
Abstract
Random coil chemical shifts are necessary for secondary chemical shift analysis, which is the main NMR method for identification of secondary structure in proteins. One of the largest challenges in the determination of random coil chemical shifts is accounting for the effect of neighboring residues. The contributions from the neighboring residues are typically removed by using neighbor correction factors determined based on each residue’s effect on glycine chemical shifts. Due to its unusual conformational freedom, glycine may be particularly unrepresentative for the remaining residue types. In this study, we use random coil peptides containing glutamine instead of glycine to determine the random coil chemical shifts and the neighbor correction factors. The resulting correction factors correlate to changes in the populations of the major wells in the Ramachandran plot, which demonstrates that changes in the conformational ensemble are an important source of neighbor effects in disordered proteins. Glutamine derived random coil chemical shifts and correction factors modestly improve our ability to predict 13C chemical shifts of intrinsically disordered proteins compared to existing datasets, and may thus improve the identification of small populations of transient structure in disordered proteins.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Secondary structure in proteins affects the chemical shifts of the nuclei in the peptide backbone, and can thus be identified by the deviations of the chemical shifts from those of a random coil. The difference between experimental chemical shifts and random coil chemical shifts is called the secondary chemical shift and has become the main method for identification of transient structure in disordered proteins. The backbone chemical shifts of a protein can be measured accurately, but the random coil chemical shifts can only be approximated using a number of assumptions. Accordingly, the main source of uncertainty in secondary chemical shifts is not the chemical shift measurement itself, but the reference values used to represent the random coil state. Recently, the interest in random coil chemical shifts has been renewed (De Simone et al. 2009; Tamiola et al. 2010; Kjaergaard et al. 2011), due to the importance of secondary chemical shift analysis of intrinsically disordered proteins (Dyson and Wright 2002; Eliezer 2007).
Random coil chemical shifts have typically been determined in two ways: Either using a series of small disordered peptides (Kjaergaard et al. 2011; Wishart et al. 1995a; Schwarzinger et al. 2000; Richarz and Wuthrich 1978; Bundi and Wuthrich 1979; Jimenez et al. 1986; Braun et al. 1994; Thanabal et al. 1994; Merutka et al. 1995; Plaxco et al. 1997; Bienkiewicz and Lumb 1999) or using a database of protein NMR chemical shifts assignments (De Simone et al. 2009; Tamiola et al. 2010; Zhang et al. 2003; Wishart et al. 1991; Peti et al. 2001). Both methods have advantages: In peptide studies parameters such as temperature, pH and co-solvents can be tightly controlled, whereas databases use data from a large number of proteins and are thus less likely to contain specific artifacts from the choice of peptide series. The random coil chemical shifts depend on the sequence of the neighboring residues, which complicates the process of determining accurate random coil chemical shifts. A serine in its random coil state thus has different chemical shifts depending on whether it is preceded by for example an alanine or an aspartate residue. Schwarzinger et al. proposed an elegant way to correct for the effect of the neighboring residues based on chemical shift measurements of a peptide series with the sequence Ac-GGXGG-NH2 (Schwarzinger et al. 2001). The chemical shifts of the glycine residues are used to calculate the neighbor correction factors of the central residue, which is then added to the random coil chemical shifts. The correction factors determined for glycine are subsequently used for all residue types assuming that they are affected equally by their neighbors. The assumption that glycine correction factors are representative for all residue types can be tested using data from two peptide series with the sequences Ac-GGX(A/P)GG-NH2 (Wishart et al. 1995a). These peptide series contain all residue types preceding either an alanine or a proline residue and thus allow determination of the proline correction factor (Wishart et al. 1995a). The Cα chemical shifts of the X residue differ by approximately 2 ppm between these two peptide series, suggesting a sequence correction factor of approximately this magnitude. One notable exception is glycine, where the chemical shift difference is only 0.6 ppm. Thus, glycine is an outlier and the neighbor correction factor measured for this residue is clearly not representative for the other residue types. The residue preceding proline has a particularly large correction factor, in part because the residue is excluded from the α-helical region of the Ramachandran plot due to a steric clash between the Cδ of the proline and the Cβ of the preceding residue (MacArthur and Thornton 1991). This is not the case for glycine that does not have a Cβ, which explains why the correction factor is much smaller for glycine than the remaining residues.
The large correction factor of proline is in part due to the large change in the Ramachandran distribution of the preceding residue. This suggests that the smaller changes in the Ramachandran distribution caused by the remaining residues may also contribute to the neighbor correction factors. Conformational studies using a coil database assembled from high resolution protein structures show that the Ramachandran distribution varies significantly depending on the identity of the neighboring residues (Ting et al. 2010). The neighbor-dependence of the Ramachandran distribution of glycine differs significantly from all the other residues due to the absence of a side chain. For this reason, glycine is the least suited residue type for the determination of generally applicable neighbor correction factors in terms of conformational effects.
In the following, we present random coil chemical shifts and correction factors determined using a peptide series with the sequence Ac-QQXQQ-NH2. Glutamine was chosen based on a study of the neighbor-dependence of the Ramachandran distribution, where it was found to be the most representative residue type (Ting et al. 2010). How well glutamine represents the other residues depends on the similarity of the side chains. Like most of the amino acids, glutamine has only a single heavy γ-atom and accordingly glutamine is most representative for this group. Glutamine is moderately representative for alanine and the β-branched amino acids, whereas glycine and proline are conformationally abnormal and should be treated as special cases. In addition to their conformational representativeness, glutamine-rich peptides are likely to be structurally random as glutamine is associated with intrinsic disorder in proteins (Romero et al. 2001). Finally, glutamine is sufficiently hydrophilic to ensure the solubility of the peptides. The neighbor correction factors derived here from glutamine chemical shifts correlate to neighbor-dependent changes in the Ramachandran distribution. This shows that neighbor-dependent changes in the conformational ensemble contribute to the random coil chemical shifts. The glutamine derived random coil chemical shifts and correction factors improve the ability to predict the 13C chemical shifts of intrinsically disordered proteins modestly compared to existing datasets, and may thus improve the sensitivity of secondary chemical shift analysis for identifying small populations of transient structure in disordered proteins.
Materials and methods
Peptides with the sequence Ac-QQXQQ-NH2, where X was each of the 20 common amino acids, were purchased from Peptide 2.0 (Chantilly, VA). The peptides were at least 95% pure as estimated by reversed phase HPLC and their identities were confirmed by mass spectroscopy. NMR samples were prepared by dissolving 2–3 mg peptide in 500 μL 20 mM sodium phosphate buffer pH 6.5 containing 5% (v/v) D2O, 3 mM NaN3, and 1 mM DSS. pH was adjusted to 6.5 by addition of small quantities of HCl or NaOH. The Ac-QQCQQ-NH2 peptide sample contained 10 mM DTT to reduce the thiol group.
All NMR spectra were acquired on a Varian Unity 800 MHz spectrometer equipped with a cold probe. All NMR spectra were recorded at 5°C to minimize exchange of the amide protons with the solvent at high pH and to avoid overlap between the Hα and the water signals. Chemical shifts were referenced to internal DSS as described previously (Wishart et al. 1995b). The temperature controller was calibrated using a thermocouple mounted in an NMR tube and inserted into the spectrometer. For each sample the following spectra were acquired: 1H–15N HSQC (Kay et al. 1992), 1H–13C HSQC, a 1Hα–13C′ HSQC (Kjaergaard et al. 2011) and a 2D Watergate TOCSY experiment. NMR data were processed using NMRPipe (Delaglio et al. 1995) and analyzed in CCPNMR Analysis (Vranken et al. 2005). For the peptides containing aspartate, glutamate and histidine, a titration was performed in steps of approximately 0.5 pH units around the expected pKa value of the side chain. The chemical shifts were assumed to be the linear combination of the chemical shift of a fully protonated and deprotonated species and thus follow the equation:
δHA and δA represent the random coil chemical shifts of the fully protonated and fully deprotonated species respectively, and Ka is the acid dissociation constant of the side chain. The chemical shifts were fitted to (1) using IGOR PRO (WaveMetrics) where Ka was treated as a global fitting parameter.
The sequence correction factors were calculated by subtracting the chemical shifts of the glutamines of the Ac-QQQQQ-NH2 peptides from the Ac-QQXQQ-NH2 peptides, where A is the chemical shift difference for Q1, B for Q2 etc. The random coil chemical shifts of a residue can thus be calculated using the equation:
where B(i+1) is the B correction factor of the subsequent residue etc. and “a” is the temperature coefficient. For the analysis of the 14 intrinsically disordered proteins temperature coefficients from (Kjaergaard et al. 2011) were used.
Results and discussion
The random coil chemical shifts of Cα, Cβ, C′, N, Hα and HN were measured using the peptide series Ac-QQXQQ-NH2, where X is each of the 20 common amino acids. Previously, the effect of denaturant on random coil chemical shifts has caused concern (Tamiola et al. 2010), so denaturants were avoided. For each of the 20 peptides, the following four 2D experiments were recorded: 15N HSQC, 13C HSQC, Hα–C′ HSQC and a watergate TOCSY (Fig. 1). Sequential assignments were performed using the 1Hα–13C′ HSQC experiment as described previously (Kjaergaard et al. 2011) and expanded to HN and Hβ using the TOCSY experiment resulting in unambiguous determination of the random coil chemical shifts of all residue types (Table 1). Despite of the poor dispersion, the signals from the glutamines could be separated, which allowed determination of the sequence correction factors up to ±2 residues (Table 2). The random coil chemical shifts change with the protonation state of the side chains, and to allow the random coil chemical shifts to be used at a range of pH values, pH titrations were performed for aspartate, glutamate and histidine (Fig. 2). The pH dependence of the chemical shifts were fitted to (1), which describes the random coils chemical shifts by a linear combination of the chemical shift for the fully protonated and the fully deprotonated state as a function of pH (Table 3).

NMR spectra of the Ac-QQNQQ-NH2 peptide. Random coil chemical shifts were determined from a 15N HSQC (a), a Hα–C′ HSQC (b) and a 13C HSQC spectrum (c). Sequential connvities were established based on the Hα–C′ HSQC spectrum (red line) and extrapolated to HN and Hβ using a 2D TOCSY experiment

The pH dependence of the chemical shifts of Asp, Glu and His were determined in a titration series
The central hypothesis in this study is that changes in the Ramachandran distribution contribute to the sequence correction factors. To test this hypothesis, we compared the sequence correction factors from the glutamine peptides with the neighbor-dependent changes observed in the population of the α-helical well in the Ramachandran plot. We choose the α-helical population as this is the secondary structure element with the largest secondary chemical shifts (Zhang et al. 2003). Figure 3 shows that for three of the backbone nuclei: Cα, C′ and N the changes in the Ramachandran distribution correlate to the correction factors. The correlations vary from very strong with a Pearson coefficient of up to 0.91 in the case of Cα(i − 1) to no correlation. For Cα and C′ the changes are correlated, whereas for N the changes are anticorrelated, i.e. a high population in the α-helical well corresponds to an increase in the Cα and C′ chemical shift and a decrease in the N chemical shift. This is consistent with the signs of the secondary chemical shifts of α-helices for the different nuclei. For the (i − 1) correction factors, proline has a much larger correction factor than the other residue types due to the steric clash between the Cδ of the proline and the Cβ of the preceding residue, and accordingly this effect dominates the correlations. Accordingly, Pearson coefficients were calculated without proline. For both Cα and N, however, the correlation between the change in the α-helical well and the correction factors remained significant as suggested by Pearson coefficients of 0.76 and 0.66, whereas for C′ the correlation disappeared. For the C′(i − 1) correlation plot, there appears to be a cluster of outliers consisting among others of the aromatic residues (Fig. 3). The aromatic residues are likely to be outliers due to an additional chemical shift contribution from the aromatic ring in their side chains. The remaining residues in the cluster are aspartate, asparagine and serine, which all contain hydrogen bonding active side chains close to the backbone. This suggests that the carbonyl correlation coefficients may contain additional contributions from changes in the hydrogen bonding patterns due to the functional groups in the side chains. For the (i + 1) correction factors, the aromatic residues were again found to be outliers possibly due to the ring current effects (Fig. 3). When the aromatic residues were excluded, the correlation coefficients were similar to those for the (i − 1) correction factors.

Correlation between sequence correction factors and the neighbor dependent changes in the population of the α-helical well in the Ramachandran plot. The glutamine-derived sequence correction factors of Cα, C′ and N correlate moderately to strongly to the change in the population of the α-helical region determined in a database derived from high resolution protein structures (Ting et al. 2010). The correction factors of the residues preceding proline dominate the correlation and thus Pearson coefficients were calculate with and without proline. Aromatic residues are outliers in the (i + 1) plots, presumably due to ring current effects and Pearson coefficients improve in their absence. No convincing correlation was observed for the correction factors of Cβ, HN and Hα. The correlations demonstrate that the neighbor-dependence of the Ramachandran distribution contributes to the sequence correction factors and suggests that glutamine-derived correction factors can be used to correct for this effect
The correlation between neighbor correction factors and changes in the Ramachandran distribution suggests that the neighbor-dependence of the random coil chemical shifts is in part due to neighbor-dependent changes in the populations of the major wells in the Ramachandran plot. Several other mechanisms exist by which a neighboring residue can affect the random coil chemical shifts, e.g. changes in the hydrogen bonding pattern, ring current effects or directly by modifying the polarization of the covalent bond and thus modifying the nuclear shielding. The relative importance of these factors is likely to differ between nuclei as suggested by the variation in the correlation coefficients. For the Cα chemical shifts, it appears that changes in the Ramachandran distribution may be the dominant source of the neighbor-dependence.
The optimal approach for determining peptide derived neighbor correction factors is to determine the correction factor for each residue-type for each type of neighbor residue. This approach is unpractical as it requires a large number of peptides and instead it is common to choose one residue type to represent several other types of residues. While neighbor effects due to ring currents or polarization of the covalent bonds may be well represented by glycine correction factors, changes in the conformational distribution are almost certainly not. Based on coil databases, it has been suggested that glutamine has the most representative dependence on the neighbors in terms of the conformational distribution. The question is thus whether glutamine derived correction factors are representative for all residue types? In the correlation plots in Fig. 3, the correction factors are correlated to the changes in the coil library. The coil library used is not specific for glutamine, but is an average for all residue types. Accordingly, the correlations not only show that conformational changes contribute to the correction factors, but since the coil library is for all residue types, it also demonstrates that the glutamine correction factors can reasonably well be used to represent the conformational changes due to the neighbors for all residue types, possibly excluding the conformational abnormal residues glycine and proline.
To compare the secondary chemical shifts obtained using different random coil chemical shift datasets, we calculated the secondary chemical shifts using sequence corrected random coil chemical shifts from two recent database derived approaches (De Simone et al. 2009; Tamiola et al. 2010) and one based on glycine rich peptides under similar solvent conditions (Kjaergaard et al. 2011; Fig. 4). The experimental data was recorded on the activation domain of ACTR, which is intrinsically disordered but contains transiently formed α-helices (Demarest et al. 2004; Ebert et al. 2008; Kjaergaard et al. 2010). The secondary chemical shifts calculated using different random coil chemical shifts agree for most nuclei, but the random coil chemical shifts from the Ac-GGXGG-NH2 peptides are outliers for HN and C′ and the random coil chemical shifts reported by Tamiola et al. are outliers for Hα. The random coil values by Tamiola et al. have a large deviation for Cα of residue G1037 (Fig. 4). This glycine is followed by a proline residue and thus represents the special case discussed in the introduction. If the glutamine-derived correction factors reported here are used for glycine residues, it results in a similar large deviation. Instead, for glycine residues we recommend using the correction factors determined on the Ac-GGXGG-NH2 peptides (Kjaergaard et al. 2011), which removes the spike in the secondary chemical shifts. All datasets show a transiently formed α-helix in the unbound state of ACTR that corresponds to the first α-helix in the complex structure of this domain (1045–1057; Demarest et al. 2002). The two smaller α-helices in the complex (1061–1070 and 1073–1077) appear to be partially formed using the new dataset in agreement with the database random coil chemical shifts (De Simone et al. 2009; Tamiola et al. 2010) and a previous study using referencing to a urea denatured stated of the same protein (Kjaergaard et al. 2010). These transient α-helices are, however, not obvious from the secondary chemical shifts obtained using the random coil chemical shifts determined on glycine containing peptides. There is currently no golden standard that determine which of the random coil chemical shifts are more correct, but it appears that the secondary chemical obtained using database derived chemicals shifts are in better agreement with chemical shifts from the glutamine- than the glycine-containing peptides.

Secondary chemical shifts of the activation domain of ACTR at pH 6.5, 5°C calculated with four recent random coil datasets. The secondary chemical shifts were calculated using two database derived random coil dataset (De Simone et al. 2009) and (Tamiola et al. 2010) and two peptide based random coil values using a Ac-GGXGG-NH2 peptides series (Kjaergaard et al. 2011) or a Ac-QQXQQ-NH2 peptide series (present study). Ac-GGXGG-NH2 derived random coil values are outliers for C′ and HN, whereas the dataset by Tamiola et al. is an outlier for Hα
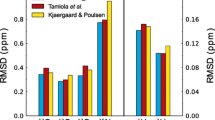
To systematically compare the performance of different random coil datasets, we calculated the secondary chemical shifts for a set of 14 intrinsically disordered proteins assembled previously (Tamiola et al. 2010) using several different random coil datasets (Fig. 5). As these proteins are highly disordered, the deviation from zero can be used as an estimate of the quality of the random coil dataset, where small deviations are used as an indicator of quality of the random coil dataset. For the two nuclei that are arguably the most important for secondary chemical shift analysis, Cα and C′, the present dataset has the smallest RMSDs in this analysis, although for C′ the dataset of Tamiola et al. performs equally well (Fig. 5). The improvement for these nuclei is consistent with the correlations between the correction factors and the changes in the Ramachandran distributions suggesting that conformational changes contribute significantly to the neighbor effect. For N, the present dataset is worse than several previously published datasets despite of the correlations in Fig. 3. The present peptide series was designed to represent the conformational effects of neighboring residues; however, it is likely that other factors are important for other nuclei. Unlike glycine, glutamine has an amide group in the side chain that may form hydrogen bonds to the backbone. The undesired contributions from hydrogen bonding interactions from the glutamine side chains may explain why the N chemical shifts perform worse than glycine-based and database-derived random coil chemical shifts. Thus for N, we recommend using the values reported by Tamiola et al. instead. For the remaining three nuclei, the new dataset did not improve the RMSD consistent with the lack of correlation between correction factors and the changes in the Ramachandran distribution. It is surprising that the Cβ RMSD does not improve relative to the glycine derived random coil chemical shifts (Kjaergaard et al. 2011) since they do not include sequence correction factors for this nucleus.

Comparison of the predictive accuracy of random coil datasets for intrinsically disordered proteins. The chemical shift were predicted using each dataset and compared to experimental values for a test set consisting of 14 intrinsically disordered proteins compiled by Tamiola et al. (2010). The new dataset modestly improves the predictive accuracy of the two most important nuclei Cα and C′. For the remaining nuclei, the performance is equal to or in the case of N worse than existing datasets
In summary, we have demonstrated that sequence correction factors determined on random coil peptides correlate to neighbor-dependent changes in the Ramachandran distribution for the nuclei Cα, C′ and N. This demonstrates that changes in the conformational ensemble caused by the neighboring residues contribute to the neighbor-dependence of the random coil chemical shifts. The prediction of random coil chemical shifts of the important C′ and Cα nuclei was modestly improved by using a random coil peptide series designed to be representative for conformational changes due to the neighbors. The application of this dataset will lead to more precise determinations of transient secondary structure in disordered proteins. Furthermore, this work demonstrates that random coil chemical shifts can be improved by understanding the physical factors underlying the neighbor-dependence, rather than simply trying to remove it. Ultimately, correction factors are residue specific as suggested by Tamiola et al., and this study suggests that in future random coil datasets parameters such as the neighbor-dependent Ramachandran distribution may contribute to the sequence correction factors.
References
Bienkiewicz EA, Lumb KJ (1999) Random-coil chemical shifts of phosphorylated amino acids. J Biomol NMR 15:203–206
Braun D, Wider G, Wuthrich K (1994) Sequence-corrected N-15 random coil chemical-shifts. J Am Chem Soc 116:8466–8469
Bundi A, Wuthrich K (1979) H-1-Nmr parameters of the common amino-acid residues measured in aqueous-solutions of the linear Tetrapeptides H-Gly-Gly-X-L-Ala-Oh. Biopolymers 18:285–297
De Simone A, Cavalli A, Hsu ST, Vranken W, Vendruscolo M (2009) Accurate random coil chemical shifts from an analysis of loop regions in native states of proteins. J Am Chem Soc 131:16332–16333
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) Nmrpipe—a multidimensional spectral processing system based on Unix pipes. J Biomol NMR 6:277–293
Demarest SJ, Martinez-Yamout M, Chung J, Chen H, Xu W, Dyson HJ, Evans RM, Wright PE (2002) Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature 415:549–553
Demarest SJ, Deechongkit S, Dyson HJ, Evans RM, Wright PE (2004) Packing, specificity, and mutability at the binding interface between the p160 coactivator and CREB-binding protein. Protein Sci 13:203–210
Dyson HJ, Wright PE (2002) Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. Adv Protein Chem 62:311–340
Ebert MO, Bae SH, Dyson HJ, Wright PE (2008) NMR relaxation study of the complex formed between CBP and the activation domain of the nuclear hormone receptor coactivator ACTR. Biochemistry 47:1299–1308
Eliezer D (2007) Characterizing residual structure in disordered protein States using nuclear magnetic resonance. Methods Mol Biol 350:49–67
Jimenez MA, Nieto JL, Rico M, Santoro J, Herranz J, Bermejo FJ (1986) A study of the Nh Nmr signals of Gly-Gly-X-Ala Tetrapeptides in H2o at low-temperature. J Mol Struct 143:435–438
Kay LE, Keifer P, Saarinen T (1992) Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc 114:10663–10665
Kjaergaard M, Norholm AB, Hendus-Altenburger R, Pedersen SF, Poulsen FM, Kragelund BB (2010) Temperature-dependent structural changes in intrinsically disordered proteins: formation of alpha-helices or loss of polyproline II? Protein Sci 19:1555–1564
Kjaergaard M, Brander S, Poulsen FM (2011) Random coil chemical shift for intrinsically disordered proteins: effects of temperature and pH. J Biomol NMR 49:139–149
MacArthur MW, Thornton JM (1991) Influence of proline residues on protein conformation. J Mol Biol 218:397–412
Merutka G, Dyson HJ, Wright PE (1995) ‘Random coil’ 1H chemical shifts obtained as a function of temperature and trifluoroethanol concentration for the peptide series GGXGG. J Biomol NMR 5:14–24
Peti W, Smith LJ, Redfield C, Schwalbe H (2001) Chemical shifts in denatured proteins: resonance assignments for denatured ubiquitin and comparisons with other denatured proteins. J Biomol NMR 19:153–165
Plaxco KW, Morton CJ, Grimshaw SB, Jones JA, Pitkeathly M, Campbell ID, Dobson CM (1997) The effects of guanidine hydrochloride on the ‘random coil’ conformations and NMR chemical shifts of the peptide series GGXGG. J Biomol NMR 10:221–230
Richarz R, Wuthrich K (1978) High-field 13C nuclear magnetic resonance studies at 90.5 MHz of the basic pancreatic trypsin inhibitor. Biochemistry 17:2263–2269
Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK (2001) Sequence complexity of disordered protein. Proteins 42:38–48
Schwarzinger S, Kroon GJ, Foss TR, Wright PE, Dyson HJ (2000) Random coil chemical shifts in acidic 8 M urea: implementation of random coil shift data in NMRView. J Biomol NMR 18:43–48
Schwarzinger S, Kroon GJ, Foss TR, Chung J, Wright PE, Dyson HJ (2001) Sequence-dependent correction of random coil NMR chemical shifts. J Am Chem Soc 123:2970–2978
Tamiola K, Acar Bi, Mulder FAA (2010) Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J Am Chem Soc 132:18000–18003
Thanabal V, Omecinsky DO, Reily MD, Cody WL (1994) The C-13 chemical-shifts of amino-acids in aqueous-solution containing organic-solvents—application to the secondary structure characterization of peptides in aqueous trifluoroethanol solution. J Biomol NMR 4:47–59
Ting D, Wang G, Shapovalov M, Mitra R, Jordan MI, Dunbrack RL Jr (2010) Neighbor-dependent Ramachandran probability distributions of amino acids developed from a hierarchical Dirichlet process model. PLoS Comput Biol 6:e1000763
Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas P, Ulrich EL, Markley JL, Ionides J, Laue ED (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59:687–696
Wishart DS, Sykes BD, Richards FM (1991) Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol 222:311–333
Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD (1995a) 1H, 13C and 15 N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J Biomol NMR 5:67–81
Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD (1995b) 1H, 13C and 15 N chemical shift referencing in biomolecular NMR. J Biomol NMR 6:135–140
Zhang H, Neal S, Wishart DS (2003) RefDB: a database of uniformly referenced protein chemical shifts. J Biomol NMR 25:173–195
Acknowledgments
This work was supported by The John and Birthe Meyer Foundation, the Carlsberg Foundation grant number 2008-01-0368 and The Danish Natural Research Council grant numbers 272-08-0500 (F.M.P). We thank Kamil Tamiola and Frans Mulder (University of Groningen) for sharing their data for comparison of the random coil data sets and Gitte Wolfsberg Haxholm and Birthe B. Kragelund for valuable discussions and critical comments to the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kjaergaard, M., Poulsen, F.M. Sequence correction of random coil chemical shifts: correlation between neighbor correction factors and changes in the Ramachandran distribution. J Biomol NMR 50, 157–165 (2011). https://doi.org/10.1007/s10858-011-9508-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-011-9508-2