
Abstract
Biomorphous, highly porous hydroxyapatite (HA) ceramics have been prepared by a combination of a novel biotemplating process and a sol–gel method, using natural plants like cane and pine as biotemplates. A HA sol was first synthesized from triethylphosphate and calcium nitrate used as the phosphorus and calcium precursors, respectively, and infiltrated into the biotemplates, and subsequently they were sintered at elevated temperatures to obtain porous HA ceramics. The microstructural changes, phase and chemical composition evolutions during the biotemplate-to-HA conversion were investigated by scanning electron microscopy (SEM), X-ray powder diffraction (XRD), and Fourier-transform infrared (FT-IR) spectroscopy. The XRD and FT-IR analysis revealed that the dominant phase of the product was HA, which contained a small amount of mixed A/B-type carbonated HA, closely resembling that of human bone apatite. Moreover, the appearance of a small amount of secondary phase CaCO3 seemed unavoidable. The HA was not transformed to the other calcium phosphate phases up to 1400°C, but it contained a trace amount of CaO when sintered at above 1100°C. The possible transformation mechanism was proposed. The SEM observation and mechanical property test showed that as-produced HA ceramics retained the macro-/micro-porous structures of the biotemplates with high precision, and possessed acceptable mechanical strength, which is suggested to be potential scaffolds for bone tissue engineering.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Bone tissue engineering, involving the fabrication of a porous scaffold, has become an alternative approach to promote the repair and regeneration of diseased or damaged bone tissue [1]. Ideal porous scaffolds for bone tissue engineering should exhibit some necessary properties, including excellent osteoconductivity and bioactivity, biocompatibility, controllable biodegradability and good absorbability, three dimensionally interconnected highly porous structure as a template favorable for new bone ingrowth, and irregular shape fabrication ability [2, 3], etc. Many studies reported that porous hydroxyapatite (Ca10(PO4)6(OH)2, HA) ceramics met most of the above criteria. Particularly, HA can induce the formation of bone-like apatite layer in vitro when exposed to SBF, which can strongly bond to living bone [4], stimulate new bone formation and growth [5] and exert a positive effect on the expression of genes regulating osteogenesis [6]. So, synthetic HA similar to bone mineral in chemical composition, biology and crystallography [7, 8], has attracted special interest in the field of bone tissue engineering over past two decades. Compared with other techniques such as wet chemical precipitation technique [9], hydrothermal method [10], solid-state reaction [11], mechanochemical synthesis [12] and combustion preparation [13], sol–gel derived HA ceramics exhibit higher bioactivity, biocompatibility, osteoconductivity and better cell responses because of their poorly crystalline and presence of carbonate ions in the crystal lattice [14, 15].
It has been recognized that the pore structure is one of the decisive factors affecting the biological function of scaffolds [16, 17]. To imitate the porous structure of spongy bone, many techniques have been developed, such as polymeric sponge replication [18], rapid prototyping techniques [19], electrospinning [20], phase-separation [21], particulate leaching [22], sacrificial filler [23], freeze drying [24], solvent casting [25] and gel-casting techniques [26]. These techniques endued scaffolds with a variety of porous microstructures to satisfy different applications. Recently, special attention is paid to some biomorphic bone substitutes and scaffolds for bone regeneration, originating from biological tissues and natural materials like cuttlefish [10], seastar [27], bovine bone [28], coral [29], seashell [30], and red algae [31], because their unique morphology and inter-connective highly porous structure similar to human bone may improve the solubility of the implant and facilitate cellular activity and faster bone in-growth [32, 33].
In recent years, a novel replication method, i.e. biotemplating technology based on wood, has been applied to manufacture biomorphic porous ceramics with woodlike structures via substitution or transformation processing, including carbide (SiC, TiC) [34], oxide (Al2O3, Cr2O3) [35] and nitride (TiN) [36], etc. Wood, a natural biopolymeric composite mainly comprised of hemicellulose, cellulose and lignin, exhibits a complex micro-/macro-structure and hierarchical microcellular architecture featuring honeycomb-like microchannels, which is very similar to that of cancellous bone. Many natural plants such as cane and wood have highly open trabecular structures, and have been used as sacrificial templates for porous ceramics. Some benefits of innovative wood-based biomorphic materials are the versatility for the fabrication of complex shapes, sufficient biomechanical properties [37], and intrinsic three-dimensional interconnected porous structure [38]. These merits allow this kind of biomorphic porous materials for biomedical applications like bone implants and scaffolds in bone reconstitution. Coating biomorphic ceramics with bioactive materials can confer them excellent bone-bonding ability [39]. P González et al. [40] proposed wood-based biomorphic SiC ceramics coated with bioactive glass by Pulsed Laser Deposition as a very promising device for dental and orthopaedic applications. Recently, they [41] tested the in vitro cytotoxicity of the biomorphic SiC ceramics uncoated or coated with bioactive glass, using MG-63 human osteoblast-like cells. Their results revealed that the biological response of the cells on the biomorphic SiC ceramics was similar to the one exhibited by well-known implant materials like Ti6Al4 V and bulk bioactive glass, which makes it possible to be applied in bone implantology like load bearing protheses.
In fact, there have been some observations on bone ingrowth into Clematis alba—derived charcoal [42] and birch wood [43] implants since the 1980 s. In another paper, bone ingrowth and appositional growth in small prosthesis implants of juniper, pretreated by a boiling procedure, was reported [44]. In a recent paper, heat-treated deciduous wood (birch) was directly used as replacement material for osteochondral bone defects in the knee joint of rabbit [45]. As a result, the natural porous channel structure of wood made it serve as a porous bioactive scaffold, which allowed ongrowth of bone and cartilage differentiation on its surface and ingrowth the porous structure (with a diameter of 100–200 μm) of the wood, demonstrating osteoconductive and chondroconductive contact. Then, wood and wood-derived materials might become a promising tissue engineering scaffold strategy.
Much interest within tissue engineering is paid to the design of the scaffolds to ensure good tissue growth. However, a common problem encountered when using scaffolds for tissue engineering is the rapid formation of tissue on the outer edge, leading to the development of a necrotic core, due to the limitations of cell penetration, and oxygen and nutrient exchanges [46]. To overcome this issue, several different strategies have been developed, such as adopting dynamic culture systems [47], incorporating chemotactic growth factors [48] and choosing an appropriate composition [49, 50]. It is suggested that the issue may also be solved by an alternative approach, i.e., tailoring the porous structure and morphological properties of scaffolds. For example, the incorporation of macro- and/or micro-channels within scaffolds can not only enhance cell penetration and tissue infiltration but also maintain suitable living cell, nutrient and oxygen concentrations throughout the scaffolds in vitro and in vivo [51]. Moreover, such scaffolds possess higher strength than the scaffolds with the same porosity [17]. Some methods have been developed to produce microchannels in scaffolds, including novel rapid-prototyping techniques [52], coextrusion process [53], freeze-dry processing [54], fiber templating technique [55], and molding process using acupuncture needles as mandrels [56], etc. However, it is difficult for them to obtain microchannels with less than 100 μm.
To the best of our knowledge, there has been no report on the preparation of biomorphic HA from natural plant templates. In the present study, we attempt to utilize the combination of this biotemplating method and sol–gel processing, for the first time, to develop novel biomorphic HA ceramic scaffolds from natural plants like wood and cane. The main objective is to probe the influence of the thermal treatment on the morphological, structural and phase changes during the biotemplate-to-scaffold conversion. The final goal is to create an ideal scaffold for bone tissue engineering application.
2 Experimental
2.1 HA sol synthesis
The aim of this work is to fabricate biomorphic HA ceramics by a biotemplating process, involving the infiltration of a HA sol into biotempates, e.g. cane and pine (Local plants, Purchased from Xi’an Wood Company, Xi’an, China), and subsequent sintering at elevated temperatures. The HA sol was prepared by a sol–gel route using calcium nitrate tetrahydrate (Ca(NO3)2 · 4H2O, AR, purity > 99.9%, Chongqing Rearents Company, Chongqing, China) and triethyl phosphate (OP(OC2H5)3, TEP, CP, purity > 99.5%, Kunshan Kunhua Group Company, Kunshan, China) as calcium and phosphorous sources, respectively. Other reagents are analytical reagents and purchased from Xi’an Chemical Plant (Xi’an, China). Briefly, TEP was first mixed with distilled water and ethanol, and hydrolyzed for 6 h under vigorous stirring using a magnetic stirrer to form a 0.6 M solution. Then, a 1 M aqueous solution of Ca(NO3)2 · 4H2O was slowly added dropwise into the above-mentioned hydrolyzed TEP solution, according to a stoichiometric molar ratio for the formation of HA. The pH value of the mixed solution is adjusted to 8 using ammonium hydroxide [NH4OH, 17% in H2O]. After the reactants were continuously stirred for additional 12 h at room temperature, a clear liquid was obtained, namely, HA sol. Finally, the sol was kept in a sealed container to allow to age for 5 days.
2.2 Fabrication of biomorphic HA ceramics
To prepare biomorphic HA ceramics, the biotemplates were immersed in HA sol in glass beaker in a self-made equipment. After the equipment was vacuumized and held for 2 h, the interior pressure was raised to 3–5 atmospheric pressure and maintained for 1 h, ensuring the infiltrate of sol into the biotemplates. And then, the impregnated samples were gelled and dried at 120°C for 24 h in an oven. The HA amount in the biotemplates can be increased by repeating the above infiltration procedure. After drying, the organic biotemplates were burned out at 550°C for 2 h with a heating rate of 1°C/min in order to minimise damage to porous HA structure. Once the organic biotemplates were removed, the temperature was raised up to 700, 1,100, 1,200, 1,300 and 1,400°C with a heating rate of 15°C/min and kept for 3 h to increase the density of the struts and to improve mechanical properties. The experimental procedure is shown in the flow chart given in Fig. 1.

Flow chart of fabricating biomorphic, porous HA ceramics by the biotemplating process
2.3 Materials characterization
The morphologies of the fracture surfaces of original templates and template-derived scaffolds were observed by S-2700 scanning electron microscopy (SEM, Hitachi, Japan). The samples were mounted on copper stubs, coated in vacuum with gold using a sputter coater, and observed by SEM at an accelerating voltage of 20 kV.
The effects of sintering temperature on the crystalline phase and chemical structure evolutions were investigated by powder X-ray diffraction (XRD) and Fourier transform infrared spectroscopy (FT-IR). The scaffolds were first ground into a powder and 1 g of the powder was collected for XRD analysis. The XRD experiments were performed on a Philips X’Pert MPD diffractometer in the 10° ≤ 2θ ≤ 80° range with a step size of 0.033° for 2θ and a scanning speed of 0.14°/min, employing Cu Kα (λ = 1.54056 Å) radiation generated at 40 kV and 40 mA. The crystalline phases were determined from a comparison of the registered standard JCPDF cards (HA: 09-0432, CaCO3: 05-0586, CaO: 37-1497, Ca(NO3)2: 07-0204, β-TCP: 09-0169) available in the system software with the obtained powder diffraction files. To characterize the chemical composition and molecular structure, FT-IR spectra were recorded on a Shimadzu IR Prestige 21 spectrometer in the range 4,000–400 cm−1 in transmission mode with 1 cm−1 resolution using a typical KBr pellet technique. In order to allow comparisons, the spectra were normalized from the ν4 band of the phosphate group at 602 cm−1 according to a classical procedure [57].
The crystallinity of the as-prepared HA was estimated from the XRD data using the following Eq. 1 [58].
in which Xc is the degree of crystallinity, V 112/300 the intensity of the hollow between (112) and (300) diffraction peaks, and I 300 the intensity of the (300) peak. The crystallite size in HA was evaluated according to Scherrer’s formula as follows [59]:
where Xs is the crystallite size (nm), λ the wavelength of the monochromatic X-ray beam (λ = 0.154 056 nm for CuKα radiation), FWHM the full width at half-maximum for the diffraction peak under consideration (rad), and θ the diffraction angle (deg). The (002) and (300) diffraction peaks were chosen for calculating the crystallite size.
The relationship between lattice spacing (d) and lattice parameters (a, b, and c) of the hexagonal structure is expressed as
where h, k, l are the Miller indices of the reflection planes. The (002) and (300) reflections were chosen for the lattice parameters calculation [60].
The samples’ porosity was measured by water displacement method based on the Archimedean principle. For the compressive strength test, cylindrical samples with dimensions of Φ6 mm × height 8 mm were loaded with an Instron 1195 universal testing machine (Instron Corp., USA) at a crosshead speed of 0.5 mm/min. Loading direction was parallel to that of the honeycomb-like pores. More than five samples were tested to obtain the average value along with its standard deviation.
3 Results and discussion
3.1 Morphological characteristics of biomorphic HA ceramics
The SEM micrographs in Fig. 2 show the morphologic structures of the cross-sections of biotemplate cane and cane-derived HA ceramics. As shown in Fig. 2a, b, cane shows a typical honeycomb-like porous structure with a bimodal pore diameter distribution. A lot of parallel long tubular channels exhibit along the direction parallel to the stem. The macrochannels with pore diameter of 50–120 μm are surrounded by lots of microchannels with an average pore size of about 10 μm. Moreover, the channels are interconnected by the inherent pits of cane. The cell wall thickness is about 1 μm.

SEM micrographs of cane (a and b) and cane-derived HA ceramics (c and d) obtained at 1,200°C for 3 h. The cross-sections are perpendicular to stem direction
It can be seen from Fig. 2c, d that, after the removal of organic biotemplates and 3 h of sintering at 1,200°C in air, the hierarchical pore structure of the initial template was preserved intact to cane-derived HA ceramics. The resulting HA is of a rougher wall surface morphology, and many micropores with irregular shape probably originating from the pits of cane and/or deficient sintering clearly exist in walls among the elongated channel-like pores, which makes the resultant HA ceramics exhibit an interconnecting porous network. In addition, it is also noted that the overall pore size observed in HA ceramics decreased lightly in comparison with that of original biotemplates.
Figure 3 shows representative the SEM images of pine and typical pine-like HA ceramics sintered at 1,200°C for 3 h in air. It can be seen that pine, different from cane, has a characteristic texture of a unimodal macropore diameter distribution with an average pore diameter of about 20 μm. The SEM micrographs in Fig. 3 indicate that the channeled porous microstructure of pine was also perfectly transformed into HA ceramics after heat treatment. Moreover, the walls of pine-derived HA ceramics appear to be denser than those of cane-derived HA ceramics due to the difference in natural microstructure between pine and cane. The result indicates that pine-derived HA ceramics exhibited a good sintering feature, and the grain size in the cell wall is about 2 μm.

SEM micrographs of pine (a and b) and pine-derived HA ceramics (c, d and e) obtained at 1,200°C for 3 h. The cross-sections are perpendicular to stem direction
The above-mentioned results suggest a possible route for producing novel porous HA ceramics using natural organic templates. According to the existing reports [31, 61], such porous HA ceramics containing interconnected microchannels will permit the circulation of the physiological fluid throughout them, induce protein adhesion, stimulate significantly cell growth and promote bone ingrowth, and modify their resorption behavior, when they are used as bone implants and/or scaffolds for bone tissue engineering.
3.2 Chemical composition changes
The FT-IR spectra of cane-derived HA ceramics sintered at 700, 1,100, 1,200, 1,300, and 1,400°C for 3 h are given in Fig. 4. The FT-IR spectra of pine-derived HA ceramics are not given here, because they are the same as those of the cane-derived HA ceramics. The broad absorption band between 3,700 and 3,100 cm−1 assigned to the O–H stretch vibration of hydrogen bonded O–H groups, together with the absorption peak at 1,636 cm−1 ascribed to O–H bending mode, are evidences of the presence of adsorbed water in the products [14]. The two sharp peaks observed at 3,571 and 632 cm−1, which are generated by unhydrogen bonded free O–H stretch, are attributed to the characteristic stretching and librational modes of structural O–H groups in HA crystallites [62], respectively. The peak at 3,642 cm−1 has been reported to be due to the OH− stretching vibrations of Ca(OH)2 [63].

FTIR spectra of HA ceramics sintered at different temperatures for 3 h. (a) 700°C, (b) 1,100°C, (c) 1,200°C, (d) 1,300°C, (e) 1,400°C
The main characteristic bands of PO4 3− tetrahedral apatite’s structure [59, 64] are clearly observed, given as follows. The peak at 472 cm−1 is attributed to v 2 O–P–O bending. The double peak at 571 cm−1 and 602 cm−1 with a high resolution belongs to v 4 O–P–O antisymmetric and symmetric bending modes [62, 65], and 962 cm−1 for v 1 P–O symmetric stretching. The peaks at 1,040 and 1,088 cm−1 are due to v 3 P–O antisymmetric stretching modes [66]. Moreover, the v 1 and v 3 peaks overlap to give a broad band in the wavenumber interval of 900–1,200 cm−1 [67]. The group of weak intensity bands in the 1,950–2,200 cm−1 region is a reflection of overtones and combinations of the v 3 and v 1 PO4 3− modes [33, 68]. These results clearly indicate the formation of a typical HA structure. The double peaks at 2,365 and 2,344 cm−1 are due to CO2 in air [69]. In the present study, however, the characteristic bands of β-TCP at 1,118 cm−1 and 945 cm−1 [33] were not observed for all the sintered samples.
It should be noted that the obvious absorption bands at 1,600–1,400 and 875 cm−1 confirm the presence of carbonate group [70]. The bands centered at 1,420 cm−1 and 1,500 cm−1 correspond, respectively, to v 3 asymmetric and symmetric C–O stretching modes [71], which is different from a single characteristic peak (1,415 cm−1) of free CO3 2− or carbonates, indicating the presence of at least two carbonated phases [72, 73]. The peaks at 875 cm−1 and 714 cm−1 correspond to the v 2 out-of-plane deformation and v 4 in-plane deformation O–C–O bending modes in CO3 group [15, 74], respectively. The FT-IR band at 1,420 cm−1 together with the XRD peak at 2θ = 29.42° supports the existence of a CaCO3 phase. The peaks at 2,515 cm−1 and 1,794 cm−1 are attributed to v 1 + v 3 and v 1 + v 4 combination bands of CaCO3, respectively. The weak bands at 1,560, 1,510 and 1,460 cm−1 correspond to the incorporation of CO3 2− groups at the OH position (A-type) [67], and those at 1,460 and 1,420 cm−1 are ascribed to the substitution of CO3 2− ions for PO4 3− sites in the apatite structure (B-type) [75]. The band at 875 cm−1 is assigned to three types of CO3 2− groups.
As sintering temperature was increased from 700°C to 1,200°C, the characteristic peaks (3,571 cm−1 and 632 cm−1) of OH groups became stronger in intensity and better in resolution. However, when sintering temperature was further increased from 1,200°C to 1,400°C, the intensity of the OH− peaks started to decrease. In contrast, the PO4 3− bands located at 602, 962, 900–1,300 and 1,950–2,200 cm−1 significantly increased in intensity and became broader with the increasing sintering temperature, indicating a deteriorating molecular arrangement. Such a behavior is in accordance with the XRD analysis which indicated partial dehydration and decomposition of HA phase. The fast reduction in the intensity of CO3 2− bands at 1,420 and 875 cm−1 with temperature up to 1,400°C was seen, which is strongly indicative of the decomposition of CaCO3. In general, the decomposition of CaCO3 occurs at 580°C or higher [76], so the stretching modes of CO3 2− groups should be absent in the spectra of the samples obtained at the present sintering temperatures. However, the remnant presence of the carbonate peaks are still evident in these spectra, even if the sample was heat-treated at 1,400°C, which further proves the inclusion of CO3 2− groups in the apatite structure [72]. Surprisedly, it is found that there seems be more CO3 2− ions incorporated into HA crystal lattice structure at 1,300 and 1,400°C than those at 1,100 and 1,200°C. But, this is accordance with the fact that the bands of PO4 groups became broader with increasing heat-treatment temperature.
3.3 Phase evolution
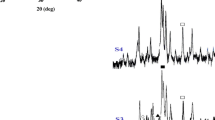
Structural information obtained by XRD is essential for the interpretation of the mechanisms taking place during sintering. The XRD patterns of cane-derived HA ceramics sintered at 700, 1,100, 1,200, 1,300 and 1,400°C for 3 h are shown in Fig. 5. The XRD patterns of pine-derived HA ceramics are not given here, because they are the same as those of the cane-derived HA ceramics. In all the XRD patterns, most diffraction peaks are identical to the characteristic patterns of stoichiometric HA of a hexagonal phase structure (JCPDF 09-0432) in terms of both intensity and position of the peaks, indicating the formation of apatitic structure. The pronounced XRD diffraction peaks observed at 2θ = 29.42, 35.98, 39.4, 43.16, 47.55 and 48.54° suggest the formation of calcite (rhombohedral CaCO3, PDF 05-0586) as a secondary phase. However, in the XRD patterns of the sample heat treated at 700°C, the two weak peaks located at 2θ = 27.78 and 30.80° indicate the formation of a trace amount of β-tricalcium phosphate (β-Ca3(PO4)2, β-TCP, JCPDF 09-0169), which originally presented in lower temperature calcined gels [10] and was attributed to the decomposition of a nonstoichiometric HA. At the same time, no CaO and Ca(NO3)2 crystalline phases were not detected. This phenomenon suggests that aging time is enough. β-TCP disappeared at above 700°C, suggesting a result caused by thermally activated reactions. The possible reason is that the existing CaCO3 restrained the decomposition of HA, and synchronously reacted with β-TCP to produce HA. For the latter, a possible reaction is grossly represented as follows:

XRD patterns of HA ceramics sintered at different temperatures for 3 h ▼: HA; ∇: β-Ca3(PO4)2; ◊: CaO; ♦: Ca(OH)2; ●: CaCO3
It was previously reported that apatite could evolve from the reactions among the impurity phases such as Ca2P2O7, Ca3(PO4)2, CaCO3, and Ca(NO3)2 present in lower temperature calcined HA [14].
The XRD diffraction peak at 2θ = 18.09° is identified to hexagonal Ca(OH)2 (PDF 04-0733), which is confirmed by another evidence of 3,642 cm−1 peak in the FT-IR spectra. It was also noticed that the additional two weak peaks at 2θ = 37.4º and 53.9º were seen when the samples were sintered at 1,300°C or above, which indicates the presence of a new crystalline phase. The two peaks are identified to the characteristic (200) and (220) reflections of cubic CaO phase (lime). We believe, in fact, that Ca(OH)2 phase detected in the samples obtained at 1,100°C should be the product of the reaction between CaO and H2O upon exposure to air (Eq. 5) during the storage period because of the hygroscopic nature of CaO.
As indicated in the reports till date on the sol–gel-derived HA from the same or similar raw materials, CaO was an unavoidable major impurity phase in the synthesized HA [14]. However, the formation of CaO was usually considered to be originated from the decomposition of partial Ca(NO3)2 molecules which had no opportunities to react with TEP sol to form Ca–OP(OH) x (OC2H5)3−x complex [77, 78]. However, in present reaction system, CaO may originate from the decomposition of CaCO3 and/or partial HA, and the corresponding reactions are shown in Eq. 6 and Eq. 7, respectively.
Particularly noteworthy, as sintering temperature increased, the diffraction patterns showed a continuous evolution of the structure. The diffraction peaks due to HA exhibited a considerable increase in the intensity, and became stronger and narrower, which suggests further nucleation/growth of hexagonal HA mostly from the transformation of the amorphous Ca–P intermediate. This was confirmed by the occurrence of some new peaks (301), (312) and (322), etc. This implies that there must be orderly transport of mass to the central growing crystal from the neighboring ones. In contrast, the relative intensity of calcite peaks remarkably decreased and even disappeared. At the same time, CaO/Ca(OH)2 amount gradually increased. This signifies that calcite decomposed to form CaO. For the samples sintered at above 1,200°C, HA became dominant phase. The CaCO3 amount in the HA ceramics sintered at different temperatures was semi-quantitatively estimated from the XRD peak height ratio of HA (211) vs. CaCO3 (104) [79], and the results are given in Table 1. All peak heights can be obtained directly from XRD patterns.
It can be seen from Table 1 that the (h k l = 211) peak intensity of HA decreased with the increase in temperature above 1,300°C. This indicates that the decomposition of partial HA has occurred via the reaction equations (Eq. 7 and/or Eq. 8). The dehydroxylation behavior of HA may lead to the formation of a nonstoichiometric phase.
Though this dehydroxylation process had no significant effect on the crystal structure of HA [80], it decreased content of OH groups, which is in good agreement with the FT-IR results. Moreover, the loss of OH− groups could lead to the contraction of the HA lattice [81], which was also confirmed by data in Table 2. According to the existing literature [33, 53, 82, 83], β-TCP should also be one of the decomposed products of HA. However, its diffraction peaks were not observed in the XRD patterns. The possible reason for this is that as-produced β-TCP reacted with CaCO3 to reform HA, and the proposed reaction is shown in Eq. 4.
The reasons for the formation of CaCO3 in sol–gel derived HA may be classified into two groups according to the existing reports. One group suggests that CaCO3 originated from the reaction between CO2 in air and Ca-containing compounds in system [65, 84]. In this case, insufficient aging, i.e. incomplete reaction between HPO(OC2H5)2−x (OH) x and Ca(NO3)2, is considered to be main reason, and can cause the appearance of impurity phases, such as CaO, Ca(OH)2, Ca3(PO4)2 or CaCO3 [85, 86]. A higher solution temperature together with a prolonged time period (of several days) is needed to form HA phase because of the poor hydrolysis activity of triethyl phosphate [87]. Another group believes that CaCO3 was a product of a series of complex reactions between calcium in the precursors and carbon-containing radicals-arising from the pyrolysis of residual organics in precursors during heat treatments [88, 89]. We believe that CaCO3 in HA in the present study might most possibly be a product of the reaction between Ca-containing compounds and some active organic radicals containing carbon produced during the pyrolysis of the organic biotemplates. The radicals were of high affinity with Ca [72], which led to the formation of carbonate groups. Moreover, from the phenomenon that CaCO3 phase is dominant in the present system below 1,200°C, it can be concluded that the organic biotemplates favored the formation of CaCO3. The formation of CaCO3 can be accounted for according to the following equation:
In addition, the gases released during the removing biotemplates, such as water, CO, CO2, etc., may also become incorporated into the HA lattice to form carbono-apatite [90].
According to Hwang et al’s discussion on the changes in crystal lattice parameters caused by carbonate substitution in HA [90], it can be deduced from the results in Table 2 that a mixed B-type and A-type carbonate substitution occurred, because both the a axis and the c axis expanded except the sample sintered at 1,400°C. It was further confirmed by the phenomenon, in which the (hk0) peaks and (00l) peak shifted to higher angles and to a lower angle than those of standard HA [91], respectively (data not given here). The result is in agreement with the previous investigations [59, 72, 76], in which the formation of B- or A/B-type carbonated HA was often observed in sol–gel-derived HA ceramics, especially in the sol–gel process involving organic reagents [92]. Then, β-TCP formed during the heat-treatment at 700°C may be due to the decomposition of carbonated HA, and other products should include HA and CaO [84].
There is a variation in intensity and width of the HA peaks in the XRD data among the different products, and this variation is closely related to the crystallinity and crystallite size. Table 3 presents the crystallinity and crystallite size along the a-axis (300 plane) and the c-axis (002 plane) of HA sintered at different sintering temperatures. It can be seen that the crystallinity increased with sintering temperature up to 1,200°C and then decreased. The crystallite size along c-axis increased from 82.75 nm to 124.11 nm as sintering temperature increased from 700°C to 1,100°C, and then nearly did not change with further increase in sintering temperature. The same trend was observed along a-axis. Moreover, the HA crystal grew along the c-axis direction preferentially.
3.4 Porosity and mechanical properties of biomorphic HA ceramics
To be used as scaffolds for bone tissue engineering, porous biomaterials should possess high porosity and appropriate mechanical strength. Table 4 presents the porosity and compressive strength of cane- and pine-derived HA ceramics sintered at 1,100, 1,200, 1,300 and 1,400°C for 3 h. It can be seen that the porosity of both cane-derived and pine-derived HA ceramics decreases as the sintering temperature increase from 1,100°C to 1,400°C, and the compressive strength gradually increases. The reason is that the elevating sintering temperature favors the sintering among the HA particles from HA sol, which leads to high strength and reduces porosity. At the same time, it is also noted that cane-derived HA ceramics have higher porosity and lower compressive strength than pine-derived HA ceramics obtained under the same experimental condition. The porosity difference between them is caused by the inherent different macro- and micro-structures of the biotemplates cane and pine. Compared with pine, the bimodal pore diameter distribution of cane probably easily causes the uneven HA coating on the strut surfaces of organic template during the infiltration process, which together with higher porosity results in lower mechanical strength. In addition, since the strength of HA ceramics sintered at 700°C is too low to be measured, the corresponding porosity and compressive strength can not be given here. Further studies are needed about the mechanical properties.
4 Conclusions
It has been proven that the novel biotemplating method presented in this study, in combination with a sol–gel technique, may be employed to prepare biomorphic HA ceramic scaffolds from the biotemplates like wood and cane. The inherent pore structure of the templates, both multimodal and monomodal pore size distribution, can be transferred to the final HA scaffolds with high precision. The interconnectivity among channel-like macropores in HA ceramic scaffolds varies with the kind of biotemplates. The extensive densification of the macro-pore walls occurred in the pine-derived HA scaffolds. In contrast, cane-derived HA ceramics exhibited an interconnecting porous network. The FT-IR and XRD results show the dominant crystal phase is identified as HA with a small amount of a mixed A and B type carbonated HA, where hydroxyl or phosphate groups are partially replaced by carbonate ones, whilst the appearance of CaCO3 is unavoidable. Under the same experimental condition, cane-derived HA ceramics have higher porosity and lower compressive strength than pine-derived HA ceramics because of their inherent different macro- and micro-structures. The ideal sintering condition for HA scaffolds should be 1200°C/3 h in terms of the purity and appropriate strength of the products. This study reveals the potential of natural plants in fabricating biomorphic porous HA ceramic scaffolds suitable for bone tissue engineering application.
References
L.L. Hench, J.M. Polak, Science 295, 1014 (2002). doi:10.1126/science.1067404
Q.Z. Chen, I.D. Thompson, A.R. Boccaccinni, Biomaterials 27, 2414 (2006). doi:10.1016/j.biomaterials.2005.11.025
Y.S. Park, K.N. Kim, K.M. Kim, S.H. Choi, C.K. Kim, R.Z. Legeros et al., J. Mater. Sci. 41, 4357 (2006). doi:10.1007/s10853-006-6261-0
L.L. Hench, J. Am. Ceram. Soc. 74, 1487 (1991). doi:10.1111/j.1151-2916.1991.tb07132.x
J. Moura, L.N. Teixeira, C. Ravagnani, O. Peitl, E.D. Zanotto, M.M. Beloti et al., J. Biomed. Mater. Res. 82A, 545 (2007). doi:10.1002/jbm.a.31165
G.A. Silva, O.P. Coutinho, P. Ducheyne, I.M. Shapiro, R.L. Reis, Biomaterials 28, 326 (2007)
W. Xu, W.Y. Hu, M.H. Li, C.E. Wen, Mater. Lett. 60, 1575 (2006). doi:10.1016/j.matlet.2005.11.072
H.W. Kim, Y.M. Kong, C.J. Bae, Y.J. Noh, H.E. Kim, Biomaterials 25, 2919 (2004). doi:10.1016/j.biomaterials.2003.09.074
S. Ramesh, C.Y. Tan, S.B. Bhaduri, W.D. Teng, Ceram. Int. 33, 1363 (2007). doi:10.1016/j.ceramint.2006.05.009
S. Kannan, J.H.G. Rocha, S. Agathopoulos, J.M.F. Ferreira, Acta Biomater. 3, 243 (2007). doi:10.1016/j.actbio.2006.09.006
R.R. Ramachandra, H.N. Roopa, T.S. Kannan, J. Mater. Sci. Mater. Med. 8, 511 (1997). doi:10.1023/A:1018586412270
C. Mochales, H. Ei Briak-Benabdeslam, M.P. Ginebra, A. Terol, J.A. Planell, P. Boudeville, Biomaterials 25, 1151 (2004). doi:10.1016/j.biomaterials.2003.08.002
E.M. Rivera, M. Araiza, W. Brostow, V.M. Castano, J.R. Diaz-Estrada, R. Hernandez et al., Mater. Lett. 41, 128 (1999). doi:10.1016/S0167-577X(99)00118-4
H. Eshtiagh-Hosseini, M.R. Housaindokht, M. Chahkandi, Mater. Chem. Phys. 106, 310 (2007). doi:10.1016/j.matchemphys.2007.06.002
J.C. Elliott, D.W. Holcomb, R.A. Young, Calcif. Tissue Int. 37, 372 (1985). doi:10.1007/BF02553704
E. Jabbarzadeh, T. Jiang, M. Deng, L.S. Nair, Y.M. Khan, C.T. Laurencin, Biotechnol. Bioeng. 98, 1094 (2007). doi:10.1002/bit.21495
M.M.C.G. Silva, L.A. Cyster, J.J.A. Barry, X.B. Yang, R.O.C. Oreffo, D.M. Grant et al., Biomaterials 27, 5909 (2006). doi:10.1016/j.biomaterials.2006.08.010
C. Vitale-Brovarone, E. Verné, L. Robiglio, P. Appendino, F. Bassi, G. Martinasso et al., Acta Biomater. 3, 199 (2007). doi:10.1016/j.actbio.2006.07.012
J.M. Williams, A. Adewunmi, R.M. Schek, C.L. Flanagan, P.H. Krebsbach, S.E. Feinberg et al., Biomaterials 26, 4817 (2005). doi:10.1016/j.biomaterials.2004.11.057
H.W. Kim, H.E. Kim, J.C. Knowles, Adv. Funct. Mater. 16, 1529 (2006). doi:10.1002/adfm.200500750
W. Helen, C.L.R. Merry, J.J. Blaker, J.E. Gough, Biomaterials 28, 2010 (2007). doi:10.1016/j.biomaterials.2007.01.011
D. Tadic, F. Beckmann, K. Schwarz, M. Epple, Biomaterials 25, 3335 (2004). doi:10.1016/j.biomaterials.2003.10.007
C.V. Brovarone, E. Verné, P. Appendino, J. Mater. Sci. Mater. Med. 17, 1069 (2006). doi:10.1007/s10856-006-0533-8
S. Sánchez-Salcedo, A. Nieto, M. Vallet-Regí, Chem. Eng. J. 137, 62 (2008). doi:10.1016/j.cej.2007.09.011
M. Shin, H. Abukawa, M.J. Troulis, J.P. Vacanti, J. Biomed. Mater. Res. 84A, 702 (2008). doi:10.1002/jbm.a.31392
H.R. Ramay, M.Q. Zhang, Biomaterials 24, 3293 (2003). doi:10.1016/S0142-9612(03)00171-6
M. Martina, G. Subramanyam, J.C. Weaver, D.W. Hutmacher, D.E. Morse, S. Valiyaveettil, Biomaterials 26, 5609 (2005). doi:10.1016/j.biomaterials.2005.02.011
A. Worth, M. Mucalo, G. Horne, W. Bruce, H. Burbidge, Clin. Oral Implants Res. 16, 379 (2005). doi:10.1111/j.1600-0501.2005.01113.x
R. Murugan, S. Ramakrishna, Biomaterials 25, 3073 (2004). doi:10.1016/j.biomaterials.2003.09.089
K.S. Vecchio, X. Zhang, J.B. Massie, M. Wang, C.W. Kim, Acta Biomater. 3, 910 (2007). doi:10.1016/j.actbio.2007.06.003
P.J. Walsh, F.J. Buchanan, M. Dring, C. Maggs, S. Bell, G.M. Walker, Chem. Eng. J. (2007). doi:10.1016/j.cej.2007.10.016
B. Ben-Nissan, A. Milev, R. Vago, Biomaterials 25, 4971 (2004). doi:10.1016/j.biomaterials.2004.02.006
C.Y. Ooi, M. Hamdi, S. Ramesh, Ceram. Int. 33, 1171 (2007). doi:10.1016/j.ceramint.2006.04.001
J.M. Qian, J.P. Wang, G.Y. Hou, G.J. Qiao, Z.Z. Jin, Scr. Mater. 53, 1363 (2005). doi:10.1016/j.scriptamat.2005.08.029
C.R. Rambo, H. Sieber, Adv. Mater. 17, 1088 (2005). doi:10.1002/adma.200401049
M. Luo, J.Q. Gao, X. Zhang, G.Y. Hou, J.F. Yang, D. Ouyang et al., J. Mater. Sci. 42, 3761 (2007). doi:10.1007/s10853-006-0425-9
A.H. Murdoch, K.J. Mathias, D.E.T. Shepherd, Biomed. Mater. Eng. 14, 1 (2004)
A. Herzog, U.F. Vogt, S. Siegmann, O. Beffort, Adv. Eng. Mater. 8, 980 (2006). doi:10.1002/adem.200600121
C.R. Rambo, F.A. Muller, L. Muller, H. Sieber, I. Hofmann, P. Greil, Mater. Sci. Eng. C 26, 92 (2006). doi:10.1016/j.msec.2005.06.003
P. González, J. Serra, S. Liste, S. Chiussi, B. León, M. Pérez-Amor et al., Biomaterials 24, 4827 (2003). doi:10.1016/S0142-9612(03)00405-8
A. de Carlos, J.P. Borrajo, J. Serra, P. González, B. León, J. Mater. Sci. Mater. Med. 17, 523 (2006). doi:10.1007/s10856-006-8935-1
J. Colville, P. Baas, V. Hoikka, K. Vainio, IAWA Bull. 1, 3 (1979)
H. Kristen, P. Bosch, H. Bednar Jr, PLENK. Arch. Orthop. Unfallchir. 89, 1 (1977). doi:10.1007/BF00414821
K.A. Gross, E. Ezer`Ietis, J. Biomed. Mater. Res. 64A, 672 (2003). doi:10.1002/jbm.a.10437
A.J. Aho, J. Rekola, J. Matinlinna, J. Gunn, T. Tirri, P. Viitaniemi et al., J. Biomed. Mater. Res. 83B, 64 (2007)
S.N. Nazhat, E.A. Abou Neel, A. Kidane, I. Ahmed, C. Hope, M. Kershaw et al., Biomacromolecules 8, 543 (2007). doi:10.1021/bm060715f
I. Martin, D. Wendt, M. Heberer, Trends Biotechnol. 22(2), 80 (2004). doi:10.1016/j.tibtech.2003.12.001
F. Rose, Q.P. Hou, R.O.C. Oreffo, J. Pharm. Pharmacol. 56(4), 415 (2004). doi:10.1211/0022357023312
J.J.A. Barry, D. Howard, K.M. Shakesheff, S.M. Howdle, M.R. Alexander, Adv. Mater. 18, 1406 (2006). doi:10.1002/adma.200502719
P.X. Ma, Adv. Drug Deliv. Rev. 60, 1848 (2008). doi:10.1016/j.addr.2007.08.041
J.M. Taboas, R.D. Maddox, P.H. Krebsbach, S.J. Hollister, Biomaterials 24, 181 (2003). doi:10.1016/S0142-9612(02)00276-4
F.C.G. de Sousa, J.R.G. Evans, J. Am. Ceram. Soc. 86, 517 (2003). doi:10.1111/j.1151-2916.2003.tb03332.x
Y.H. Koh, H.W. Kim, H.E. Kim, J.W. Halloran, J. Am. Ceram. Soc. 85, 2578 (2002). doi:10.1111/j.1151-2916.2002.tb00500.x
S. Stokols, M.H. Tuszynski, Biomaterials 27, 443 (2006). doi:10.1016/j.biomaterials.2005.06.039
T.T. Yu, M.S. Shoichet, Biomaterials 26, 1507 (2005). doi:10.1016/j.biomaterials.2004.05.012
A.J. Wang, Q. Ao, W.L. Cao, M.Z. Yu, Q. He, L.J. Kong et al., J. Biomed. Mater. Res. 79A, 36 (2006). doi:10.1002/jbm.a.30683
C. Rey, B. Collins, T. Goehl, I.R. Dickson, M.J. Glimcher, Calcif. Tissue Int. 45, 157 (1989). doi:10.1007/BF02556059
E. Landi, A. Tampieri, G. Celotti, S. Sprio, J. Eur. Ceram. Soc. 20, 2377 (2000). doi:10.1016/S0955-2219(00)00154-0
H.W. Kim, L.H. Li, Y.H. Koh, J.C. Knowles, H.E. Kim, J. Am. Ceram. Soc. 87, 1939 (2004). doi:10.1111/j.1151-2916.2004.tb07493.x
Y.X. Sun, G.S. Guo, D.L. Tao, Z.H. Wang, J. Phys. Chem. Solids 68, 373 (2007). doi:10.1016/j.jpcs.2006.11.026
J.C. Hornez, F. Chai, F. Monchau, N. Blanchemain, M. Descamps, H.F. Hildebrand, Biomol. Eng. 24, 505 (2007). doi:10.1016/j.bioeng.2007.08.015
A. Stoch, W. Jastrzębski, E. Długoń, W. Lejda, B. Trybalska, G.J. Stoch et al., J. Mol. Struct. 744–747, 633 (2005). doi:10.1016/j.molstruc.2004.10.080
S.R. Ramanan, R. Venkatesh, Mater. Lett. 58, 3320 (2004). doi:10.1016/j.matlet.2004.06.030
C.B. Baddiel, E.E. Berry, Spectrochim Acta [A] 22, 1407 (1966)
S. Chakraborty, S. Bag, S. Pal, A.K. Mukherjee, J. Appl. Cryst. 39, 385 (2006). doi:10.1107/S0021889806010351
A. Sinha, S. Nayar, A. Agrawal, D. Bhattacharyya, P. Ramachandrarao, J. Am. Ceram. Soc. 86, 357 (2003). doi:10.1111/j.1151-2916.2003.tb00024.x
B. Su, G.Q. Zhang, X.D. Yu, C.T. Wang, J. Univ. Sci. Technol. Beijing 13(5), 469 (2006)
M. Markovic, B.O. Fowler, M.S. Tung, J. Res. Natl. Inst. Stand. Technol. 109, 553 (2004)
Y.J. Wang, X.J. Liu, K. Wei, S.H. Zhang, L. Ren, N.R. Zhao et al., J. Chin. Ceram. Soc 35, 1200 (2007)
J. Chakraborty, M.K. Sinha, D. Basu, J. Am. Ceram. Soc. 90, 1258 (2007). doi:10.1111/j.1551-2916.2007.01596.x
A. Bigi, E. Boanini, K. Rubini, J. Solid State Chem. 177, 3092 (2004). doi:10.1016/j.jssc.2004.05.018
M. Manso, M. Langlet, C. Jiménez, J.M. Martínez-Duart, Biomol. Eng. 19, 63 (2002). doi:10.1016/S1389-0344(02)00012-6
Q.X. Zhu, J.Q. Wu, J. Chin. Ceram. Soc 35(7), 866 (2007)
W.H. Emerson, E.E. Fischer, Arch. Oral Biol. 7, 671 (1962). doi:10.1016/0003-9969(62)90116-4
C.F. Li, F.T. Meng, Mater. Lett. 62, 932 (2008). doi:10.1016/j.matlet.2007.07.013
D.M. Liu, Q.Z. Yang, T. Troczynski, W.J. Tseng, Biomaterials 23, 1679 (2002). doi:10.1016/S0142-9612(01)00295-2
X.S. Dai, S. Shivkumar, Mater. Sci. Eng. C 28, 336 (2008). doi:10.1016/j.msec.2007.04.010
I.S. Kim, P.N. Kumta, Mater. Sci. Eng. B 111, 232 (2004). doi:10.1016/j.mseb.2004.04.011
M.F. Hsieh, L.H. Perng, T.S. Chin, H.G. Perng, Biomaterials 22, 2601 (2001). doi:10.1016/S0142-9612(00)00448-8
J.X. Zhang, H. Tanaka, F. Ye, D.L. Jiang, M. Iwasa, Mater. Chem. Phys. 101, 69 (2007). doi:10.1016/j.matchemphys.2006.02.016
P.E. Wang, T.K. Chaki, J. Mater. Sci. Mater. Med. 4, 150 (1993). doi:10.1007/BF00120384
S. Raynaud, E. Champion, D. Bernache-Assollant, P. Thomas, Biomaterials 23, 1065 (2002). doi:10.1016/S0142-9612(01)00218-6
T.K. Anee, M. Ashok, M. Palanichamy, S.N. Kalkura, Mater. Chem. Phys. 80, 725 (2003). doi:10.1016/S0254-0584(03)00116-0
J.P. Lafon, E. Champion, D. Bernache-Assollant, J. Eur. Ceram. Soc. 28, 139 (2008). doi:10.1016/j.jeurceramsoc.2007.06.009
D.M. Liu, T. Troczynski, W.J. Tseng, Biomaterials 23, 1227 (2002). doi:10.1016/S0142-9612(01)00242-3
G. Kordas, C.C. Trapalis, J. Sol–Gel Sci. Technol. 9, 17 (1997)
D.M. Liu, T. Troczynski, W.J. Tseng, Biomaterials 22, 1721 (2001). doi:10.1016/S0142-9612(00)00332-X
J.A.M. Van Der Houwen, G. Cressey, B.A. Cressy, E. Valsami-Jones, J. Cryst. Growth 249, 572 (2003). doi:10.1016/S0022-0248(02)02227-3
M.F. Hsieh, L.H. Perng, T.S. Chin, Mater. Chem. Phys. 74, 245 (2002). doi:10.1016/S0254-0584(01)00474-6
K. Hwang, J. Song, B. Kang, Y. Park, Surf. Coat. Tech. 123, 252 (2000). doi:10.1016/S0257-8972(99)00512-5
R.Z. Legeros, O.R. Trautz, J.P. Legeros, E. Klein, W.P. Shirra, Science 155, 1409 (1967). doi:10.1126/science.155.3768.1409
D.M. Liu, Q.Z. Yang, T. Troczynski, Biomaterials 23, 691 (2002). doi:10.1016/S0142-9612(01)00157-0
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 50603020 and No. 50773062) and the Program for New Century Excellent Talents in University (NCET-07-0673).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Qian, J., Kang, Y., Zhang, W. et al. Fabrication, chemical composition change and phase evolution of biomorphic hydroxyapatite. J Mater Sci: Mater Med 19, 3373–3383 (2008). https://doi.org/10.1007/s10856-008-3475-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10856-008-3475-5