
Abstract
Precipitates formed at an early stage (during the first 6 h) of the hydroxyapatite crystallization of a solution were studied. A nitrous synthesis was used (0.583M (NH4)2HPO4 and 0.35 M Ca(NO3)2·4H2O solutions at pH 11–12, 21°C, fast mixing, lyophilization of aliquots). Although XRD patterns indicated an amorphous calcium phosphate (ACP), IR spectra revealed apatite nanocrystals in the precipitates. Some amount of free calcium was found in the mother solution by mass spectrometrical analysis of the aliquots. This amount considerably decreased as the synthesis proceeded, however, the decrease had a slight effect on the crystallinity of the precipitates. A new suggestion on the nature of delayed crystallization (under conditions as those in the present study) was proposed. The free calcium adsorbed by the nanoparticles from the solution formed a shell around a particle because the calcium diffusion into the bulk was poor at the low synthesis temperature. As such, the encapsulation delayed the crystallization of the nanoparticles. Evidence for this suggestion was given. New possibilities were proposed for preparation of bioactive materials of desired composition based on the structural and compositional peculiarities of the X-ray diffractionamorphous calcium phosphates.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Amorphous calcium phosphate (ACP) has been of great interest in the last few decades. ACP was revealed to be a precursor of hydroxyapatite (HA) crystallization in reactions of wet synthesis [1, 2]. Because ACP was found in living tissues, the processes of ACP formation and conversion to HA at an early stage of these reactions has been studied as those mimicking bone mineralization [3–7]. ACP has also gained much attention due to its applications. For example, as an effective absorber, a catalyst or packing agent [8], component in hydraulic bone cements [9], carrier of medicine [6, 9] and nucleic acid into living cells [10] or as a best third-generation biomaterial owing to its excellent bioactivity and high biodegradation [6, 11].
In all above cases, the compositional, structural and impurity states of ACP are of paramount importance. However, despite many studies, there is contradictory data for these characteristics. The composition of ACP, namely the Ca/P ratio, beside the classic value of 1.5 [2], was found to vary within 1.3–2.0 [8, 12, 13]. Employment of novel high resolution technique resulted in doubt of the existence of this substance in an amorphous state [14, 15]. Strong impurity effects on the properties of ACP have been known in earlier studies [16–19]. However, the origin and position of the impurities in ACP and modes of their effect on the ACP to HA conversion (particularly, for such important pieces as H2O and CO3 2−) are still the subject of much debate [20–24].
It was recently shown [11, 25] that the final product (a powder) was amorphous if, beside the appropriate key factors of the synthesis (concentration of reagents, pH value and temperature of the mother solution), a high addition rate and obligatory freeze-drying of the precipitates was employed. Considering this, some structural and impurity particularities of the ACP associated with the contradictory data above was studied.
2 Materials and methods
Chosen conditions of precipitation were similar to those under which the majority of the contradictory data on ACP was obtained. The nitrous method was employed [8] based on the reaction:
The variables of the synthesis were as follows: high concentrations of the parent solutions (95.6 g Ca(NO3)2·4H2O in 300 ml and 32.1 g (NH4)2HPO4 in 900 ml of freshly distilled water) and the Ca/P ratio in their mixture of 1.67, i.e. that in HA. Each of the solutions was adjusted to pH 12 by adding NH4OH in order to maintain high basic values even after the dropping of pH values during the precipitation; the synthesis temperature was 21°C. A moderate and a fast addition rates was used. In the first case, the phosphate solution was added to the nitrate solution through a measuring buret with a dropping cock which allowed to accurately measure the addition rate as 23 ml min−1. During the fast mixing, the phosphate solution was added through a tube of about 1 cm2 cross section during 1 min; the addition rate was about 900 ml min−1. Continuous stirring of the mixtures was employed in both cases.
Aliquots of the fast mixed mother solution were withdrawn immediately after visible formation of the precipitate in the reaction vessel and every hour for six hours after the beginning of the addition. Thereafter aliquots were removed at the end of one week and one month. The aliquots were immediately frozen at −23°C to stop the reaction. This preliminary freezing also resulted in the separation of the precipitate from the mother solution since during usual centrifugation, taking considerable time, the synthesis proceeded further. The preliminary freezed aliquots were slowly defrosted, the mother liquor was decanted, the rest of the water was extracted with blotting paper, and the precipitates (separated from liquid but still wet) were immediately freezed again for drying. Since in the separation process an icebox was used, the temperature of the samples never exceeded 1–2°C. The wet aliquots were freeze-dried at −23°C for around 3 weeks (the results were already reproducible in a week drying). Finally, dry friable powders were obtained. During storage in a closed desiccator at room temperature, the powders manifested no diffraction signs of crystallinity for at least a week.
The powders were studied by traditional methods of XRD (DRON-2, USSR, CuKα radiation), IR (Specord 75IR, Germany; KBr procedure), TG-DTA (Q-Derivatograph, Hungary) and thermo-desorptive mass-spectrometry (MSTA; a home-made vacuum apparatus similar to the one proposed earlier [26] and supplied by a MX-7304 mass-spectrometer, Sumy, Ukraine). The rate of rising temperature in the last two methods was the same, about 5°/min.
3 Results and discussion
Because the addition rate and the drying procedure were expected to affect the properties of the product, which was the result of an early stage of precipitation under other conditions maintained invariable, the role of these two factors were studied first.
3.1 Moderate addition rate
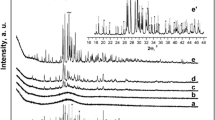
The dripping lasted 40 min, therefore, the first XRD pattern of a centrifugalized wet aliquot paste was taken 2 h after the mixing started. The pattern (Fig. 1) showed a typical diffusion maximum of an amorphous substance at about 28° of the diffraction angle 2θ. The first poor diffraction peaks appeared in a day of drying the paste in air. The intensities of the diffraction peaks increased and the intensity of the diffusion maximum (the square under the maximum) accordingly decreased with drying time. The peaks reached maximal intensities after 4 days drying the paste when it seemed dry. The XRD pattern of the seemingly dry paste was similar to that of a nanocrystalline HA powder. However, the most impressive fact was that the wet centrifugalized aliquots of the precipitate, simultaneously aged in the mother solution (the reaction vessel was closed to limit the CO2 absorbtion) without stirring at 21°C to equalize the other conditions in the solution and the drying paste, manifested a worse extent of crystallinity even after 2 month of aging (Fig. 2).

XRD patterns of a centrifugalized wet aliquot 2 h after beginning the synthesis and after drying in air at room temperature for 1–4 days

XRD patterns of the centrifugalized wet aliquots of the precipitate aged in the mother solution for different time
Two conclusions were made. The crystallization process detected by the XRD likely happened in the mother solution in a few weeks after the mixing. The synthesis temperature was known to greatly affect the ACP to HA conversion. For instance, it took about 25 h to reach full conversion at 25°C [27]. Consequently, it could be expected that at 21°C, which was a considerably lower temperature with regard to the process, the conversion would take a longer period. The second conclusion was much more important. The structural state of a conventionally dried aliquot may not adequately represent the stage of the precipitation process in the mother solution, even though structural alterations in a drying aliquot is an interesting subject of study by itself (e.g. novel information on structural evolution in the hydrated layer on the nanoparticles was obtained from such experiments [28]).
3.2 Fast addition
True ACP’s were prepared at relatively low temperatures (not exceeded 20°C) and stabilized for some time [11, 13, 25, 29]. However, one of the most stable ACP was obtained (without stabilization) at a temperature as low as 5°C coupled with a high addition rate of about 60 ml min−1 [11]. This implied that in the synthesis at 21°C in the present study a much higher addition rate was needed. It was increased to about 900 ml min−1 (Sect. 2), i.e. the addition rate was about 15 times higher as at 5°C.
XRD and IR patterns of the freeze-dried powders are in Figs. 3 and 4. The XRD pattern of the powder obtained after 2 min of precipitation manifested a diffusion maximum at about 30°2θ (Fig. 3a). Earlier, the maximum was attributed to the formation of an amorphous substance, namely of ACP [1, 2]. However, further examination of the lyophilized samples before their crystallization (in the sense of the appearance of diffraction peaks) revealed an interesting tendency: the 2θ value increased with the lifetime of the precipitate during 5 h of the synthesis, dropped to about 29°2θ for the 6 h precipitate and negligibly changed for more aged ACP’s (Table 1, Fig. 3b–d).

XRD patterns of the lyophilized aliquots of the precipitate in the a 2 min, b 5 h, c 6 h stages of synthesis; d in 1 month aging in the solution

IR spectra of the samples corresponding to those in Fig. 3 (a–c) and of the reference NH4NO3 (d)
The 2θ values were determined by applying the full profile analysis method [30] to the diffusion maxima. Therefore, though the 2θ changes were very small, it was suspected that the tendency might have reflected a real process happening in the precipitates. Moreover, shifting the diffusion maximum to the higher 2θ side was clearly seen in the XRD patterns in a recent work on the ACP problem [24] performed under conditions similar to those in the present study. However, the authors paid no attention to this fact.
The IR spectra showed for all samples strong bands of PO4 3− in ν4 (500–700 cm−1) and ν1, ν3 (900–1200 cm−1) domains with very poor resolution of the possible peaks (Fig. 4a–c). Nevertheless, using enlarged fragments of the spectra, two very weak peaks at 1020 and 1040 cm−1 with poor and very poor shoulders at 755, 965, 1005, 1040, 1055 and 1070 cm−1 in ν1, ν4 regions and two very weak peaks at 553 and 560 cm−1 were detected together with very poor shoulders at 520, 545, 590 and 600 cm−1 in ν4 domain in the spectrum of the aliquot of the 6 h aged precipitate (the 6 h precipitate). The number of these peaks and shoulders increased with the lifetime of the precipitate, particularly, in the spectrum of 6 h precipitate compared to that of 5 h precipitate. This showed the PO4 3− ions to be in some short-ordered environment because a true ACP was known to give featureless bands in these regions [31, 32]. Hence, according to the IR-study, the precipitates were mainly amorphous, however, containing nanocrystalline calcium phosphate areas growing with the precipitation time, especially after the 5–6th h stage of the process.
Additional valuable information was derived from MSTA data. The most intense peaks in the spectra of the lyophilized samples was recorded at mass numbers, M’s, of 17, 18 (H2O), 30 (NO), 32 (O2), 16 (NH2 +), 2 (H2), 28 (N2), 46 (NO2), and 44 (CO2, N2O). This set of mass numbers was found in each spectrum of the samples during 2 min to 6 h lifetime of the ACP. The origin of the peaks and alteration in their intensities during heating were recently clarified with a similar sample in vacuo [33]. In particular, the peak at 17 M was considerably higher in intensity than expected for OH− ions of the evolved water, especially at 150°C. Beside the water, this mass number indicated the liberation of NH3 + ions, particularly since the mass numbers of 16 (NH2 +) and 15 (NH+) also showed clear maxima at the same temperature; consequently, ammonia release was also detected at 150°C. As is known, nitrous oxides, NOx, form as a result of thermal destruction of NO3 − ions [34]. However, the NOx might have resulted from the destruction of NO3 − groups of two salts present in a lyophilized sample, i.e. of Ca(NO3)2 and NH4NO3. The first of these salts was used as a parent reagent, the second formed as the by-product in the synthesis reaction (Sect. 2). To solve the dilemma, the MSTA of both salts was conducted. The thermal vacuum decomposition of an NH4NO3 sample (a reagent grade referent substance was taken) occurred at 250–500°C with a maximum at about 440°C. The main gases released were NO, H2O, NH3, N2, and N2O. The decomposition of the Ca(NO3)2·4H2O (used as a parent reagent) happened in a higher temperature interval of 450–650°C with a maximum at 520°C. It was also found that NO2 (46 M) released in both cases and appeared as a single peak well separated in the temperature scale: at 300°C of NH4NO3 and at about 500°C of Ca(NO3)2·4H2O (Fig. 5a, b). This finding gave a very convenient way of mass-spectometrical detection of the two salts in the lyophilized aliquots.

Mass-spectra of NO2 (M = 46) released during the thermal decomposition of reference NH4NO3 (a) and Ca(NO3)2 (b) and of aliquots at different aging time of the precipitate: c 2 min, d 1 h, e 4 h
As the precipitation proceeded, the intensity ratio of the two peaks at 46 M’s changed. The 46 M intensity of Ca(NO3)2 was higher than that of NH4NO3 in the beginning of the precipitation, and the ratio changed in the opposite direction with time (Fig. 5c–e). These changes were consistent with the alteration in the 30 M intensity of Ca(NO3)2 during the first 6 h of the precipitation (Fig. 6). This was also seen in the corresponding IR spectra. Considering the MSTA data, it became clear that the intensity of the 1385 cm−1 absorbtion in the IR spectra was a result of superimposition of the NO3 − bands of both salts present in the samples. However, this absorbtion represented more the NO3 − ions of the Ca(NO3)2 in the sample at the beginning of the precipitation, and that of NH4NO3 as the reaction proceeded, and the amount of the by-product increased. The last was convincingly seen from an increase in the intensity of the NH4 + band at about 3160 cm−1 with time (Fig. 4; for convenience, the spectrum of the reference NH4NO3 used in the MSTA is also shown, d). Hence, some Ca2+ ions remained in the mother solution immediately after the precipitate was formed. These ions partly reacted with NO3 − groups and yielded calcium nitrate in the lyophilized aliquots. The Ca2+ amount became less with the duration of the synthesis. The decrease was not gradual. An abrupt drop in the amount of the Ca(NO3)2 in the samples, resulted from a drop of the Ca2+ concentration in the solution since the ions had reacted, was clearly seen in the 5–6 h period of the process. The amount of reacting Ca2+ ions in the solution further changed slightly: the 30 M intensity only decreased by about 5% to 1 month aging (Fig. 6)––including that for the crystallization of the precipitate detected by the XRD (appearance of the first weak reflections 3 weeks after the reaction was initiated).

NO (M = 30) released from the aliquots during the early stage of precipitation
Heating the freeze-dried aliquots in air up to 1000°C resulted in formation of an HA apatite structure and some traces of CaO in the samples for all length of the precipitation time (Fig. 7). However, the most surprising was the results of heating the similar samples in vacuo (10−4 Pa). Actually, the XRD patterns were taken from the samples subjected to the MSTA. Except the 2 min sample, all others manifested a single HA phase. Additionally, the longer the lifetime of the precipitate the higher the crystallinity of the phase (Fig. 8). This was a very unexpected result, since it seemed that in the HA crystallization the water molecules of the samples were involved (most likely, of those entrapped into the bulk of the precipitate) which transformed into OH− ions during heating to a high temperature (1000°C).

The phase composition in the aliquots of the precipitate at different lifetimes heated to 1000°C in air

XRD patterns of the aliquots (similar to those heated in air, Fig. 7) after mass-spectrometrical analysis
3.3 On the mode of crystallization
Temperature is usually considered the decisive factor in the ACP formation and its conversion to HA under conditions used in this study [19, 24, 27]. However, the temperature turned out to be too high to obtain a completely amorphous phase. IR spectra revealed a nanocrystalline or, most likely, a biphasic mixture of a nanocrystalline and an amorphous substance (Fig. 4). During the first 6 h of the synthesis, though very slowly, the nanocrystalline state progressed. This was supported by the shift of the diffusion maximum which likely was caused by the superimposition of the ACP diffusion maximum at about 29°2θ and the broadened principal reflexes of HA at about 32°2θ increasing in intensities with the precipitation time, and the slight increase in splitting of the PO4 3− absorption bands (Table 1; Figs. 3, 4). The developing crystallinity was associated with the formation of crystal planes and, consequently, with an increase in specific surface area [35]. The drop of calcium nitrate content in the samples (Fig. 6) could be seen as resulting from the adsorption of the free Ca2+ ions onto the progressively developing crystalline planes in the nanoparticles in the solution. However, on the other hand, the temperature was too low for the adsorbed Ca2+ ions to diffuse from the surface into the bulk of a nanoparticle. As a result, the nanoparticles became encapsulated by a shell of calcium; the shell, likely, was covered by a hydrated layer [28]. While drying, the hydrated layer was being destroyed, and the calcium pieces of the shell were reacting with NO3 − groups. This resulted in dried nanoparticles, covered by Ca(NO3)2·nH2O molecules.
Considering the hypothetical picture proposed above, all the results of this study could be explained. First, the calcium ions of the shell negligibly diffused into the bulk and also hampered the penetration of OH− ions from the solution into a nanoparticle. Therefore, they retarded the development of crystallization both in the period of the abrupt drop in the Ca2+ concentration in the solution and during further aging of the precipitate for a long time. Second, heating the freeze-dried samples in vacuo resulted in well crystallized HA’s because the appropriate calcium amount was present in the sample (it consisted of the parts in the core and in the surface shell; the given Ca/P ratio in the solution was 1.67). Also, the OH− ions formed from the entrapped water. However, heating similar samples in air yielded poor crystallized HA’s, likely, calcium-deficient HA’s, because a part of the calcium adsorbed on the particles was oxidized into CaO (a CaO layer was recently observed on the surface of the nanoparticles by HRTEM [25]). Third, a small amount of Ca(NO3)2 was often detected by the MSTA in a fully nanocrystalline sample (both lyophilized or dried in air after conventional repeated centrifugation and washing). IR spectra of such samples heated to temperatures higher than that of the complete decomposition of Ca(NO3)2 (680°C), always manifested the 1385 cm−1 band of NO3 − groups. Probably, these groups originated from Ca(NO3)2 molecules formed by the surface adsorbed calcium and were incorporated into the defective lattice during the heating to high temperatures.
3.4 Some practical consequences
Thus, the amount of free Ca2+ was highest in the mother solution immediately after mixing, which after gradually decreased until the abrupt drop. From Fig. 6, considering that the curve represented alteration in this amount, it was found that the free Ca2+ concentration in the solution only decreased by about 2.5% in the first hour of the precipitation (this was the part of the entire decrease in intensity for 6 h). This result was a starting point of a few simple preparation routes of calcium phosphate materials with desired Ca/P ratios based on the initial nanoprecipitates:
-
(a)
The principal features of the first route were as those described above. Fast mixing and immediate lyophilization of the precipitate followed by heating in vacuo to 1000°C. The route repeatedly resulted in a well crystallized HA (Fig. 8) having the Ca/P ratio close to 1.67.
-
(b)
Fast mixing followed by centrifugation and drying the precipitate in air at RT for 3 days. Heating the dried powder in air to 1000°C resulted in a biphasic HA/β-TCP material (Fig. 9a). The Ca/P ratio in the initial powder derived from the phase composition of the material [36] gave a value of 1.59. Hence, the centrifugation removed from the precipitate of \( {\frac{1.67 - 1.59}{1.67}} \approx 4.8\% \) of calcium.
Fig. 9 
XRD patterns of a an aliquot subjected to the usual centrifugation-washing-drying procedure after heating to 1000°C (a HA/β-TCP mixture) and b an aliquot centrifugalized and dried in air (for 3 days) after heating to 820°C (β-TCP)
-
(c)
The main steps in the third route were also outlined above. Mixing by dripping for 40 min, 4-time centrifugation and washing, 3-day drying of the precipitate in air at RT followed by heating in air to 820°C. This yielded a pure well crystallized β-TCP (Fig. 9b). Removing the by-product of the reaction (NH4NO3) via this conventional route resulted in a loss of \( {\frac{1.67 - 1.5}{1.67}} \approx 10\% \) of calcium. This was expected because the purification procedure was much intensive than that in the case of only centrifugation.

The results of the three preparation routes also evidenced the above suggestions on the mode of precipitation under conditions of the present study. Additionally, they presented the ACP at the early stage of synthesis (i.e. containing a small amount of nanocrystalline substance) as a very versatile and promising initial substance for the preparation of a wide range of bioactive calcium phosphate materials of the desired composition.
4 Conclusions
Precipitates at the early stage (during the first 6 h) of the reaction of 0.583 M (NH4)2HPO4 and 0.35 M Ca(NO3)2 solutions having a Ca/P ratio of 1.67 and high pH value of 11–12, at a relatively low temperature of 21°C and high addition rate of 900 ml min−1 were mixtures of an ACP and a nanocrystalline apatitic phase despite the XRD examination of the precipitates only detected an amorphous substance.
Some amount of free calcium was found in the mother solution immediately after mixing the reagents. The amount first gradually and then (in 5 h after the mixing) abruptly decreased. However, the decrease slightly affected the crystallinity of the precipitates.
The nascent nanoparticles adsorbed the free calcium from the solution. The adsorbed calcium formed a shell around the nanoparticles and isolated them from the exchange reactions in the solution. This resulted in a considerable delay (of a few weeks) of the crystallization of the encapsulated nanoparticles in the solution.
It was also found that depending on the initial state of a dried precipitate (powder) its thermal treatment yielded different products. The powder containing all the calcium designed for the reaction (it resulted from a lyophilized aliquot) transformed into HA after heating it to 1000°C both in air and in vacuo (the vacuum treatment revealed the entrapped water to be enough for the HA crystallization in the powder). During the conventional removing of the by-product of the reaction (NH4NO3), some calcium was also extracted from the precipitate. The powder of a calcium lost precipitate, upon a high temperature treatment, resulted in a biphasic HA/β-TCP product (if the precipitate was centrifugalized and freeze-dried) or in β-TCP (if the precipitate was subjected to the traditional centrifugation–washing–drying procedure). These findings are initial data for the development of new processing routes of novel bioactive materials using early-stage nanoprecipitates.
References
Bachra BN, Trauts OR, Simon SL. Precipitation of calcium carbonates, phosphates. I. Spontaneous precipitation of calcium carbonates, phosphates under physiological conditions. Arch Biochem Biophys. 1963;103:124–38.
Eanes ED, Gillessen IH, Posner AS. Intermediate states in the precipitation of hydroxyapatite. Nature. 1965;208:365–7.
Termine JD, Posner AS. Infrared analysis of rat bone: age dependency of amorphous and crystalline mineral fractions. Science. 1966;153:1523–5.
Termine JD, Posner AS. Amorphous/crystalline interrelationships in bone mineral. Calcif Tissue Res. 1967;1:8–23.
Posner AS, Betts F. Synthetic amorphous calcium phosphate and its relation to bone mineral structure. Bone Miner Struct. 1975;8:273–81.
Ratner BD. Biomaterials science: an introduction to materials in medicine. Elsevier Academic Press; 2004.
Epple M, Baeuerlein E, editors. Biomineralisation: medical and clinical aspects. Weinheim: Wiley-VCH; 2007.
Narasaraju TSB, Phebe DE. Some physico-chemical aspects of hydroxylapatite. J Mater Sci. 1996;31:1–21.
Dorozhkin SV. Calcium orthophosphate cements for biomedical application. J Mater Sci. 2008;43:3028–57.
Sokolova V, Kovtun A, Prymak O, Meyer-Zaika W, Kubareva EA, Romanova EA, et al. Functionalisation of calcium phosphate nanoparticles by oligonucleotides and their application for gene silencing. J Mater Chem. 2007;17:721–7.
Tadic D, Peters F, Epple M. Continuous synthesis of amorphous carbonated apatite. Biomaterials. 2002;23:2553–9.
Termine JD, Eanes ED. Comparative chemistry of amorphous and apatitic calcium phosphate preparations. Calcif Tissue Res. 1972;10:171–97.
Li Y, Weng W. In vitro synthesis and characterization of amorphous calcium phosphates with various Ca/P atomic ratios. J Mater Sci Mater Med. 2007;18:2303–8.
Suvorova EI, Buffat PA. Electron diffraction and high resolution transmission electron microscopy in the characterization of calcium phosphate precipitation from aqueous solutions under biomineralization conditions. Eur Cells Mater. 2001;1:27–42.
Suvorova EI, Buffat P-A. Size effect in X-ray and electron diffraction patterns from hydroxyapatite particles. Crystallogr Rep. 2001;46:722–9.
Termine JD, Posner AS. Calcium phosphate formation in vitro. I. Factors affecting initial phase separation. Arch Biochem Biophys. 1970;140:307–17.
Termine JD, Peckauskas RA, Posner AS. Calcium phosphate formation in vitro. II. Effects of environment on amorphous-crystalline transformation. Arch. Arch Biochem Biophys. 1970;140:318–25.
Greenfield DJ, Eanes ED. Formation chemistry of amorphous calcium phosphates prepared from carbonate containing solutions. Calcif Tissue Res. 1972;9:152–62.
Boskey AL, Posner AS. Conversion of amorphous calcium phosphate to microcrystalline hydroxyapatite. A pH-dependent, solution-mediated, solid-state conversion. J Phys Chem. 1973;77:2313–7.
Greenfield DJ, Termine JD, Eanes ED. A chemical study of apatites prepared by hydrolysis of amorphous calcium phosphates in carbonate-containing aqueous solutions. Calcif Tissue Res. 1974;14:131–8.
Blumenthal NC, Betts F, Posner AS. Stabilization of amorphous calcium phosphate by Mg and ATP. Calcif Tissue Res. 1977;23:245–50.
Brečević L, Hlady V, Füredi-Milhofer H. Influence of gelatin on the precipitation of amorphous calcium phosphate. Colloids Surf. 1987;28:301–13.
Abbona F, Baronnet A. A XRD and TEM study on the transformation of amorphous calcium phosphate in the presence of magnesium. J Cryst Growth. 1996;165:98–105.
Kim S, Ryu H–S, Shin H, Jung HS, Hong KS. Direct observation of hydroxyapatite nucleation from amorphous phase in a stoichiometric calcium/phosphate aqueous solution. Chem Lett. 2004;33:1292–3.
Urch H, Vallet-Regi M, Ruiz L, Gonzalez-Calbet JM, Epple M. Calcium phosphate nanoparticles with adjustable dispersability and crystallinity. J Mater Chem. 2009;19:2166-71.
Anderson CW, Beebe RA, Kittelberger JS. Programmed temperature dehydration studies of octacalcium phosphate. J Phys Chem. 1974;78:1631–5.
Liu C, Huang Y, Shen W, Cui J. Kinetics of hydroxyapatite precipitation at pH 10 to 11. Biomaterials. 2001;22:301–6.
Eichert D, Sfihi H, Banu M, Cazalbou S, Combes C, Rey C. Surface structure of nanocrystalline apatites for bioceramics and coatings. CIMTEC 2002 proceedings, 10th inter. ceramics congress and 3rd forum of new materials, Florence, Italy, Juillet 14–18, 2002.
Harries JE, Hukins DWL, Holt C, Hasnain SS. Conversion of amorphous calcium phosphate into hydroxyapatite investigated by EXAFS spectroscopy. J Cryst Growth. 1987;84:563–70.
Rietveld HM. Line profiles of neutron powder-diffraction peaks for structure refinement. Acta Crystallographica. 1967;22:151–2.
Gadaleta SJ, Paschalis EP, Betts F, Mendelson R, Boskey AL. Fourier transform infrared spectroscopy of the solution-mediated conversion of amorphous calcium phosphate to hydroxyapatite: new correlations between X-ray diffraction and infrared data. Calcif Tissue Int. 1996;58:9–16.
Termine JD, Posner AS. Infra-red determination of the percentage of crystallinity in apatitic calcium phosphates. Nature. 1966;211:268–70.
Zyman Z, Epple M, Rokmistrov D, Glushko V. On impurities and the internal structure in precipitates occurring during the precipitation of nanocrystalline calcium phosphate. Mat-wiss u Werkstofftech. 2009;40:297–301.
The Chemical Encyclopedic Vocabulary. In: Knunyanc IL editor. Soviet Enciclopedia Press, Moscow, 1983.
Ruys AJ, Sorrell CC, Brandwood A, Milthorpe BK. Hydroxyapatite sintering characteristics: correlation with powder morphology by high-resolution microscop. J Mater Sci Lett. 1995;14:744–7.
Ishikawa K, Ducheyne P, Radin S. Determination of the Ca/P ratio in calcium-deficient hydroxyapatite using X-ray diffraction analysis. J Mater Sci: Mater Med. 1993;4:165–8.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zyman, Z.Z., Rokhmistrov, D.V. & Glushko, V.I. Structural and compositional features of amorphous calcium phosphate at the early stage of precipitation. J Mater Sci: Mater Med 21, 123–130 (2010). https://doi.org/10.1007/s10856-009-3856-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10856-009-3856-4