analysis of the NMR profile includes two key steps. First, the number of peaks observed is compared with the number of backbone amide group and tryptophan indole group peaks expected from the amino acid sequence (Fig. 3a), showing whether or not we observe the complete polypeptide chain. For uniformly 13C,15N-labeled YP_926445.1 the expected 108 15N–1H cross peaks, which include 3 indole ring signals, were observed in the 2D [15N,1H]-HSQC spectrum (Fig. 3b). Second, the signal intensities in the micro-coil 700 MHz 2D [15N,1H]-COSY spectrum are related to those in the experiments used to obtain polypeptide backbone chemical shift assignments. In the present work these were the APSY-NMR experiments listed in Fig. 1, which were recorded with a 5 mm cryogenic probehead at 600 MHz. From the NMR-profile of YP_926445.1 generated with the data of Fig. 3b we concluded that the set of three APSY experiments listed in Fig. 1 would provide sequential connectivities for 103 of the 108 residues, i.e., for all residues with signal intensities above the broken horizontal line in Fig. 3c. Overall, screening with NMR profiles enables to select NMR structure-quality protein solutions based on microscale production of 15N-labeled protein, and to predict the extent of polypeptide backbone chemical shift assignments that can be obtained with the use of a given selection of NMR experiments.
APSY-based backbone chemical shift assignment using UNIO-MATCH
Analysis of the three APSY-NMR data sets listed in Fig. 1 with the program GAPRO (Hiller et al. 2005) yielded one 4-dimensional and two 5-dimensional peak lists as input for the software UNIO-MATCH (Volk et al. 2008). UNIO-MATCH generates a list of higher-dimensional generic spin systems, which are then assigned to their sequence locations by an evolutionary algorithm (Volk et al. 2008). For YP_926445.1, UNIO-MATCH provided chemical shifts for 92 % of the atoms Hα, Cα, HN, N, C′ and Cβ (Fig. 2). Complete assignments of all six chemical shifts were obtained for 89 residues, for 16 additional residues at least the chemical shifts needed to establish the sequential connectivities were assigned, and for 9 residues no sequential connectivities were established. UNIO-MATCH failed to assign His 8 and Leu 73, which are located between prolines and for which no connectivities are available from APSY-NMR (Hiller et al. 2008), the prolines, and three of the residues with 15N–1H signal intensities below the cut-off indicated in Fig. 3c. Interactive completion of the backbone assignments (Fig. 1) resulted in extension of the assignment to 98 % of the aforementioned chemical shifts. There remained four gaps in the sequential connectivity pathway at the amide groups of residues Gln10, Leu16, Gly21 and Cys92, but all residues exhibited at least one sequential connectivity.
Chemical shift adaptation and automated UNIO-ATNOS/ASCAN side chain chemical shift assignment
The input for automated side chain chemical shift assignment consisted of the 3D 15N-, 13Cali- and 13Caro-resolved [1H,1H]-NOESY spectra and the previously derived backbone chemical shifts. As a first step, the backbone chemical shifts in the NOESY spectra were interactively adapted to the corresponding shifts in the APSY data sets. Thereby the 1H and 15N chemical shifts in the 3D 15N-resolved [1H,1H]-NOESY spectrum were adjusted until all the (HN,HN,15N) diagonal peaks and (Hα,HN,15N) cross peaks appeared at the positions defined by the high-precision chemical shifts derived from the APSY-NMR data. A corresponding procedure was applied to the Hα and 13Cα chemical shifts in the 3D 13Cali-resolved [1H,1H]-NOESY spectrum. It then turned out that the same calibration for 13C–1H fragments could be applied for the aliphatic region and the 3D 13Caro-resolved [1H,1H]-NOESY data. The backbone chemical shift list and the thus chemical shift-calibrated NOESY spectra were used as the input for the software UNIO-ATNOS/ASCAN to obtain side chain chemical shift assignments. For YP_926445.1, 73 % of the non-labile hydrogen atoms were thus automatically assigned.
Automated UNIO-ATNOS/CANDID NOE assignment and calculation of ‘Structure A’
The input for a first round of seven cycles of NOESY peak picking and NOE assignments with UNIO-ATNOS/CANDID (Herrmann et al. 2002a, b) in combination with structure calculations using the simulated annealing routine of CYANA (Güntert et al. 1997) consisted of the validated chemical shift assignments for the polypeptide backbone, the UNIO-ATNOS/ASCAN output of side chain chemical shift assignments, and the three NOESY data sets listed in Fig. 1. The resulting bundle of twenty NMR conformers, representing the Structure A of YP_926444.1 (Fig. 1), is shown in Fig. 4a, and the statistics of the structure determination are given in Table 1.

NMR structures of the protein YP_926445.1 at different stages of the J-UNIO protocol (Fig. 1). a Stereo view of the bundle of 20 NMR conformers representing Structure A, which was obtained based on using the side chain chemical shift assignments from the automated UNIO-ATNOS/ASCAN routine and the validated backbone chemical shift assignments. These chemical shifts were included in the input for automated NOESY peak picking and NOE assignment with UNIO-ATNOS/CANDID in combination with structure calculation using the CYANA simulated annealing routine. Residues located in α-helices and β-strands are colored red and green, respectively, where the identification of the regular secondary structures was taken from Structure V in b. b Stereo view of the bundle of 20 NMR conformers representing Structure V, which was obtained after interactive validation and extension of the side chain chemical shift assignments obtained from UNIO-ATNOS/ASCAN. c All-heavy-atom stereo view of the conformer closest to the mean coordinates of the bundle of 20 Structure V conformers, with the side chains color-coded following their global displacement values: green <0.4 Å, blue 0.4–0.9 Å, red >0.9 Å. d Stereo ribbon representation of the same conformer as in c. The red balls indicate side chains of residues for which UNIO-ATNOS/ASCAN yielded erroneous chemical shift assignments that were then part of the input for the determination of Structure A (see text)
Interactive extension and validation of the side chain chemical shift assignments, and calculation and validation of ‘Structure V’
The chemical shift list obtained with UNIO-ATNOS/ASCAN was corrected and extended by interactive examination of the 3D 15N-, 13Cali- and 13Caro-resolved [1H,1H]-NOESY spectra with the software CARA (Keller 2004). The thus updated chemical shift assignments were used in the input for a new round of NOE assignments with UNIO-ATNOS/CANDID and structure calculation with the simulated annealing routine of CYANA. As indicated in Fig. 1, this step may be performed repeatedly in order to obtain the “final” Structure V (Table 1; Fig. 4b, c). In our practice this includes that all chemical shift assignments are at this stage checked by a spectroscopist who has not been involved in the previous structure determination steps. If errors in the chemical shift or NOE assignments are detected, a new structure calculation is performed. The resulting Structure V is validated using an in-house combination of tools, as described in the “Appendix”. Selected validation parameters are included in the Table 1, where a column has been added that lists our current validation cut-offs. The “Appendix” describes additional procedures that we use to monitor the course of the automated structure calculation. We also check relations between Structure V and some raw NMR data, such as the agreement of secondary 13C chemical shifts and patterns of medium-range 1H–1H-NOEs with the locations of regular secondary structures in Structure V, and comparison of observed ring current shifts with ring current shifts calculated using the atom coordinates of Structure V. The thus validated Structure V is deposited in the PDB (accession code for the protein YP_926445.1: 2l6o).
Results and discussion
NMR structure of YP_926445.1 determined with J-UNIO
The data for the Structure V (PDB accession code 2l6o) in Table 1 show that the automated J-UNIO procedure (Fig. 1) yielded a high-quality NMR structure, which is comparable to structures determined by conventional interactive approaches. Comparison of the panels (a) and (b) in Fig. 4 documents that the Structures A and V have the same global fold, but that the Structure V is defined with much higher precision (Table 1). The improved precision is primarily due to the interactive expansion of the side chain chemical shift assignments, which resulted in a larger number of long-range NOE constraint identifications by UNIO-ATNOS/CANDID when preparing the input for the calculation of Structure V (Table 1).
The molecular architecture of YP_926445.1 contains a 5-stranded β sheet and three α-helices, with the regular secondary structures in the sequential order β1–β2–α1–β3–β4–β5–α2–α3 (Figs. 3, 4). There are three long polypeptide segments devoid of regular secondary structure, i.e., the N-terminal tetradecapeptide segment and two loops of residues 40–50 and 90–101. The protein forms a globular architecture with a precisely defined core of primarily hydrophobic residues and a surface layer of significantly less well-defined side chains (Fig. 4c). Comparison of the YP_926445.1 structure with the deposits in the Protein Data Bank indicated that this protein adopts a novel fold. Therefore, after the NMR structure was deposited in the PDB, the amino acid sequence of YP_926445.1 was used to generate a new Pfam protein family, PF13642 (alternatively included as DUF4144 in the list of “domains of unknown function”). PF13642 presently includes 82 members from 52 different bacterial species, with YP_9264451.1 as the only representative with known three-dimensional structure.
Applications of J-UNIO with JCSG target proteins
The Table 2 lists metrics about J-UNIO structure determinations for 17 JCSG target proteins, which all have been investigated as described in the preceding section for YP_926445.1 (during the past few months J-UNIO was used to determine an additional 10 protein structures of targets from various PSI:biology projects, with similar results as described here). In the following we discuss the data of Table 2 in the order of the individual steps of the J-UNIO protocol (Fig. 1).
In implementing J-UNIO (Fig. 1) we gave due consideration to the fact that screening of potential targets and the preparation of protein solutions for NMR structure determination (or of diffracting crystals for X-ray structure determination) is by far the most work-intensive part of each project, which also imposes the main limitations on the number of structures solved. For each successful sample preparation we were therefore very liberal when deciding on the measurement times for the individual NMR data sets. Both the APSY-NMR and NOESY data sets could have been obtained with shorter total recording times than used here. However, the improved signal-to-noise ratio and spectral resolution achieved with the generously selected recording times contributed significantly to high reliability of the results of the automated steps in J-UNIO (Fig. 1). In future applications one might also consider to select longer NOE mixing times than 65 ms, as used here, which could result in further improved, “cleaner” NOESY data sets (Wüthrich 1986).
The preparation of the YP_926445.1 NMR sample is described at the outset of the “Methods” section. This biochemical work was started after observing that YP_926445.1 represented a “hit” in a microscale screen of potential targets (Page et al. 2005; Peti et al. 2005; B. Pedrini et al., in preparation). For all the proteins in Table 2, a structure-quality protein solution was similarly obtained and used for the recording of the seven NMR data sets listed in Fig. 1. Based on the NMR-profile it was also known from the start (Fig. 1) to which extent the polypeptide chain would be observable in the experiments used for the chemical shift assignments, which resulted in further improved efficiency.
Backbone chemical shift assignments with the software UNIO-MATCH yielded results for between 75 and 100 % of the amino acid residues, with all but three proteins being in the range 81–96 % (Table 2). Interactive validation based on the [1H,1H]-NOESY spectra confirmed that with input from APSY-NMR experiments, UNIO-MATCH may yield incomplete assignments but very rarely generates errors in its output when used with the recommended standard set of parameters (Volk et al. 2008). Obtaining nearly complete correct backbone chemical shift assignments, including the Cβ atoms, by interactive supplementation of the results from UNIO-MATCH is of key importance with regard to both the subsequent automated amino acid side chain chemical shift assignment with UNIO-ATNOS/ASCAN and the automated NOE assignment with UNIO-ATNOS/CANDID. Since the large majority of the chemical shifts are known from the output of UNIO-MATCH, the extension of the assignments has been achieved with only a few hours of interactive work for each of the proteins in Table 2.
There are important advantages of the presently used APSY-NMR techniques when compared with conventional triple-resonance experiments: (1) Savings of instrument time. For the proteins in Table 2 the three APSY-NMR data sets (Fig. 1) were recorded with total measurement times of 6–96 h (B. Pedrini et al., to be published). (2) Higher digital resolution (Hiller et al. 2008). (3) 4- and 5-dimensional APSY-NMR experiments generate data of outstanding quality as input for automated chemical shift assignment with UNIO-MATCH (Volk et al. 2008). This is due to the high accuracy of the chemical shifts in 4- and 5-dimensional APSY data sets, which enables almost complete correct spin system identification by UNIO-MATCH. This key intermediate result is the basis for the high completeness of the assignments obtained by the subsequent optimization scheme for placing the thus identified spin systems into their positions in the protein sequence. This contrasts with the experience gained when using conventional triple-resonance data, which typically yield extensive degeneration of spin systems and consequently less complete and less reliable resonance assignments.
With the use of 1H–1H-NOE data for both, the amino acid side chain chemical shift assignment and as the major source of constraints for the structure calculation input, the J-UNIO protocol is reminiscent of earlier attempts at NMR structure determination based on the fact (Wüthrich 1986) that 1H–1H-NOE experiments contain, in principle, all the information needed to determine a protein structure (Ikeya et al. 2011; Kraulis 1994). However, in contrast to this earlier work, supplementing the NOESY data with verified chemical shift assignments for the polypeptide backbone and the 13Cβ positions makes J-UNIO robust and nonetheless highly efficient, since with the use of APSY-NMR the polypeptide backbone assignments are a small part of the overall effort.
When evaluating the extent of the automated side chain chemical shift assignments (Table 2), one has to consider that the percentage of assignment completeness after UNIO-ATNOS/ASCAN does not have the same weight as the assignment completeness reported for automated or interactive procedures based on NMR experiments that delineate through-bond connectivities. UNIO-ATNOS/ASCAN assigns chemical shifts for side chain hydrogen atoms involved in 1H–1H-NOE connectivities that yield NOE signal intensities above a user-defined threshold for assignment acceptance (Fiorito et al. 2008). Therefore, the side chain atoms with chemical shift assignments from UNIO-ATNOS/ASCAN will subsequently generate meaningful distance restraints. On the other hand, hydrogen atoms at or near the protein surface may be left unassigned or possibly even be erroneously assigned.
In the present study, UNIO-ATNOS/ASCAN provided assignments for 67–89 % of the atoms, with all but three proteins in the range from 72 to 89 % (Table 2). Interactive validation and extension of these assignments resulted on the one hand in an increased extent of the assignments to 90–96 % of the atoms for the individual proteins (Table 2), and on the other hand revealed that the results from the automated UNIO-ATNOS/ASCAN procedure contained up to 5 % erroneous assignments, depending on the protein. As shown previously (Fiorito et al. 2008), most of these erroneous assignments are highly permissive with regard to the outcome of the structure calculation, and the extent and quality of the UNIO-ATNOS/ASCAN assignments was for all proteins sufficient to achieve the correct fold in the Structure A (Figs. 1, 4a). The small impact of the erroneous assignments on the global fold can be rationalized from the observation that they are located almost exclusively on peripheral, solvent-accessible side chains (Fig. 4d). Overall, the NOE-based side chain chemical shift assignment strategy is thus highly efficient in providing nearly complete assignments for those hydrogen atom positions which are important for the definition of the three-dimensional protein structure. It further ensures efficient use of NMR instrument time, and requires minimal chemical shift calibrations when compared to using experiments that delineate through-bond connectivities for obtaining the side chain chemical shift assignments (Cavanagh et al. 2007).
The structure calculations with UNIO-ATNOS/CANDID and the simulated annealing routine of CYANA converged well, and the quality of the resulting protein structures compares favorably with the results of structure determinations based on interactive analysis of the NMR data. The J-UNIO protocol is an addition to a rapidly growing collection of procedures with more or less extensive automation of protein structure determination by NMR (for example, Atreya et al. 2000; Bartels et al. 1997; Crippen et al. 2010; Lemak et al. 2008; Lescop and Brutscher 2009; Moseley et al. 2001; Schmucki et al. 2008; Staykova et al. 2008; Zimmermann et al. 1997), which all result in improved efficiency and reduced bias when compared to interactive procedures. J-UNIO differs from most of the other presently available procedures in that automation starts with the peak picking of the raw NMR spectra, rather than with interactively prepared peak lists. It will now be of interest to observe which ones of the presently available procedures, or possibly upcoming new additions, will find broader application over the coming years.
References
Atreya HS, Sahu SC, Chary KVR, Govil G (2000) A tracked approach for automated NMR assignments in proteins (TATAPRO). J Biomol NMR 17:125–136
Bartels C, Güntert P, Billeter M, Wüthrich K (1997) GARANT—a general algorithm for resonance assignment in multidimensional nuclear magnetic resonance spectra. J Comput Chem 18:139–149
Cavanagh J, Fairbrother WJ, Rance M, Palmer AG III, Skelton NJ (2007) Protein NMR spectroscopy: principles and practice, 2nd edn. Elsevier Academic Press, Amsterdam
Crippen GM, Rousaki A, Revington M, Zhang Y, Zuiderweg ERP (2010) SAGA: rapid automatic mainchain NMR assignment for large proteins. J Biomol NMR 46:281–298
DeMarco A, Wüthrich K (1976) Digital filtering with a sinusoidal window function: an alternative technique for resolution enhancement in FT NMR. J Magn Reson 24:201–204
Elsliger MA, Deacon A, Godzik A, Lesley S, Wooley J, Wüthrich K, Wilson IA (2010) The JCSG high-throughput structural biology pipeline. Acta Cryst F 66:1137–1142
Fiorito F, Herrmann T, Damberger FF, Wüthrich K (2008) Automated amino acid side-chain NMR assignment of proteins using 13C- and 15N-resolved [1H,1H]-spectra. J Biomol NMR 42:23–33
Güntert P, Mumenthaler C, Wüthrich K (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol 273:283–298
Herrmann T, Güntert P, Wüthrich K (2002a) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol 319:209–227
Herrmann T, Güntert P, Wüthrich K (2002b) Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J Biomol NMR 24:171–189
Hiller S, Fiorito F, Wüthrich K, Wider G (2005) Automated projection spectroscopy (APSY). Proc Natl Acad Sci USA 102(31):10876–10881
Hiller S, Wider G, Wüthrich K (2008) APSY-NMR with proteins: practical aspects and backbone assignment. J Biomol NMR 42:179–195
Ikeya T, Jee J-G, Shigemitsu Y, Hamatsu J, Mishima M, Ito Y, Kainosho M, Güntert P (2011) Exclusively NOESY-based automated NMR assignment and structure determination of proteins. J Biomol NMR 50:137–146
Jaudzems K, Geralt M, Serrano P, Mohanty B, Horst R, Pedrini B, Elsliger MA, Wilson IA, Wüthrich K (2010) NMR structure of the protein NP_247299.1: comparison with the crystal structure. Acta Cryst F 66:1367–1380
Keller R (2004) CARA: computer aided resonance assignment. http://cara.nmr.ch/
Koradi R, Billeter M, Wüthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14:51–55
Kraulis PJ (1994) Protein three-dimensional structure determination and sequence-specific assignment of 13C and 15N-separated NOE data. J Mol Biol 243:696–728
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK—a program to check the stereochemical quality of protein structures. J Appl Cryst 26:283–291
Lemak A, Steren CA, Arrowsmith CH, Llinas M (2008) Sequence specific resonance assignment via multicanonical Monte Carlo search using an ABACUS approach. J Biomol NMR 41:29–41
Lescop E, Brutscher B (2009) Highly automated protein backbone resonance assignment within a few hours: the «BATCH» strategy and software package. J Biomol NMR 44:43–57
Lesley S, Kuhn P, Godzik A, Deacon A, Mathews I, Kreusch A, Spraggon G, Klock H, McMullan D, Shin T, Vincent J, Robb A, Brinen L, Miller M, McPhillips T, Miller M, Scheibe D, Canaves J, Guda C, Jaroszewski L, Selby T, Elsliger MA, Wooley J, Taylor S, Hodgson K, Wilson IA, Schultz P, Stevens R (2002) Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. Proc Natl Acad Sci USA 99:11664–11669
Lüthy R, Bowie J, Eisenberg D (1992) Assessment of protein models with three-dimensional profiles. Nature 356:83–85
Metzler W, Constantine K, Friedrichs M, Bell A, Ernst E, Lavoie T, Mueller L (1993) Charecterization of the three-dimensional solution structure of human profilin: 1H, 13C, and 15N NMR assignments and global folding pattern. Biochemistry 32:13818–13829
Mohanty B, Serrano P, Pedrini B, Jaudzems K, Geralt M, Horst R, Herrmann T, Elsliger ME, Wilson IA, Wüthrich K (2010) NMR structure of the protein NP_247299.1: comparison with the crystal structure. Acta Cryst F 66:1381–1392
Moseley HN, Monleon D, Montelione GT (2001) Automatic determination of protein backbone resonance assignments from triple resonance nuclear magnetic resonance data. Meth Enzym 399:91–108
Page R, Peti W, Wilson IA, Stevens RC, Wüthrich K (2005) NMR screening and crystal quality of bacterially expressed prokaryotic and eukaryotic proteins in a structural genomics pipeline. Proc Natl Acad Sci USA 102(6):1901–1905
Peti W, Page R, Moy K, O’Neil-Johnson M, Wilson IA, Stevens RC, Wüthrich K (2005) Towards miniaturization of a structural genomics pipeline using macro-expression and microcoil NMR. J Struct Funct Genomics 6:259–267
Schmucki R, Yokohama S, Güntert P (2008) Automated assignment of NMR chemical shifts using peak-particle dynamics simulation with the DYNASSIGN algorithm. J Biomol NMR 43:97–109
Serrano P, Pedrini B, Geralt M, Jaudzems K, Mohanty B, Horst R, Herrmann T, Elsliger MA, Wilson IA, Wüthrich K (2010) Comparison of NMR and crystal structures highlights conformational isomerism in protein active sites. Acta Cryst F 66(10):1392–1405
Staykova DK, Fredriksson J, Bermel W, Billeter M (2008) Assignment of protein NMR spectra based on projections, multi-way decomposition and a fast correlation approach. J Biomol NMR 42:87–97
Volk J, Herrmann T, Wüthrich K (2008) Automated sequence-specific protein NMR assignment using the memetic algorithm MATCH. J Biomol NMR 41:127–138
Wishart D, Sykes B (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR 4:135–140
Wüthrich K (1986) NMR of proteins and nucleic acids. Wiley, New York
Wüthrich K (2010) NMR in a crystallography-based high-throughput protein structure-determination environment. Acta Cryst F 66:1365–1366
Zimmermann DE, Kulikowski CA, Huang Y, Feng W, Tashiro M, Shimotakahara S, Chien C, Powers R, Montelione GT (1997) Automated analysis of protein NMR assignments using methods from artificial intelligence. J Mol Biol 269:592–610
Acknowledgments
The following financial support is acknowledged: Swiss National Science Foundation and ETH Zürich through the NCCR Structural Biology; Swiss National Science Foundation for a Fellowship to BP (PA00A–104097/1); NIH, National Institute of General Medical Services, Protein Structure Initiative, Grants U54 GM094586 and U54 GM074898 (the content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Science or the National Institutes of Health). KW is the Cecil H. and Ida M. Green Professor of Structural Biology at The Scripps Research Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Pedro Serrano and Bill Pedrini contributed equally to this work.
Appendix: Validation of J-UNIO NMR structures
Appendix: Validation of J-UNIO NMR structures
Our validation strategy makes use of quantitative criteria to qualify the Structure V (Fig. 1), including the publically available tools Procheck (Laskowski et al. 1993), Verify3D (Lüthy et al. 1992) and the PDB validation suite. In-house threshold values for acceptance of the individual criteria (Table 1) were established based on past high-quality interactive protein structure determinations in our laboratory. Furthermore, some qualitative tools are used for initial checks of the final Structure V, in order to guide the spectroscopist during the early stages of the validation procedure, and additional tools are used to monitor the course of the automated structure determination. In the following we comment on the validation tools represented in Table 1, and then on the additional criteria.
A first criterion considered in Table 1 enables an evaluation of the input for the protein structure calculation, i.e., we request that the number of long-range NOE constraints per residue must be higher than the threshold of five. In our experience, satisfying this sole criterion is sufficient to document that nearly complete chemical shift assignments have been obtained and that there is also a dense network of sequential and medium-range NOE distance constraints, thus qualifying an input for the structure calculation that is of high overall quality.
A second group of criteria is used to document acceptable convergence of the structure calculation, with small residual violations of the experimental input data and small distortions of the covalent structure geometry. These are the residual target function value, the number of residual NOE distance constraint violations, the number of residual dihedral angle violations, and the RMSD from standard covalent structure geometry.
In a third group of criteria, the precision of the Structure V (Fig. 1) is characterized by RMSDs to the mean coordinates of the bundle of conformers (Fig. 4b) calculated for the backbone heavy atoms and all heavy atoms, respectively. In addition, we introduce the “core precision” as the all-heavy-atom RMSD calculated for all the residues with solvent accessibility below 15 %. Initial experience with this parameter indicates that it is useful for comparison of the core packing in different protein structure types. The overall quality of the Structure V is monitored also by the PROCHECK global quality score, the Verify3D raw score, and the side chain planarity Z-score, with the acceptance threshold values listed in Table 1. In addition, a structure is accepted only if all criteria of the PDB validation suite are satisfied.
Additional qualitative criteria for structure validation are used to directly assess the agreement between selected raw experimental NMR data and corresponding data derived from the Structure V bundle of conformers (Fig. 4b). First, comparison of the structure-derived and the observed ring current shifts provides qualitative checks on possible local errors in amino acid side chain arrangements. The Fig. 5 shows a plot of the observed methyl hydrogen ring current shifts (RCSobs) versus the corresponding ring current shifts calculated from the atomic coordinates of the NMR structure (RCSpre) for the protein YP_926445.1. Prior to structure validation with the tools listed in Table 1, methyl groups with entries located far from the diagonal in this presentation would be singled out for further interactive analysis until a satisfactory fit is attained, or a rationale is found to explain the apparent discrepancy. Second, comparison of the regular secondary structures in Structure V and those predicted from the 13Cα and 13Cβ chemical shift values (Fig. 6) afford a check of the agreement between experimental NMR data for the polypeptide backbone and the final Structure V (Wishart and Sykes 1994), and the same applies to analysis of the agreement between experimental patterns of sequential and medium-range 1H–1H-NOEs and the locations of regular secondary structures in Structure V (Fig. 7) (Wüthrich 1986). Similar to the aforementioned handling of the ring current shift data, apparent discrepancies between the locations of regular secondary structures, the corresponding 13Cα and 13Cβ chemical shift values and/or the NOE patterns are followed up prior to the structure validation reported in Table 1.

Plot of observed methyl hydrogen ring current shifts (RCSobs) for the protein YP_926445.1 versus the corresponding ring current shifts calculated from the atomic coordinates of the NMR structure (RCSpre). RCSobs is the difference between corresponding observed and random coil chemical shifts. RCSpre was computed with the Johnson–Bovey model implemented in the software MOLMOL (Koradi et al. 1996), and the average over the 20 NMR conformers of Structure V (Fig. 4b, c) is given

Sum of the secondary 13Cα and 13Cβ chemical shifts, Δδi, in the protein YP_926445.1 plotted versus the amino acid sequence. The Δδi value for residue i represents the average over the three consecutive residues i − 1, i and i + 1: Δδi = 1/3 (ΔδC αi−1 + ΔδC αi + ΔδC αi+1 + ΔδC βi−1 + ΔδC βi + ΔδC βi+1 ) (Metzler et al. 1993). The ΔδCα and ΔδCβ values were determined with the program package UNIO-ATNOS/CANDID (Herrmann et al. 2002a, b) by subtracting the random coil shifts from the experimentally determined chemical shifts. Positive Δδi values indicate that the residue i is located in a helical structure, while a negative value indicates a location in a β-strand. The positions of the regular secondary structures in the Structure V are indicated at the top of the figure

Sequential and medium-range 1H–1H NOE constraints observed for YP_926445.1. The amino acid sequence and the regular secondary structures identified by MOLMOL (Koradi et al. 1996) in Structure V are indicated at the top. Residues are included in the regular secondary structures if the criteria are satisfied for at least 15 conformers in the bundle of 20 conformers (Fig. 4b). In the notations for the 1H–1H NOEs on the left, N, α and β indicate the HN, Hα and Hβ atoms, respectively. Sequential NOEs are indicated by continuous horizontal lines extending over the connected polypeptide segments, where thick and thin lines represent strong and weak NOEs, respectively. Medium-range NOEs are indicated by horizontal lines linking the two residues that are connected by the NOE (Wüthrich 1986)
The Table 3 lists the three principal criteria that we use to monitor the course of the calculation of Structure V with the software UNIO-ATNOS/CANDID and the simulated annealing routine of CYANA (for the initial round of calculations which result in Structure A, we only evaluate the final result obtained after cycle 7 (Herrmann et al. 2002a), since the criteria of Table 3 would be dominantly affected by the obvious limitations of the input used, as is described in the main text). The CYANA target function value must be below the threshold of 300 Å2 after the first cycle, should then monotonously adopt smaller values after cycles 2–6, and be below the threshold of 10 Å2 after cycle 7. The percentage of covalent NOEs assigned (Herrmann et al. 2002b) is automatically recorded by the ATNOS module in UNIO-ATNOS/CANDID. Obtaining high completeness of these “covalent assignments” assures robustness of the 1H–1H-NOE-based approach used by J-UNIO. Finally, checking the extent to which the NOE cross peaks in the three NOESY data sets (Fig. 1) have been assigned serves primarily to evaluate the success of the effort made for the interactive completion of the assignments from the automated routines. Rationales for choosing the rather permissible threshold of <20 % are given in the main text.
Rights and permissions
About this article
Cite this article
Serrano, P., Pedrini, B., Mohanty, B. et al. The J-UNIO protocol for automated protein structure determination by NMR in solution. J Biomol NMR 53, 341–354 (2012). https://doi.org/10.1007/s10858-012-9645-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-012-9645-2