
Abstract
The NMR structure of the 206-residue protein NP_346487.1 was determined with the J-UNIO protocol, which includes extensive automation of the structure determination. With input from three APSY-NMR experiments, UNIO-MATCH automatically yielded 77 % of the backbone assignments, which were interactively validated and extended to 97 %. With an input of the near-complete backbone assignments and three 3D heteronuclear-resolved [1H,1H]-NOESY spectra, automated side chain assignment with UNIO-ATNOS/ASCAN resulted in 77 % of the expected assignments, which was extended interactively to about 90 %. Automated NOE assignment and structure calculation with UNIO-ATNOS/CANDID in combination with CYANA was used for the structure determination of this two-domain protein. The individual domains in the NMR structure coincide closely with the crystal structure, and the NMR studies further imply that the two domains undergo restricted hinge motions relative to each other in solution. NP_346487.1 is so far the largest polypeptide chain to which the J-UNIO structure determination protocol has successfully been applied.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The NMR solution structure of a member of the haloacid dehalogenase (HAD) protein superfamily (Aravind et al. 1998; Burroughs et al. 2006; Hisano et al. 1996), the two-domain 206-residue putative phosphoglycolate phosphatase NP_346487.1, was determined with the J-UNIO protocol, which includes extensive automation of protein structure determination in solution (Serrano et al. 2012). J-UNIO is used routinely in our laboratory for NMR structure determination of proteins with sizes up to about 150 amino acid residues. Here, we want to explore how J-UNIO can deal with the increased spectral complexity of a significantly larger protein, which also has somewhat broader linewidths of the NMR signals. As a target for this study we selected the protein NP_346487.1, for which a crystal structure (PDB code 2go7) was available for validation of the NMR structure determination. Additional interest comes from the fact that four molecules of this two-domain protein are contained in the crystallographic unit cell, affording the opportunity to compare structural variations among these four molecules with the arrangement of the two domains in solution.
Methods
Protein expression and purification
The NP_346487.1 gene in the plasmid vector pSpeedET as obtained from the JCSG Crystallomics Core, was amplified and inserted into a modified pET-28b vector containing an engineered TEV-protease cleavage site between NdeI and HindIII restriction sites, and the resulting plasmid pET-28b-TEV-NP_346487.1 was used to transform the E. coli strain BL21(DE3) (Novagen). The protein was expressed in M9 minimal medium containing 1 g/L of 15NH4Cl and 4 g/L of [13C6]-d-glucose (Cambridge Isotope Laboratories) as the sole sources for nitrogen and carbon. Standard procedures were used for the expression and purification of the protein. The yield of purified NP_346487.1 was 40 mg per liter of culture. NMR samples were prepared by adding 5 % (v/v) D2O, 0.03 % (w/v) NaN3 and complete protease inhibitor cocktail (Roche) to 500 μL of a 1.5 mM solution of either 15N-labeled or 13C,15N-labeled NP_346487.1 in NMR buffer (20 mM sodium phosphate, 50 mM NaCl, pH 6.5).
NMR spectroscopy
NMR experiments for resonance assignment and collection of distance restraints were conducted at 298 K on a Bruker Avance 600 MHz spectrometer equipped with a TXI HCN z-gradient cryoprobe, and an Avance 800 MHz instrument with a TXI 1H-13C/15N room temperature probe with xyz-gradient. 5D APSY-HACACONH, 4D APSY-HACANH and 5D APSY-CBCACONH data sets (Hiller et al. 2005, 2008) were recorded at 600 MHz with 52, 57 and 46 projections, respectively, each containing 2048 × 64 acquisition points. 3D [1H,1H]-NOESY-15N-HSQC, 3D [1H,1H]-NOESY-13C(ali)-HSQC and 3D [1H,1H]-NOESY-13C(aro)-HSQC data sets were recorded at 800 MHz with a mixing time of 60 ms. Chemical shifts are relative to DSS, and were calibrated against the water signal (4.77 ppm at 298 K). Spectra were processed with Topspin 2.1 (Bruker Biospin).
Resonance assignment and structure calculation
The J-UNIO procedure (Serrano et al. 2012) was followed. The automated backbone assignments obtained using the APSY-NMR results as input for the software UNIO-MATCH (Volk et al. 2008) was extended to near-completion by interactive analysis of the 3D 15N-resolved [1H,1H]-NOESY spectrum, using the software CARA (Keller 2004). The backbone assignments and the aforementioned three NOESY data sets were then used as input for obtaining side chain assignments with UNIO-ATNOS/ASCAN (Herrmann et al. 2002a; Fiorito et al. 2008), which was followed by automated NOE assignment with UNIO-ATNOS/CANDID 1.0.4 (Herrmann et al. 2002a, b) and structure calculation with CYANA 3.0 (Güntert et al. 1997), using the standard 7-cycle protocol (Herrmann et al. 2002b). The two-domain architecture of NP_346487.1 previously observed in the crystal structure (PDB code 2go7) was found also in solution. During refinement, the structures of the two domains were separately calculated, using as input the unambiguous intra-domain constraints from cycle 7 of the calculation for the intact protein. The 40 conformers with the lowest residual CYANA target function values after cycle 7 for the intact protein, and after cycle 8 for the two individual domains, respectively, were energy-minimized in a water shell with the program OPALp (Luginbühl et al. 1996; Koradi et al. 2000). The 20 conformers with the lowest target function values that satisfied all validation criteria (Serrano et al. 2012) were selected to represent the NMR structure. The structures were analyzed and figures were generated using MOLMOL (Koradi et al. 1996).
The chemical shift assignments have been deposited in the BioMagResBank (Accession No. 25127; http://www.bmrb.wisc.edu), and atomic coordinates were deposited in the Protein Data Bank for bundles of 20 NMR conformers of the complete protein (accession code 2msn) and the two separately refined (see text above) individual domains (accession codes 2mu1 and 2mu2).
Results
Considering the high expression yield of the protein NP_346487.1 (see ‘Methods’), all exploratory NMR experiments were performed in 5 mm NMR tubes (Fig. 1), rather than with microscale experiments as in the standard J-UNIO protocol (Serrano et al. 2012). These experiments showed that a 1.5 mM “structure-quality” solution of the protein (Pedrini et al. 2013) could be prepared for the structure determination.

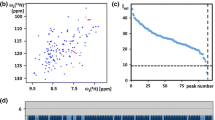
Characterization of the solution of the protein NP_346487.1 used for the NMR structure determination and survey of the polypeptide backbone assignments. a Amino acid sequence (residues-2 and -1 originate from the expression tag), extent of the automated backbone assignments obtained with the software UNIO-MATCH, which are based on APSY-NMR data alone (brown underline; for some residues only part of the backbone chemical shifts were assigned, as described in the text), and backbone assignments after interactive validation and extension of the automatic assignments with the use of 3D heteronuclear-resolved [1H,1H]-NOESY data (green underline; green dots identify residues with partial assignments of the backbone chemical shifts). Above the sequence, β-strands are indicated by cyan arrows and helices by red bars. b 600 MHz 2D [15N,1H]-HSQC spectrum of a 1.5 mM solution of uniformly 13C,15N-labeled NP_346487.1 in 20 mM sodium phosphate buffer at pH 6.5 and T = 298 K. The backbone amide group assignments are indicated by the sequence numbers. c Expansion of the central spectral region indicated by broken lines in (b)
Automated NMR assignment
The standard J-UNIO automated assignment with UNIO-MATCH (Volk et al. 2008) yielded chemical shifts for the 1HN, 15N, 13C′ and 13Cα atoms of 158 residues (77 %, Fig. 1a), the 13Cβ atoms of 145 among these residues (74 %), and the 1Hα chemical shifts of 171 residues (83 %). Interactive validation and extension of the automated backbone assignments using the 3D 15N-resolved and 3D 13C(ali)-resolved [1H,1H]-NOESY spectra resulted in the identification and correction of three erroneous assignments (backbone atoms of Phe136, 13Cβ of Ala96 and Thr205), and in obtaining complete backbone assignments for a total of 200 residues (97 %). For the six remaining residues, Leu10, Asp11, Val50, His108, Lys139 and Gly189, the backbone 15N–1H groups were not observed, but part of the 1Hα and 1Hβ signals could be assigned based on sequential NOEs. The final assignments are documented in a 2D [15N,1H]-HSQC spectrum (Fig. 1b, c). Automated side-chain assignment with the program UNIO-ATNOS/ASCAN (Herrmann et al. 2002a; Fiorito et al. 2008) resulted in 77 % of the expected assignments (88 % of all non-labile hydrogens). Interactive inspection showed that 92 % of the ASCAN assignments were correct. The erroneous assignments were identified and corrected, and the chemical shift lists for side chains with incomplete assignments could be expanded, resulting in about 88 % of the expected assignments.
Automated NOE assignment and structure calculation
A total of 4222 NOE upper distance limits were collected with the combined use of UNIO-ATNOS/CANDID (Herrmann et al. 2002a, b) and CYANA (Güntert et al. 1997; Table 1). A two-domain structure was obtained, with a “core domain” of residues 1–10 and 88–206, and a “cap domain” of residues 17–80, which are linked by two short polypeptide segments (residues 11–16 and 81–87). The relative orientation of the two domains is variable among the bundle of NMR conformers (Fig. 2), but the Fig. 2b, c clearly show that the core domain and the cap domain are individually well defined (see also Table 1). To further investigate the convergence of the structure calculation for the intact protein (Fig. 2a), we computed the structures of the individual domains by adding an 8th cycle of structure calculation to the standard J-UNIO protocol, which used an input of intra-domain constraints only (Table 1). The resulting domain structures (Fig. 3) are closely similar to those resulting from the calculation with the intact protein (Figure S1), with RMSDs between the mean coordinates of the corresponding bundles of 20 conformers in Figs. 2b, c, 3a, c, of 0.58 Å for the backbone heavy atoms and 1.03 Å for all heavy atoms of the cap domain, and 0.59 and 1.00 Å for the core domain. This shows that the computational tools used in J-UNIO handled the structure calculation of the intact protein quite well in spite of the implicated plasticity of the structure.

NMR structure of NP_346487.1 in phosphate buffer calculated for the complete two-domain protein and comparison with the crystal structure (PDB 2go7). Bundles of 20 NMR conformers were superimposed for best fit with their mean coordinates, and the mean crystal structure was superimposed for best fit with the mean NMR atom coordinates. a Structure presentation by a bundle of 20 conformers superimposed for best fit of residues 3–205 (green). The crystal structure is represented by a superposition of the four molecular structures per unit cell (black). b Superposition of the polypeptide backbone heavy atoms for best fit of the core domain residues 3–10 and 88–205. c Best fit superposition for the cap domain residues 17–80

NMR structures calculated separately for the two domains of the protein NP_346487.1 from data collected with the intact protein (same data as for Fig. 2, see Table 1; the structures are closely similar to the structures of the individual domains in Fig. 2, see Figure S1 and the text) and comparison with the crystal structure. a Core domain (residues 1–10 and 88–206). 20 NMR conformers (brown) and the four crystal structures (black) are superimposed for best fit of the polypeptide segments 3–10 and 88–205. b Stereo ribbon diagram of the NMR conformer closest to the mean coordinates of the bundle of conformers in a. Color code β-strands, cyan; helices, red/yellow; non-regular secondary structure, grey. The individual regular secondary structures are identified, and the two chain ends are marked with N and C. c, d Cap domain residues 17–80. Same presentations as in a and b, with best-fit superposition of residues 17–80
No NOEs between the two domains could be identified, but the hydrogen atoms in the two linker polypeptide segments are involved in 80 NOE constraints (Table 1), i.e., 13 sequential NOEs, 9 intra-chain medium-range NOEs, and 58 long-range NOEs with hydrogen atoms in the other linker peptide segment or one of the two domains. The NOEs with the linker polypeptides significantly restrict the accessible relative domain orientations (Fig. 2b, c), as was verified by a structure calculation for the intact protein based on an input without these 80 linker-associated NOE constraints.
NMR structure of NP_346487.1
The NMR structure of the core domain (residues 1–10 and 88–206) consists of a strongly twisted six-stranded parallel β-sheet (β3–β2–β1–β4–β5–β6) formed by the polypeptide segments 126–130, 102–106, 5–8, 159–163, 178–181 and 190–192. The β-sheet is flanked on one side by the helices α6, α7 and α10, and by α8 and α9 on the other side, and there are three 310-helical turns at both ends of β3 and preceding β4 (Figs. 1a, 3b).
The NMR structure of the cap domain (residues 17–80) shows an antiparallel four-helix bundle with α1, α2, α3 and α4, which is typical for the subfamily I of phosphohydrolases (Griffin et al. 2012; Strange et al. 2009). A short helix, α5, at the C-terminal end of the cap domain forms a helix–kink–helix motif with α4.
The two linker polypeptide segments (residues 11–16 and 81–87) connect the strand β1 and the helix α6 of the core domain to the helices α1 and α5 of the cap domain.
Discussion
The J-UNIO protocol for automated structure determination (Serrano et al. 2012) is used routinely in our laboratory for studies of proteins with up to about 150 amino acid residues (for example, Jaudzems et al. 2010; Mohanty et al. 2010; Serrano et al. 2010, 2014; Wahab et al. 2011). Here, the protein NP_346487.1 was selected from the list of JCSG targets to explore how J-UNIO, with the use of APSY-NMR and 3D heteronuclear-resolved [1H,1H]-NOESY experiments, would cope with the complexities of the spectra of larger proteins. As Table 1 and Figs. 1, 2, 3 show, the result is comparable to those obtained with smaller proteins. This was possible because a structure-quality protein solution (Pedrini et al. 2013) could be obtained for the NMR experiments, which is a condition that has to be met also when working with smaller proteins. Overall, the present work shows that J-UNIO can be used for structure determination of non-deuterated proteins with molecular weights up to at least 25 kDa.
While 3D heteronuclear [1H,1H]-NOESY experiments are known to yield good results for larger proteins (Horst et al. 2006), APSY-NMR has so far been used mainly with smaller proteins (Dutta et al. 2014). For proteins of similar size to NP_346487.1, Gossert et al. (2011) described an alternative approach for automated NMR assignment with APSY-NMR, which is based on the use of fractionally deuterated proteins. In addition to polypeptide backbone assignments, this protocol enabled also the chemical shift assignments for peripheral side chain methyl groups. The two protocols are complementary, since the combination of APSY-NMR with fractional deuteration has been introduced for providing chemical shift assignments needed for studies of protein–ligand interactions, whereas the present approach provides data needed for de novo protein structure determination.
The two individual domain structures of NP_346487.1 (Table 1; Fig. 3) fit near-identically with the corresponding parts of the protein in crystals. For the core domain, the backbone and all-heavy-atom RMSD values between the mean atom coordinates of the bundle of 20 NMR conformers and the bundle of four molecules in the crystallographic unit cell are 1.2 and 1.8 Å, respectively, and the corresponding values for the cap domain are 1.3 and 2.3 Å, where the somewhat larger all-heavy-atom RMSD value for the cap domain can be rationalized by its smaller size and concomitantly larger percentage of solvent-exposed amino acid residues (Jaudzems et al. 2010). Previously introduced additional criteria for comparison of crystal and NMR structures (Jaudzems et al. 2010; Mohanty et al. 2010; Serrano et al. 2010) showed that the values of the backbone dihedral angles ϕ and ψ of the crystal structure are outside of the value ranges covered by the bundle of NMR conformers for <10 residues. Both the high-precision of the individual domain structures (Table 1) and the close fit with the crystal structure document the success of the use of J-UNIO with this larger protein.
Comparison of the complete structures of NP_346487.1 in crystals and in solution shows that the range of relative spatial arrangements of the two domains is significantly larger in solution than in the crystal. The four molecules in the asymmetric crystallographic unit cell have nearly identical inter-domain orientations, as shown by the superposition of the four structures (black lines in Fig. 2). In solution, the superpositions shown in Fig. 2 indicate that the two domains undergo limited-amplitude hinge motions about the double-linker region. The limited range of these motions is due to restraints from NOEs between the linker peptide segment and the globular domains, whereas no NOEs were identified between the two domains. There are indications from line broadening of part of the linker residue signals (missing amide proton signals, see Fig. 1a) that the hinge motions are in the millisecond to microsecond time range. Measurements of 15N{1H}-NOEs showed uniform values near +0.80 for the two domains and across the linker region, documenting the absence of high-frequency backbone mobility.
Homologous proteins to NP_346487.1 have been shown to interact weakly with magnesium ions (the crystal structure of NP_346487.1 contains one magnesium ion per molecule) and phosphate ions. Exploratory studies indicated that the addition of either phosphate or Mg2+ to the NMR sample did not visibly affect the structures of the individual domains, and had at most very small effects on the plasticity of the intact NP_346487.1. These function-related ligand-binding studies will be described elsewhere (K. Jaudzems, personal communication).
A recent structure determination of a β-barrel fold 200-residue protein with an integrative approach, “resolution-adapted structural recombination (RASREC) Rosetta”, used a wide array of different NMR experiments with multiple differently isotope-labeled protein preparations measured under different solution conditions (Sgourakis et al. 2014). This result was highly acclaimed (Lloyd and Wuttke 2014) and, as was correctly stated by one of the reviewers, it should not be directly compared with the present work because Sgourakis et al. (2014) performed their experiments with a dilute protein solution of limited stability. Nonetheless it is important to demonstrate to the biochemistry community that NMR structures of this size can efficiently be determined using routine NMR experiments, provided that a structure-quality protein solution (Pedrini et al. 2013) is prepared (see also Mohanty et al. 2014). Similar to obtaining diffracting crystals for protein crystallography, obtaining structure-quality solution NMR samples may require major efforts of protein engineering and optimization of solution conditions. As illustrated in this paper and also by Mohanty et al. (2014), with the availability of a structure-quality protein sample, the J-UNIO protocol (Serrano et al. 2012) applied to a 200-residue protein requires the recording of only 7 experiments, i.e., one [15N,1H]-HSQC spectrum, three APSY-NMR data sets and three heteronuclear-resolved [1H,1H]-NOESY spectra, which can all be measured at identical solution conditions with about 10 days of spectrometer time (or <7 days when using non-uniform sampling of the NOESY spectra; to be published) and a few days of work by a spectroscopist for the data analysis.
References
Aravind L, Galperin MY, Koonin EV (1998) The catalytic domain of the P-type ATPase has the haloacid dehalogenase fold. Trends Biochem Sci 23:127–129
Burroughs AM, Allen KN, Dunaway-Mariano D, Aravind L (2006) Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J Mol Biol 361:1003–1034
Dutta SK, Serrano P, Proudfoot A, Geralt A, Pedrini B, Herrmann T, Wüthrich K (2014) APSY-NMR for protein backbone assignment in high-throughput structural biology. J Biomol NMR. doi:10.1007/s10858-014-9881-8
Fiorito F, Herrmann T, Damberger FF, Wüthrich K (2008) Automated amino acid side-chain NMR assignment of proteins using 13C- and 15N-resolved 3D [1H,1H]-NOESY. J Biomol NMR 42:23–33
Gossert AD, Hiller S, Fernández C (2011) Automated NMR resonance assignment of large proteins for protein-ligand interaction studies. J Am Chem Soc 133:210–213
Griffin JL, Bowler MW, Baxter NJ, Leigh KN, Dannatt HR, Hounslow AM, Blackburn GM, Webster CE, Cliff MJ, Waltho JP (2012) Near attack conformers dominate β-phosphoglucomutase complexes where geometry and charge distribution reflect those of substrate. Proc Natl Acad Sci USA 109:6910–6915
Güntert P, Mumenthaler C, Wüthrich K (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol 273:283–298
Herrmann T, Güntert P, Wüthrich K (2002a) Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J Biomol NMR 24:171–189
Herrmann T, Güntert P, Wüthrich K (2002b) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol 319:209–227
Hiller S, Fiorito F, Wüthrich K, Wider G (2005) Automated projection spectroscopy (APSY). Proc Natl Acad Sci USA 102:10876–10881
Hiller S, Wider G, Wüthrich K (2008) APSY-NMR with proteins: practical aspects and backbone assignment. J Biomol NMR 42:179–195
Hisano T, Hata Y, Fujii T, Liu JQ, Kurihara T, Esaki N, Soda K (1996) Crystal structure of L-2-haloacid dehalogenase from Pseudomonas sp. YL. An alpha/beta hydrolase structure that is different from the alpha/beta hydrolase fold. J Biol Chem 271:20322–20330
Horst R, Wider G, Fiaux J, Bertelsen EB, Horwich AL, Wüthrich K (2006) Proton-proton Overhauser NMR spectroscopy with polypeptide chains in large structures. Proc Natl Acad Sci USA 103:15445–15450
Jaudzems K, Geralt M, Serrano P, Mohanty B, Horst R, Pedrini B, Elsliger MA, Wilson IA, Wüthrich K (2010) NMR structure of the protein NP_247299.1: comparison with the crystal structure. Acta Cryst F 66:1367–1380
Keller R (2004) Computer-aided resonance assignment. Cantina. http://cara.nmr.ch/
Koradi R, Billeter M, Wüthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14(51–55):29–32
Koradi R, Billeter M, Güntert P (2000) Point-centered domain decomposition for parallel molecular dynamics simulation. Comp Phys Commun 124:139–147
Laskowski RA, Macarthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26:283–291
Lloyd NR, Wuttke DS (2014) Less is more: structures of difficult targets with minimal constraints. Structure 22:1223–1224
Luginbühl P, Güntert P, Billeter M, Wüthrich K (1996) The new program OPAL for molecular dynamics simulations and energy refinements of biological macromolecules. J Biomol NMR 8:136–146
Mohanty B, Serrano P, Pedrini B, Jaudzems K, Geralt M, Horst R, Herrmann T, Elsliger MA, Wilson IA, Wüthrich K (2010) Comparison of NMR and crystal structures of the proteins TM1112 and TM1367. Acta Cryst F 66:1381–1392
Mohanty B, Serrano P, Geralt M, Wüthrich K (2014) NMR structure determination of the protein NP_344798.1 as the first representative of the Pfam PF06042. J Biomol NMR (in press)
Pedrini B, Serrano P, Mohanty B, Geralt M, Wüthrich K (2013) NMR-profiles of protein solutions. Biopolymers 99:825–831
Serrano P, Pedrini B, Mohanty B, Geralt M, Herrmann T, Wüthrich K (2010) Comparison of NMR and crystal structures highlights conformational isomerism in protein active sites. Acta Cryst F 66:1393–1406
Serrano P, Pedrini B, Geralt M, Jaudzems K, Mohanty B, Horst R, Herrmann T, Wüthrich K (2012) The J-UNIO protocol for automated protein structure determination by NMR in solution. J Biomol NMR 53:341–354
Serrano P, Geralt M, Mohanty B, Wüthrich K (2014) NMR structures of the α-proteobacterial ATPase-regulating ζ-subunits. J Mol Biol 15:2547–2553
Sgourakis NG, Natajaran K, Ying J, Vögeli B, Boyd LF, Margulies DH, Bax A (2014) The structure of mouse cytomegalovirus m04 protein obtained from sparse NMR data reveals a conserved fold of the m02–m06 viral immune modulator family. Structure 22:1263–1273
Strange RW, Antonyuk SV, Ellis MJ, Bessho Y, Kuramitsu S, Shinkai A, Yokoyama S, Hasnain SS (2009) Structure of a putative beta-phosphoglucomutase (TM1254) from Thermotoga maritima. Acta Cryst F 65:1218–1221
Volk J, Herrmann T, Wüthrich K (2008) Automated sequence-specific protein NMR assignment using the memetic algorithm MATCH. J Biomol NMR 41:127–138
Wahab AT, Serrano P, Geralt M, Wüthrich K (2011) NMR structure of the Bordetella Bronchiseptica protein NP_888769.1 establishes a new phage-related protein family PF13554. Prot Sci 20:1137–1144
Acknowledgments
This work was supported by the Joint Center for Structural Genomics (JCSG) through the NIH Protein Structure Initiative (PSI) grant U54 GM094586 from the National Institute of General Medical Sciences (www.nigms.nih.gov). Kurt Wüthrich is the Cecil H. and Ida M. Green Professor of Structural Biology at The Scripps Research Institute. We thank Dr. Ian A. Wilson for a careful reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jaudzems, K., Pedrini, B., Geralt, M. et al. J-UNIO protocol used for NMR structure determination of the 206-residue protein NP_346487.1 from Streptococcus pneumoniae TIGR4 . J Biomol NMR 61, 65–72 (2015). https://doi.org/10.1007/s10858-014-9886-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-014-9886-3