
Abstract
In this work, we report a facile synthesis of mesoporous Co3O4 nanowires by solid-state thermal conversion of CoC2O4·2H2O precursors for supercapacitors. The formation mechanism of the ultra-long CoC2O4·2H2O nanowires is discussed based on a series of time-dependent experiments. Electrochemical measurements reveal that the mesoporous Co3O4 nanowires electrode exhibits a specific capacitance up to 250 F g−1 with a better capacity retention of 90.2% after 1000 continuous charge–discharge cycles at a constant current density of 5 A g−1. The good supercapacitor performance could be attributed to the unique mesoporous structure of the Co3O4 nanowires which provide fast ion and electron transfer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
As an important transition metal oxide, cobalt oxide (Co3O4) nanomaterials become one of the most attractive functional materials with intriguing physical and chemical properties [1], which is widely applied in the fields of lithium ion batteries, supercapacitors, electro-chemical sensors and energy storage devices [2]. It is well known that these properties of Co3O4 materials are strongly dependent on the structure and morphology [3]. Therefore, development of a morphologically controllable synthesis of Co3O4 nanostructures is urgently important to answer the demand for exploring the potentials of Co3O4.
Several synthesis methods including hydrothermal synthesis method, sol–gel method, precursor conversion processes, chemical vapor deposition synthesis, microwave assisted synthesis, and electrospinning technique have been reported to prepare multiple morphologies, such as nanowires, nanowalls, nanoflowers, nanorods and nanoflakes. Among these methods, a morphology-conserved transformation from precursor route has proven to be promising approach for the synthesis of Co3O4 nanostructures. Moreover, chemists and materialists demonstrated the existence of a close relationship between precursor and product through recent studies on solid-state pyrolysis of well-designed precursors into unique porous Co3O4 nanostructures [4].
As a solid precursor with easy synthesis, good structure stability, and relatively low decomposition temperature in air, cobalt oxalate is potential candidates that can be used as precursor to produce porous Co3O4 nanostructures [5]. The size and morphology of Co3O4 closely depend on that of the cobalt oxalate precursor, so developments of size and morphology controlled preparation methodologies are of great interest in materials chemistry [6]. In recent years, there has been an increasing interest in developing porous Co3O4 materials with one-dimensional (1D) nanostructures such as nanotubes, nanorods and nanowires due to their potential technological application in lithium ion batteries, supercapacitors and energy storage devices, so it remains a challenge to develop simple, fast, environmentally benign, and low-cost methods to achieve this goal.
In the present study, we first demonstrate a green method to synthesize ultra-long CoC2O4·2H2O nanowires in aqueous solution at room temperature under the assistance of sorbitan monopalmitate (Span 40). After calcinations in air, the as-prepared CoC2O4·2H2O nanowires subsequently convert to mesoporous 1D Co3O4 nanostructures while their original morphologies have been well maintained and their electrochemical properties are studied by cyclic voltammetry (CV) and galvanostatic charge–discharge techniques. The resulting mesoporous Co3O4 nanowires have been applied as electrodematerials for supercapacitors which exhibit high specific capacitances and excellent cycle stability.
2 Experimental section
2.1 Preparation of mesoporous Co3O4 nanowires
In the typical preparation, the mesoporous Co3O4 nanowires were prepared by solid-state thermal conversion of CoC2O4·2H2O precursors. Firstly, 0.291 g Co(NO3)2·6H2O and 0.05 g Span 40 were dissolved in a solution mixture of 25 ml distilled water and 25 ml ethylene glycol (EG) to form a transparent solution (A). Secondly, 0.5 g H2C2O4 was dissolved in another solution mixture of 25 ml distilled water and 25 ml EG to form a clear solution (B). Then the as-prepared solutions A and B were mixed together and stirred by a magnetic mixer at room temperature (around 25 °C) for 10 h. The pink cobalt oxalate precursors were collected by centrifugation and washed with distilled water and absolute ethanol for several times and dried in a vacuum oven at 50 °C for 6 h. Finally, the as-prepared intermediate products were calcined at 450 °C for 2 h in the air to prepare mesoporous Co3O4 nanowires. The chemical reactions involved in the formation of the mesoporous Co3O4 nanowires proceeding are as follows:
2.2 Structural and morphology characterization
The crystal structure of the products were evaluated by X-ray powder diffraction (XRD) using a D/Max-IIIA X-ray diffractometer with Cu Kα radiation (λ = 1.54056 Å) at a scanning rate of 0.02° S−1. X-ray photoelectron spectroscopy (XPS) spectrum was performed on a thermo ESCALAB 250 electron spectrometer with a monochromatic Al Kα X-ray source. Thermogravimetry and differential thermal analysis (TG-DTA) were performed on a NETZSCH-STA449 F3 simultaneous thermal analyzer under N2 atmosphere at a heating rate of 10 °C min−1 in a temperature range of 50–600 °C. Fourier transform infrared (FT-IR) spectrum was recorded with a TENSOR27 spectrometer in the range of 400–4000 cm−1 using a KBr wafer. The morphology and microstructure were characterized by scanning electron microscopy (SEM, Hitachi S4800) and transmission electron microscopy (TEM, Philips Tecnai G2 F20).
2.3 Electrode preparation and electrochemical characterization
The Co3O4 nanowires electrode was prepared by mixing active materials with acetylene black as a conductive agent, and polyvinylidene difluoride (PVDF) as binder in the proportions of 80:15:5 in N-methyl pyrrolidine (NMP). Then the mixture was homogeneously mixed and pressed into a nickel foam (1 cm × 1 cm) and vacuum dried at 80 °C for 12 h. The load weight of Co3O4 is about 10 mg cm−2. All electrochemical measurements were carried out at room temperature in a three-electrode system consisting of the Co3O4 nanowires electrode as working electrode, a saturated calomel electrode (SCE, Hg/HgCl2) as reference electrode, and a platinum plate (1 cm × 1 cm) as counter electrode. The used electrolyte was 4 mol L−1 KOH. Cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) tests were carried out at Metrohm Autolab PGSTAT302N electrochemical workstation.
3 Results and discussion
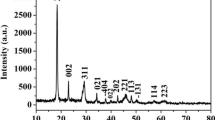
The crystalline structure of the cobalt oxalate precursor obtained at room temperature for 10 h is shown in Fig. 1. All the diffraction peaks could be indexed as orthorhombic β-CoC2O4·xH2O with lattice parameters of a = 11.87 Å, b = 5.419 Å and c = 15.624 Å, which are in good agreement with the standard card (JCPDS Card 25-0250) [7].

XRD pattern of the cobalt oxalate precursor
The composition and purity of the cobalt oxalate precursor was investigated by XPS. Figure 2a shows the XPS survey spectrum of as-synthesized cobalt oxalate. Based on peaks due to the binding energies, the elements in the compound are established. They are at 285 eV for C 1s, 531 eV for O 1s and 782 eV for Co 2p [8]. The spectrum of Co 2p (Fig. 2b) presents two main peaks of Co 2p3/2 and Co 2p1/2 binding energies at 781.2 and 797.2 eV respectively, and the other two peaks at 785.3 and 801.9 eV correspond to the satellite peaks of Co 2p3/2 and Co 2p1/2 binding energies respectively [8,9,10]. The O 1s spectrum (Fig. 2c) shows two peaks, one peak at 531.8 eV due to C=O, the other peak at 532.4 eV due to H2O [11]. The C 1s spectrum (Fig. 2d) consists of three peaks observed at around 284.5, 285.8 and 288.5 eV respectively, and they are both in a range compatible with oxalate anions. No impurities were found on the surface of the product, indicating that the as-synthesized cobalt oxalate nanowires are relatively pure.

XPS spectrum of the as-synthesized cobalt oxalate precursor: a Survey, b Co 2p, c O 1s, d C 1s
The decomposition of cobalt oxalate precursor has been investigated by TG-DSC analysis at a heating rate of 10 °C min−1 in flowing N2 atmosphere given in Fig. 3. From the TG curve, one can see that two weight loss steps in the temperature ranges 50–250 and 250–440 °C, respectively. The first one in the thermal decomposition of cobalt oxalate is a dehydration process. About 19.74% weight loss is obtained and this is in consistency with a calculated weight loss of 19.7%, which is attributed to the complete dehydration of two water molecules [12]. Therefore, the molecular formula is confirmed to be CoC2O4·2H2O. The anhydrous cobalt oxalate decomposed in the second step which indicates a weight loss of 49.68%, close to the calculated weight loss of 50.9% due to the formation of CoO in the N2 flow. However, the calcined product of the anhydrous cobalt oxalate will be Co3O4 if it is decomposed in an air flow [13]. From the DSC curve, the endothermic peaks of dehydration reaction and decomposition reaction occurred at 219 and 410 °C respectively were observed and accompanied the larger weight loss in TG curve.

TG-DSC graph for decomposition of CoC2O4·2H2O precursor
The chemical bonding of CoC2O4·2H2O was investigated by FT-IR analysis, as shown in Fig. 4. The as-prepared CoC2O4·2H2O·2H2O showed a strong absorption band at 1626 cm−1 is assigned to asymmetric γ (C–O) and the closely spaced bands at 1361 and 1317 cm−1 are assigned to symmetric γ (C–O), indicating the presence of bridging oxalates with all four oxygen atoms coordinated to the metal atoms. The asymmetric δ (O–C–O) band appears at 825 cm−1, while the broad band at 496 cm−1 is assigned to γ (Co–O) and symmetric δ (C–C–O) [14]. The band at 752 cm−1 is assigned to ν (O–Co–O) [15]. The broad band at 3367 cm−1 is assigned to the stretching vibration of the O–H group of molecular water [16]. It is therefore to confirm the chemical bonding characteristic of the obtained product. This result further confirmed the formation of CoC2O4·2H2O nanowires, in agreement with the XRD and TG-DSC results.

FT-IR spectra of the as-prepared CoC2O4·2H2O nanowires
The morphologies and structures of the as-prepared CoC2O4·2H2O precursor were examined by field emission scanning electron microscopy (FESEM) and transmission electron magnification (TEM) techniques. Figure 5a shows a representative low magnification FESEM image of the collected product. It reveals that the as-prepared cobalt oxalate precursor is composed of uniform nanowires and is in high-yield. Figure 5b is a high-magnification FESEM image for the precursor and it clearly shows that the nanowires have width of 50–250 nm and length of several micrometers. Figure 5c shows the typical TEM image of the as-prepared CoC2O4·2H2O nanowires. It was found that the sample showed nanowire structure, consistent with the SEM results. Figure 5d shows a TEM image of an individual CoC2O4·2H2O nanowire.

a Low-magnification SEM image and of the as-prepared CoC2O4·2H2O nanowires, b High-magnification SEM image of the as-prepared CoC2O4·2H2O nanowires, c Typical TEM image of CoC2O4·2H2O nanowires, d TEM image of a single nanowire
In our experiment, when no Span 40 was added, there was no CoC2O4·2H2O nanowires precipitated from the final solution. Instead, the product was aligned nanorods (Fig. 6a). Apparently, Span 40 plays a key role in the process of growing of the nanowires. The solvent also plays an important role in determining the final morphology of CoC2O4·2H2O crystals. In the absence of EG, the product was a large number of short rod-like structures (Fig. 6b). When isometric 1, 2-propylene glycol was substituted for EG, assemblies of nanorods were produced (Fig. 6c). 1, 2-propylene glycol with higher viscosity (EG 19.9 cP; 1, 2-propylene glycol 60.5 cP) restrained the diffusion of CoC2O4·2H2O nuclei and resulted in the formation of assemblies of nanorods. Comparatively, the mixture solution of water and EG favoured the reduction of the reaction rate and the formation of nanowires.

FESEM images of the cobalt oxalate precursor obtained at room temperature for 10 h: a Without Span 40, b In the absence of EG, c In the solution mixture of water and 1, 2-propylene glycol
In order to reveal the evolution process of the CoC2O4·2H2O nanowires, the intermediate products obtained at different stages were investigated by SEM analysis. Once H2C2O4 was added into the water/EG solution mixture of Co(NO3)2·6H2O and Span 40, a pink turbid suspension was formed immediately. After 10 min of reaction, the product exhibits bundle-like rods (Fig. 7a). When the reaction time reaches 20 min, the sample shows that the bundles begin to disassemble to rod-like structure (Fig. 7b). When the reaction has proceeded for 1 h, most of the products evolve into wires (Fig. 7c). After 10 h, ultra-long and uniform CoC2O4·2H2O nanowires were formed (Fig. 5a).

The evolution process monitored by FESEM technique: a 10 min, b 20 min, c 1 h, d Schematic diagram illustrating the formation process of CoC2O4·2H2O nanowires
Based on the above experimental results, a model for the formation of CoC2O4·2H2O nanowires is suggested. Figure 7d shows the schematic illustration for the possible formation process of CoC2O4·2H2O structure. The whole evolution can be divided in three main stages: (1) initial nucleating and successive formation of bundles of rod-like structures: Firstly, according to the Eq. (1), Co(NO3)2·6H2O reacted with H2C2O4 forming CoC2O4·2H2O molecular. Then the molecules formed CoC2O4·2H2O nuclei rapidly in the supersaturated solution and began to grow up which exhibited the bundles of rod-like structures. According to the literature [17], the C2O4 2− might act as a bidentate ligand in aqueous solution forming the stable complex. In the complex, the central Co atom was bonded by two C2O4 2− ions forming a planar molecule. Perpendicular to the molecular plane there were two coordinated H2O molecules. These complexes further constructed into rod-like structures. Furthermore, some rod-like structures are found in bundle-like assemblies, suggesting there exists a process of continuous nucleation attaching on the growing architecture for a period time at the early stage [18]. (2) The disassembly of the bundles of rod-like structures: As such structures were not stable, they gradually disassembled to single rod. (3) Crystal growth: With prolonged reaction time, ultra-long CoC2O4·2H2O nanowires were formed. The formation of nanowires is probably facilitated by the stabilization effect of nonionic surfactant Span 40 molecules, which is going to be along the sides of the linear structure of a single chain of cobalt oxalate dehydrate. Such mechanism is similar to the formation process of the previous report [19].
On the basis of the TG results, the temperature for calcinations of the cobalt oxalate precursor to Co3O4 was set at 450 °C for 2 h to ensure complete decomposition of the cobalt oxalate precursor.
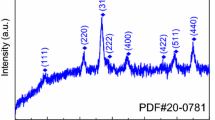
The typical XRD pattern of the as-obtained mesoporous Co3O4 nanowires after heat treatment is shown in Fig. 8. All the diffraction peaks of the XRD pattern are in accordance with the cubic phase of Co3O4 (space group Fd3m) with a = 8.080 Å (JCPDS No. 42-1467). No other impurities were detected in the XRD pattern, suggesting the high purity of the product.

XRD patterns of the as-obtained mesoporous Co3O4 nanowires from cobalt oxalate precursor
The XPS spectrum of the mesoporous Co3O4 nanowires is shown in Fig. 9. The Co 2p XPS spectrum exhibits two strong peaks at 794.4 and 779.3 eV, corresponding to Co 2p1/2 and Co 2p3/2 spin–orbit peaks of Co3O4, respectively (Fig. 9a) [20]. The deconvoluted O 1s spectrum displays one sharp peak at 529.6 eV and one broad peak at 530.9 eV, which may be assigned to the existence of –OH species adsorbed onto the surface of the Co3O4 nanowires (Fig. 9b).

XPS spectrum of as-prepared cobalt oxide: a Co 2p, b O 1s
The morphology and microstructure of the Co3O4 nanowires was observed by SEM and TEM shown in Fig. 10. It can be clearly seen that the as-obtained Co3O4 maintained the morphology of the CoC2O4·2H2O precursor and showed a 1D nanowire characteristic (Fig. 10a). The high-magnification SEM image in Fig. 10b further reveals that the Co3O4 nanowires have width of 100–200 nm and length up to several micrometers similar to those of the precursor. Moreover, the nanowires have unique mesoporous structure, which are constructed by many interconnected nanoparticles with diameter of about 20–30 nm.

a and b FESEM images of Co3O4 nanowires, c TEM image of Co3O4 nanowires, the inset is TEM image of a single Co3O4 nanowire, d The corresponding electron diffraction pattern, e SAED pattern obtained from a nanoparticle in an individual Co3O4 nanowire, f High-resolution TEM image obtained from the edge of an individual Co3O4 nanowire
Figure 10c shows the representative TEM image of the twin nanowires. The inset in Fig. 10c shows that a single Co3O4 nanowire has a diameter of 150 nm with small intercrystallite pores and rough surfaces, which agrees with SEM observations. This type of porous 1D structure is believed to be very suitable for supercapacitor applications. The corresponding SAED pattern shows that the as-prepared mesoporous Co3O4 nanowires are polycrystalline in nature (Fig. 10d). The diffraction rings in corresponding SAED pattern could be indexed as (111), (220), (311) and (400) reflections, indicating the formation of cubic phase of Co3O4, in agreement with the XRD result. The SAED pattern of a nanoparticle in the Co3O4 nanowire (Fig. 10e) clearly shows its single-crystalline structure and presents intense reflection spots of cubic structure Co3O4. Furthermore, the measured lattice fringe with the spacing of 0.285 nm is in good agreement with the (220) diffraction of cubic Co3O4 phase (Fig. 10f).
To study the specific surface area, Brunauer–Emmett–Teller (BET) gas-sorption measurements were performed. Figure 11 shows N2 adsorption–desorption isotherms of the Co3O4 nanowires carried out at 77 K, indicating the existence of pores on the surface of the nanowires. The BET surface area of the mesoporous Co3O4 nanowires is calculated to be about 18.2 m2 g−1 and a total pore volume of 0.11 cm3 g−1.

N2 adsorption–desorption isotherm of Co3O4 nanowires
The electrochemical performance of the as-prepared Co3O4 nanowire was investigated by CV, GCD and cycling life measurements. Figure 12a shows the CV curves of the mesoporous Co3O4 nanowires electrode at different scan rates of 1, 2, 5 and 10 mV s−1 in the potential range of 0–0.5 V. From CV curves, we can see that the anodic peaks shift toward large anodic potential and the cathodic peaks shift toward larger cathodic potential with the increasing of scan rate. Two typical redox peaks are clearly observed in each CV curve, which exhibit good supercapacitive behaviors of mesoporous Co3O4 nanowires, similar to previous reports [21]. In addition, the first redox couple P1/P4 corresponds to the change between Co3O4 and CoOOH, and the second redox couple P2/P3 can be ascribed to the conversion between CoOOH and CoO2 [22], which are well-defined good pseudocapacitive characteristics of the final product of Co3O4 caused by fast and reversible faradaic redox reaction of the materials. The reaction can be expressed by the following equations:

a CV curves of the mesoporous Co3O4 nanowires at various scan rates, b Average specific capacitances at various scan rates, c Galvanostatic discharge curves at various discharge current densities, d Corresponding specific capacitances at various discharge current densities, e Galvanostatic charge and discharge voltage profiles at a current density of 5 A g−1, f Cycling performance at a current density of 5 A g−1
The specific capacitance (C, F g−1) values in the CV measurement are calculated using the following equation:
where Q (C) is the average charge during anodic and cathodic scan, m (g) is the mass of the active material and △V (V) is the applied potential window. The specific capacitance can be calculated from the CV curves to be 366, 270, 165 and 101 F g−1 at the scanning rates of 1, 2, 5 and 10 mV s−1, respectively (Fig. 12b). It can be clearly seen that the specific capacitance values decrease with the increase of scan rate. As is known, at lower scan rates, the OH− ions intercalate well with the both outer and inner surface of the electrode [23]. But at high scan rates, the diffusion process restricts the OH− ions movement, so the OH− ions can only reach the outer surface of the electrode and not enter into the interior pores. Therefore the effective utilization of the electroactive material has been greatly restricted, leading to the decrease of the specific capacitance [24].
Figure 12c shows the constant-current discharge curves of the as-prepared Co3O4 nanowire electrode using 4 mol L−1 KOH as electrolyte in the potential range of 0–0.54 V at various galvanostatic current densities. The specific capacitance of the Co3O4 nanowires can be calculated based on the following equation:
where C (F g−1) is the specific capacitance, I (mA) is charge–discharge current, △t (s) is the discharge time, △V (V) is the potential drop in the galvanostatic discharge process, and m (mg) is the loading mass of active material within the electrode. Therefore, the specific capacitances calculated from each discharge curve were 271, 264, 207 and 164 F g−1 at current sensities of 2, 5, 10 and 20 A g−1, respectively (Fig. 12d). It can be seen that the discharge specific capacitance of the electrode is decreased with increase in current density, which may be due to the increased internal polarization at higher current densities [25].
Figure 12e shows the GCD test of the mesoporous Co3O4 nanowires electrode at a current density of 5 A g−1 for the first 6 cycles. The cycle stability of the electrode was investigated by repeating the GCD test for 1000 cycles shown in Fig. 12f. After 1000 cycles, the specific capacitance gradually decreased from 277 to 250 F g−1. Remarkably, the capacitance still remained 90.2% of the initial capacitance, demonstrating good electrochemical cyclability of the synthesized mesoporous Co3O4 nanowires. The cycling characteristic is much better than mesoporous Co3O4 crater-like microspheres prepared by Wang [26], which exhibits a cycling stability with 26% capacitance loss after 500 cycles at a sweep rate of 3 mV s−1. It may be due to the unique porous structure of the Co3O4 nanowires which affords a short diffusion path for both ions and electrons transport within the electrode–electrolyte interface to enhance the redox kinetics.
4 Conclusions
In summary, mesoporous Co3O4 nanowires have been successfully synthesized on a large scale via a green method. Electrochemical measurements indicate that the mesoporous Co3O4 nanowires could deliver a specific capacitance of 250 F g−1 at a current density of 5 A g−1 and offer an excellent capacitance retention of 90.2% at 1000 cycles. The encouraging results confirm that the mesoporous Co3O4 nanowires can provide large specific capacitance, good cyclic performance and high electrochemical stability, which may have potential applications as anode materials for supercapacitors. This environmentally benign methodology of mesoporous Co3O4 nanowires may be extended to the synthesis of other transitional metal oxides systems.
References
P. Poizot, S. Laruelle, S. Grugeon, L. Dupont, J.M. Tarascon, Nature 407, 496 (2000)
X.W. Xie, Y. Li, Z.Q. Liu, M. Haruta, W.J. Shen, Nature 458, 746 (2009)
H. Goesmann, C. Feldmann, Angew. Chem. Int. Ed. 49, 1362 (2010)
X. Wang, X.L. Wu, Y.G. Guo, Y.T. Zhong, X.Q. Cao, Y. Ma, J.N. Yao, Adv. Funct. Mater. 20, 1680 (2010)
H.P. Cong, S.H. Yu, Cryst. Growth Des. 9, 210 (2009)
N. Du, Y.F. Xu, H. Zhang, C.X. Zhai, D.R. Yang, Nanoscale. Res. Lett. 5, 1295 (2010)
L. Ren, P.P. Wang, Y.S. Han, C.W. Hu, B.Q. Wei, Chem. Phys. Lett. 476, 78 (2009)
B.J. Tan, K.J. Klabunde, P.M.A. Sherwood, J. Am. Chem. Soc. 113, 855 (1991)
Z. Xing, X. Ji, Y.L. Zhao, H.P. Ren, Y.C. Deng, Z.C. Ju, Q.C. Zhuang, J. Mater. Chem. A 5, 8714 (2017)
Z.C. Ju, G.Y. Ma, Y.L. Zhao, Z. Xing, Y.H. Qiang, Y.T. Qian, Part. Part. Syst. Charct. 32, 1012 (2015)
M. Bazzaoui, L. Martins, E.A. Bazzaoui, J.I. Martins, J. Electroanal. Chem. 537, 47 (2002)
M.A. Gabal, A.A. El-Bellihi, S.S. Ata-Allah, Mater. Chem. Phys. 81, 84 (2003)
X. Wang, X.Y. Chen, L.S. Gao, H.G. Zheng, Z.D. Zhang, Y.T. Qian, J. Phys. Chem. B 108, 16401 (2004)
Z.P. Xu, H.C. Zeng, Chem. Mater. 12, 3459 (2000)
H. Luo, D.B. Zou, L.G. Zhou, T.K. Ying, J. Alloys Compd. 481, L12 (2009)
H.L. Du, J.Z. Wang, B. Wang, D.Q. Cang, Powder Technol. 199, 149 (2010)
X.L. Li, J.F. Liu, Y.D. Li, Mater. Chem. Phys. 80, 222 (2003)
B.G. Wang, H. Lin, Z.G. Yin, Mater. Lett. 65, 41 (2011)
Y.D. Li, H.W. Liao, Y. Ding, Y. Fan, Y. Zhang, Y.T. Qian, Inorg. Chem. 38, 1382 (1999)
S.L. Xiong, C.Z. Yuan, X.G. Zhang, B.J. Xi, Y.T. Qian, Chem. Eur. J. 15, 5320 (2009)
D.W. Wang, Q.H. Wang, T.M. Wang, Inorg. Chem. 50, 6482 (2011)
F. Zhang, L. Hao, L.J. Zhang, X.G. Zhang, Int. J. Electrochem. Sci. 6, 2943 (2011)
U.M. Patil, K.V. Gurav, V.J. Fulari, C.D. Lokhande, O.S. Joo, J. Power Sources 188, 338 (2009)
V. Subramanian, H. Zhu, R. Vajtai, P.M. Ajayan, B. Wei, J. Phys. Chem. B 109, 20207 (2005)
H.Y. Mi, X.G. Zhang, X.G. Ye, S.D. Yang, J. Power Sources 176, 403 (2008)
L. Wang, X.H. Liu, X. Wang, X.J. Yang, L.D. Lu, Curr. Appl. Phys. 10, 1422 (2010)
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (Project Number XDJK2016C003; XDJK2016E001, for Innovation and Entrepreneurship Students), the Foundation of Chongqing Municipal Education Commission (KJ1711292), and the Ninth Undergraduate Science and Technology Innovation (Project 20162303008).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, Y., Wu, H., Wu, Y. et al. Facile synthesis of mesoporous Co3O4 nanowires for application in supercapacitors. J Mater Sci: Mater Electron 28, 16826–16835 (2017). https://doi.org/10.1007/s10854-017-7598-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-017-7598-7