
Abstract
In situ hydroxyapatite/apatite–wollastonite glass ceramic composite was fabricated by a three dimensional printing (3DP) technique and characterized. It was found that the as-fabricated mean green strength of the composite was 1.27 MPa which was sufficient for general handling. After varying sintering temperatures (1050–1300°C) and times (1–10 h), it was found that sintering at 1300°C for 3 h gave the greatest flexural modulus and strength, 34.10 GPa and 76.82 MPa respectively. This was associated with a decrease in porosity and increase in densification ability of the composite resulting from liquid phase sintering. Bioactivity tested by soaking in simulated body fluid (SBF) and In Vitro toxicity studies showed that 3DP hydroxyapatite/A–W glass ceramic composite was non-toxic and bioactive. A new calcium phosphate layer was observed on the surface of the composite after soaking in SBF for only 1 day while osteoblast cells were able to attach and attain normal morphology on the surface of the composite.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Due to its similarity in chemical composition to the main inorganic mineral constituent in bone, hydroxyapatite is osteoconductive and shows excellent biocompatibility. It has been used clinically on its own as bone implant in the form of powder, porous structure, or dense body or as a coating on other materials to enhance the bioactivity. Recently, hydroxyapatite has been directly produced by a new fabrication technology namely three dimensional printing (3DP) technique [1–5]. This is one type of freeform fabrication technology that offer the ability to readily produce complex-shaped and personalized implants suitable for individual patients. This is sometimes difficult using conventional material processing techniques [6, 7]. However, low mechanical properties and brittleness have usually been obtained from the direct fabrication approach. It was found that residual pores in the 3DP material were the main cause of such low mechanical properties. Although a porous structure is favorable for some applications i.e. scaffold, it might not be suitable for use in load bearing areas. Post-processing by infiltration has been generally employed as a means to increase mechanical properties of 3DP material [8–11]. For example, infiltration by acrylate polymer has been carried out successfully to increase the mechanical properties of 3DP hydroxyapatite [12]. However, acrylate is not bioactive so the bioactivity of the infiltrated material was reduced. Additional processing steps were also needed for such infiltration.
Several studies have reported the use of additional secondary phases as sintering aids to improve the densification and mechanical properties of synthetic hydroxyapatite fabricated by conventional ceramic pressing through liquid phase sintering [13–18]. The majority of sintering aids employed were glassy materials, for example phosphate based glass and silicate based glass. The resultant properties and behaviour varied from system to system.
The present work has studied possible improvement of mechanical properties of 3DP hydroxyapatite by employing the liquid phase sintering concept to produce in situ bioactive composite of apatite–wollastonite (A–W) glass ceramic/hydroxyapatite composite. A–W glass ceramic was selected as the reinforcing phase since it is bioactive and its mechanical properties are higher than other bioactive glasses and glass ceramics [19, 20]. To date, there has been only limited study of this composite system. Physical and mechanical properties of as-fabricated green and sintered composites were determined. Bioactivity after soaking in simulated body fluid and In Vitro cell toxicity of developed composite were also investigated.
2 Materials and methods
2.1 Materials
Hydroxyapatite powders (Taihei Chemical Industrial., Japan) and maltodextrin powders (Shandong Duqing, Inc., China) with mean particle size of 3–5 and 90–100 μm respectively were purchased. A–W glass ceramic powders were prepared as recommended by Kokubo et al. [19, 20]. This was done by melting a mixture of the nominal composition of MgO 4.6, CaO 44.9, SiO2 34.2, P2O5 16.3, CaF2 0.5 by weight ratio in a crucible at 1450°C for 2 h. The melt was then poured into a water bath and the resultant solid material ground to powder form. The mean particle size of prepared A–W powders was approximately 88 μm as determined by a mastersizer.
2.2 Specimen preparation
Slurry of hydroxyapatite and A–W glass ceramic mixture was prepared by mixing hydroxyapatite and A–W glass ceramic powders with maltodextrin and water using the ratio of hydroxyapatite: A–W glass ceramic and hydroxyapatite +A–W glass ceramic: maltodextrin at 1 and 4 w/w respectively. Darvan C (R.T. Vanderbilt Company, Inc., USA) was used as a dispersing agent. The slurry mixture was ball-milled overnight. It was then dried while stirring on a hotplate at 80°C. The solidified mixture was then crushed and ground by a high speed grinder. Green specimens (80 mm × 10 mm × 4 mm) were fabricated by a three dimensional printing machine (Z400, Z Corporation, USA) using prepared composite powders. A layer thickness of 0.1 mm was used. Water-based binder was used as a jetting media in all formulations. After building, all the specimens were left in the machine for 2 h before removal and left for drying on the table in the laboratory for 24 h. The specimens were then air blown to remove the unbound powders. In the case of sintered specimens, the green specimens were sintered in an electrical furnace at specified temperature and time in normal air atmosphere.
2.3 Particle size determination
Particle size of powders was determined by a mastersizer (Malvern Instruments, UK) using a helium–neon laser as a detector source. The sample was characterized by dry-feed technique using air as a media. Three repeated runs were carried out and the average values are reported.
2.4 Microstructure examination
Microstructures of particles and fabricated specimens were examined using a scanning electron microscope (JSM-6301F and JSM 5410, JEOL, Japan) at an accelerating voltage of 20 kV. All samples were gold or carbon sputtered prior to the observation.
2.5 Apparent porosity determination
Apparent porosity of sintered samples was determined by Archimedes principle according to ASTM C373. Dry weight (W1) was measured by a precision balance (Metler-Toledo AG 204, Switzerland). The specimen was then submerged in water and heated at 150°C for 5 h and soaked for another 24 h. Then, the weight of specimen suspended in water (W2) was measured. The specimen was taken out and dried by tissue paper to remove the excess water and immediately reweighed (W3). The porosity was calculated using the following equation:
2.6 X-ray diffraction
XRD characterization was carried out using a JEOL JDX 3530 X-ray diffractometer with Co K-alpha radiation in the range of 10–80° 2θ with step angle of 0.02°. JCPDS files were used to identify the peaks of main phases in the sample.
2.7 Mechanical testing
Flexural tests were performed on a universal testing machine (Model 55R4502, Instron Corporation, USA) equipped with a 10 kN load cell. The tests were carried out at 23°C and 50% RH. respectively. The tests were done using a three point bending method and a constant crosshead speed of 1.9 mm min−1 in the case of green specimens and 0.5 mm min−1 in the case of sintered specimens.
2.8 Bioactivity in SBF
Kokubo’s simulated body fluid (SBF) was prepared by dissolving reagent-grade NaCl, NaHCO3, KCl, K2HPO4 ∙ 3H2O, MgCl2 ∙ 6H2O, CaCl2, and Na2SO4 in distilled water, with the ion concentrations adjusted similarly to those in human blood plasma as shown in Table 1 [21]. The pH of the SBF solution was buffered at 7.4 with 1 mol/L of HCl and tris-(hydroxymethyl)-aminomethane [(CH2OH)3CNH2]. The specimens were immersed in separate containers each with 10 ml of SBF solution and incubated at 37°C. After soaking for 1, 2, 4, 16 and 32 days, the specimens were removed from the solution, rinsed with distilled water and oven-dried. No refreshment with new SBF was made during all soaking periods.
2.9 In vitro cell toxicity
3DP-HAAW composite which was sintered at 1300°C for 3 h was tested for toxicity by the direct contact method using human osteoblast cells (h-OBs). The sample was autoclaved at 121°C for 15 min prior to cell culture studies. Ten thousand cells per one milliliter of media (Fitton Jackson modified Biggers medium containing 10% FCS, glutamine 20 μl/ml, penicillin/streptomycin 10 μl/ml and 25 μl/ml Hepes, abbreviated to sFJm [22]) were added to each tissue culture dish containing samples, or to an empty tissue culture dish as a reagent control. Thermanox and Portex Control PVC were used as negative and positive control respectively. The incubation period was 6 days. After 6 days, the morphology of cells was then observed using inverted light microscopy after staining the cells with neutral red to observe to cell viability. To study cell characteristics on the surface of samples, the cultured samples were dried using a critical point dryer, gold sputtered and then observed by a scanning electron microscope (JSM-6301F, JEOL, Japan) at the accelerating voltage of 3 kV.
3 Results and discussion
3.1 Green specimens
Figure 1 shows the original morphology of the raw materials and fabricated 3DP hydroxyapatite/A–W glass ceramic composite (3DP-HAAW). As-purchased hydroxyapatite powders consist of agglomerates of needle-like crystals while A–W glass ceramic powders have irregular morphology caused by the milling process. After mixing with dextrin binder, the ground powders are irregular-shaped aggregates having average particle size of approximately 70 μm which is suitable for 3D-printing. When using these composite powders to fabricate specimens by 3DP machine, it was possible to print successfully without slippage between layers or part collapse. No shape distortion was noticed. The specimens were sufficiently strong for handling and passed through all post cleaning steps without breakage. The fabricated green 3DP specimen was found to be porous and composed of particles which were connected by binder. Table 2 shows green flexural modulus and strength of 3DP-HAAW specimens. Although the flexural properties seem not to be high, the achieved flexural strength of 1.27 MPa is greater than the previously suggested preferred level of at least about 1 MPa [23].

SEM micrographs of raw material, prepared composite powders and green 3DP fabricated composite
3.2 Sintered specimens
Figures 2 and 3 show XRD patterns of 3DP-HAAW composite after sintering at 1050–1300°C and for durations between 1 and 10 h at 1300°C. Hydroxyapatite, calcium phosphate silicate and tricalcium phosphate were the main phases detected depending on the sintering temperatures. The formation of calcium phosphate silicate and tricalcium phosphate in the sample results from the incorporation of silicate groups from A–W glass ceramics into hydroxyapatite [13, 17]. Figure 4 shows the shrinkage of specimens after sintering at 1050, 1200 and 1300°C for 3 h. It can be seen that shrinkage in all directions increase linearly with increasing sintering temperature while the shrinkage in thickness is slightly greater than the other directions. This is typical for 3DP material since the compaction is slightly lower in z direction. In practice, although the firing shrinkage could cause the final sintered structure to be smaller than the green structure, this shrinkage can be compensated during the computer design of implants prior to fabrication. Flexural properties of specimens after sintering at 1050, 1200 and 1300°C for 3 h are shown in Table 3. The specimens sintered at 1050°C were fragile and were easily broken during handling. Although this level of sintering temperature could yield reasonably strong hydroxyapatite or A–W glass ceramic specimens when prepared by conventional pressing techniques [24, 25], it proved to be not sufficiently high for hydroxyapatite/A–W glass ceramic prepared by 3D printing. In contrast to the pressing technique, the particles in green 3DP specimens are lightly packed and each particle is distantly connected by adhesive binder. The rearrangement of the powders during sintering is therefore limited and the degree of necking formation between particles is limited to the contact area that is primarily joined by binder as could be seen from the limited amount of sintering shrinkage. At 1200 and 1300°C, the sintered specimens were much stronger and could be flexurally tested. Flexural modulus and strength increased 5 and 2.6 times respectively when sintering temperature increased from 1200°C to 1300°C. This is obviously resulted from the increase in microstructure densification with increasing sintering temperature as also be seen by the increase in density and decrease in porosity. SEM micrographs in Fig. 5 clearly confirm the increase in densification of microstructure induced by increasing sintering temperature. At 1200°C, the morphology of composite consists of connected framework of A–W glass ceramic matrix with distributed hydroxyapatite particles inside and interconnected pores throughout the body. This microstructure is quite similar to the green sample indicating that the temperature is still not sufficiently high for mass transport by diffusion and coalescence of A–W glass ceramic. Hence, only short-range connections are created by liquefied A–W ceramic particles which are in contact. At 1300°C, the composite body appears denser with a reduced amount of isolated pores. This densification is due to diffusion effects and coalescence of liquefied A–W glass ceramic and formation of continuous matrix. The densification process of 3DP-HAAW composite is found to follow liquid phase sintering steps namely particle rearrangement, solution–reprecipitation and coalescence [26]. In the green state, both hydroxyapatite and A–W glass ceramic particles are bonded by dextrin binder. After sintering, the binder is burnt out while A–W glass ceramic starts to melt, diffuse and coalesce together leading to the shrinkage of the composite. This would also allow some of the hydroxyapatite particles which are surrounded by liquefied glass ceramic to come into contact. If the sintering temperature and duration is sufficient, a continuous matrix of glass ceramic develops leading to further reduction in porosity and increase in shrinkage.

XRD patterns of 3DP-HAAW composite after sintering at 1050, 1200 and 1300°C for 3 h

XRD patterns of 3DP-HAAW composite after sintering at 1300°C for 1, 3, 5 and 10 h

Influence of sintering temperature on dimensional shrinkage of 3DP-HAAW composite

Microstructure of 3DP-HAAW composite sintered at 1200 and 1300°C for 3 h
A sintering temperature of 1300°C was seen to achieve the lowest amount of porosity, hence; the sintering time was then varied at this temperature to see if there would be any further influences on the properties of composite. No influence of sintering time on phase formation was noticed. All 1300°C sintered samples show similar XRD patterns indicating the presence of calcium silicate and tricalcium phosphate, Fig. 3. Figure 6 shows that shrinkage of composites also increases with increasing sintering time. The shrinkage increases rapidly at short sintering time (<3 h), but levels off at longer sintering times (>3 h). Table 4 shows the influence of sintering time on mechanical and physical properties. One hour sintering results in the highest porosity and hence the lowest flexural modulus and strength. This sintering time is; thus, not sufficiently long for diffusion and coalescence of A–W glass ceramic as previously observed for low sintering temperature. The SEM micrograph in Fig. 7 also shows that 1 h sample contains large and elongated pores throughout the body. A 3 h sintering time gave the lowest porosity and hence the highest flexural properties and density. SEM study revealed that the shape of the pores tended to be more globular than for the 1 h sample. Longer sintering times of 5 and 10 h, do not further increase the flexural properties, but leads to the decrease in density and increase in porosity of samples. Little difference in microstructure is observed between samples sintered for 3–10 h except that the 5 and 10 h samples contain larger pores than 3 h sample. It is thought that too long sintering times cause the glass ceramic matrix to flow out of the sample leaving the pores inside the sample as could be seen from the re-increase in porosity of the samples. This is also confirmed by the observation that the 5 and 10 h samples were attached to the supporting substrate and needed force to detach them out in contrast to the 1 and 3 h samples.

Influence of sintering time at 1300°C on dimensional shrinkage of 3DP-HAAW composite

Microstructure of 3DP-HAAW composite sintered at 1300°C for 1, 3, 5 and 10 h
In comparison to pure hydroxyapatite which was fabricated by three dimensional printing technique [3–5], mechanical properties of 3DP-HAAW were observed to be much greater. This proves the effectiveness of the use of liquid phase sintering coupled with the 3D-printing technique. The highest values of modulus and strength achieved in this study are in the range of values of compact bone, 7–30 GPa and 50–150 MPa respectively [27, 28]. To demonstrate the applicability of developed composite for building personalized or customized implants for individual patients, part of a mandibular bone based on CT scan of the patient was reconstructed and three dimensionally printed using developed 3DP-HAAW composite, Fig. 8. It was observed that the implant could be fabricated and progressed through all processing steps successfully. The final size of the mandible was clearly much smaller than the fabricated green part due to the shrinkage from sintering. As discussed previously, compensation for shrinkage with reference to both sintering temperature and time must be made to all dimensions of a part so that the final dimensions are correct.

As-fabricated green and sintered 3DP-HAAW model of mandibular bone
3.3 Bioactivity and toxicity
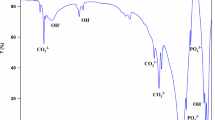
3DP-HAAW composite which was sintered at 1300°C for 3 h was selected for bioactivity and toxicity tests since this condition gave suitable flexural and physical properties. Figure 9 shows the surfaces of 3DP-HAAW composite before and after soaking in SBF for 1, 2, 4, 16 and 32 days. Prior to soaking, the morphology of composite consists of distributed calcium phosphate grains in the glass ceramic matrix. Ca, P and Si are main elements in the sample as determined by EDS. After soaking, it is observed that a newly formed layer is found on the glass ceramic matrix area which surrounds the grains after soaking for only one day. Increasing soaking duration increases the amount and the density of this new layer as it changes from porous and thin to a dense, thick layer. Furthermore, higher magnification reveals that nucleation and deposition occur on the surfaces of the individual grains as well. Figure 10 shows that the surfaces of grains are initially smooth before soaking. After soaking in SBF from day 1 to day 32, nano-sized crystals are formed on the surfaces. No significant difference in feature and amount are noted between different periods of soaking. EDS spectrum of composite after soaking for 32 days in Fig. 11 shows that the newly formed layer on glass ceramic matrix area is composed of only Ca and P. The composition is different from the matrix prior to soaking which contains Ca, P and Si. This confirms the formation of new calcium phosphate layer on the matrix area during soaking in SBF. In contrast, the EDS spectrum of the surface of grains still shows similar Ca, P and Si as composite before soaking. This implies that the deposition is thin and EDS could still detect Si element underneath. Since it was reported that resorption of A–W glass ceramic was higher than hydroxyapatite [29, 30], the release of Ca and P ions from glass ceramic would be greater and it would help to increase the nucleation and deposition rate of calcium phosphate on the glass ceramic area. Similar small and thin calcium phosphate layers on individual grains, as seen in this study, was observed previously for dense hydroxyapatite after soaking in SBF for 32 days and was attributed to the low solubility and low ion release [31]. Therefore, different deposition rates at different areas in the composite might be associated with different solubility in SBF.

Microstructure of 3DP-HAAW composite sintered at 1300°C for 3 h after soaking in SBF for various periods of time (×5,000 magnification)

Magnified surface features of hydroxyapatite grains in 3DP-HAAW composite sintered at 1300°C for 3 h after soaking in SBF for various periods of time (×10,000 magnification)

EDS spectra of a formed layer on A–W glass ceramic matrix and b surfaces of HA grain
Preliminary In vitro cell toxicity shows that 3DP-HAAW composite is non-toxic to osteoblast cells as the cells are neutral red stained indicating vital cells. Figure 12 shows the scanning electron microscope micrographs of human osteoblast cells cultured for 6 days on the surface of 3DP-HAAW composite. The cells spread, adhere and attain a normal morphology on surfaces of composite. The combination of bioactivity in SBF and in vitro cell toxicity data indicates that sintered 3DP-HAAW composite is bioactive and could potentially be used as bone replacement material.

SEM micrographs of human osteoblast cells on the surface of 3DP-HAAW composite (1300°C, 3 h) after 6 days incubation
4 Conclusions
Properties of 3DP hydroxyapatite/A–W glass ceramic composite were influenced by sintering temperatures and times. Sintering the composite at 1300°C for 3 h produced the greatest flexural modulus and strength and lowest porosity, 34.10 GPa, 76.82 MPa and 2.50% respectively. A new Ca–P layer was observed to form on the surface of composite after soaking in SBF for only 1 day. However, the deposition rate and amount of Ca–P layer formation were influenced by difference in solubility at different areas in the composite. Formation at intergranular glass ceramic regions was faster than at the grains due to the higher solubility in SBF. Osteoblast cells were observed to attach and attain normal morphology on the surface of composites. The advantages of this fabrication technique compared to others is that the implants can be customized and that there is virtually no limitation in the complexity of the shape that can be designed and fabricated.
References
J.L. Simon, T.D. Roy, J.R. Parsons, E.D. Rekow, V.P. Thompson, J. Kemnitzer, J.L. Ricci, J. Biomed. Mater. Res. 66A, 275 (2003). doi:10.1002/jbm.a.10569
T.D. Roy, J.L. Simon, J.L. Ricci, E.D. Rekow, V.P. Thompson, J.R. Parsons, J. Biomed. Mater. Res. 67A, 1228 (2003). doi:10.1002/jbm.a.20034
H. Seitz, W. Rieder, S. Irsen, B. Leukers, C. Tille, J. Biomed. Mater. Res. Part B Appl. Biomater. 74B, 782 (2005). doi:10.1002/jbm.b.30291
R. Chumnanklang, T. Panyathammaporn, K. Sitthiseripratip, J. Suwanprateeb, Mater. Sci. Eng. C 27, 914 (2007). doi:10.1016/j.msec.2006.11.004
J. Will, R. Melcher, C. Treul, N. Travitzky, U. Kneser, E. Polykandriotis, R. Horch, P. Greil, J. Mater. Sci. Mater. Med. 19, 2781 (2008). doi:10.1007/s10856-007-3346-5
K.F. Leong, C.M. Cheah, C.K. Chua, Biomaterials 24(13), 2363 (2003). doi:10.1016/S0142-9612(03)00030-9
T. Boland, A. Ovsianikov, B.N. Chickov, A. Doraiswamy, R.J. Narayan, W.Y. Yeong, K.F. Leong, C.K. Chua, Adv. Mater. Process 165(4), 51–53 (2007)
B.D. Kernan, E.M. Sachs, S.M. Allen, A. Lorenz, C. Sachs, L. Raffenbeul, A. Pettavino, J. Metallur. Mater. Transac. A 36(10), 2815 (2005). doi:10.1007/s11661-005-0278-x
J. Suwanprateeb, Polym. Int. 55(1), 57 (2006). doi:10.1002/pi.1918
J. Suwanprateeb, J. Mater. Sci. Mater. Med. 17(12), 1383 (2006). doi:10.1007/s10856-006-0614-8
J. Suwanprateeb, Rapid Prototyping J. 13(1), 48 (2007). doi:10.1108/13552540710719217
J. Suwanprateeb, R. Sanngam, W. Suwanpreuk, J. Mater. Sci. Mater. Med. 19(7), 2637 (2008). doi:10.1007/s10856-007-3362-5
S. Padilla, J. Roman, S. Sanchez-Salcedo, M. Vallet-Regi, Acta Biomater. 2, 331 (2006). doi:10.1016/j.actbio.2006.01.006
Y.E. Greish, P.W. Brown, J. Biomed. Mater. Res. 52, 687 (2000). doi:10.1002/1097-4636(20001215)52:4<687::AID-JBM13>3.0.CO;2-K
D.C. Tancred, A.J. Carr, B.A.O. McCormack, J. Mater. Sci. Mater. Med. 12, 81 (2001). doi:10.1023/A:1026773522934
G. Georgiou, J.C. Knowles, Biomaterials 22, 2811 (2001). doi:10.1016/S0142-9612(01)00025-4
G. Goller, H. Demirkiran, F.N. Oktar, E. Demirkesen, Ceram. Int. 29, 721 (2003). doi:10.1016/S0272-8842(02)00223-7
S.J. Kalita, S. Bose, H.L. Hosick, A. Bandyopadhyay, Biomaterials 25, 2331 (2004). doi:10.1016/j.biomaterials.2003.09.012
T. Kokubo, S. Ito, M. Shigematsu, S. Sakka, J. Mater. Sci. 20, 2001 (1985). doi:10.1007/BF01112282
T. Kokubo, H.M. Kim, M. Kawashita, Biomaterials 24, 2161 (2003). doi:10.1016/S0142-9612(03)00044-9
T. Kokubo, H. Takadama, Biomaterials 27, 2907 (2006). doi:10.1016/j.biomaterials.2006.01.017
R.M. Shelton, A.C. Rasmussen, J.E. Davies, Biomaterials 9, 22 (1988). doi:10.1016/0142-9612(88)90065-8
J.F. Bredt, T.C. Anderson, D.B. Russel, Three Dimensional Printing Material System and Method. US Patent Application No US 2004/0138336 A1
S. Ramesh, C.Y. Tan, I. Sopyan, M. Hamdi, W.D. Teng, Sci. Technol. Adv. Mater. 8, 124 (2007). doi:10.1016/j.stam.2006.11.002
A.S. Kosmos, L.I. Bellic, D. Susnik, FIZIKA 2, 85 (1996)
W.D. Kingery, J. Appl. Phys. 30, 301 (1959). doi:10.1063/1.1735155
I. Oh, N. Nomura, N. Masahashi, S. Hanada, Scr. Mater. 49(12), 1197 (2003). doi:10.1016/j.scriptamat.2003.08.018
M. Milosevski, J. Bossert, D. Milosevski, N. Gruevska, Ceram. Int. 25(8), 693 (1999). doi:10.1016/S0272-8842(99)00003-6
H. Oonishi, L.L. Hench, J. Wilson, F. Sugihara, E. Tsuji, M. Matsuura, S. Kin, T. Yamamoto, S. Mizokawa, J. Biomed. Mater. Res. 51(1), 37 (2000). doi:10.1002/(SICI)1097-4636(200007)51:1<37::AID-JBM6>3.0.CO;2-T
H. Teramoto, A. Kawai, S. Sugihara, A. Yoshida, H. Inoue, Key. Eng. Mater. 240/242, 269 (2003)
J. Vandiver, N. Patel, W. Bonfield, C. Ortiz, Key. Eng. Mater. 284, 497 (2005). doi:10.4028/0-87849-961-x.497
Acknowledgement
This work was supported by a grant from National Metal and Materials Technology Center, National Science and Technology Development Agency. The authors would like to extend their gratitude to P. Tesavibul for helping in SBF soaking experiment.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Suwanprateeb, J., Sanngam, R., Suvannapruk, W. et al. Mechanical and in vitro performance of apatite–wollastonite glass ceramic reinforced hydroxyapatite composite fabricated by 3D-printing. J Mater Sci: Mater Med 20, 1281–1289 (2009). https://doi.org/10.1007/s10856-009-3697-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10856-009-3697-1