
Abstract
Mesoporous α-Fe2O3 nanowires have been prepared through solid-state thermal conversion of ferrous oxalate dihydrate precursor for supercapacitor application. The possible growth mechanism of the FeC2O4·2H2O nanowires was proposed based on a series of time-dependent experiments. The specific surface area and pore size distribution of the mesoporous α-Fe2O3 nanowires were calculated to be about 70.6 m2 g−1 and 2.5 nm, respectively. Furthermore, electrochemical measurements demonstrate that the as-prepared mesoporous α-Fe2O3 nanowire electrode delivers a high specific capacitance up to 267.5 F g−1 at 2 A g−1 and good cycle performance (87% capacitance retention under 2000 cycles). The excellent supercapacitor performance of the α-Fe2O3 nanowires can be ascribed mainly to the unique mesoporous structure with large specific surface area, which provide fast electron/ion transfer path as well as large reaction surface area.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Development of alternative and sustainable energy storage and conversion devices with high efficiency, stability, and environmental friendliness has been urgently demanded in today’s human society due to the exhaustible fossil energy and serious environmental pollution [1,2,3,4,5,6,7]. As a kind of energy storage and conversion equipment, supercapacitors (SCs) with the high power density, fast charge–discharge process, long cycle stability, low cost, and good environmental friendliness have become promising candidates for portable and mobile-based applications [8], which have been widely used in the fields of notebook computers, electrical vehicles, backup power devices, and so on [9, 10].
Among various transition metal oxides, hematite (α-Fe2O3) remains one of the most promising SCs electrode materials owing to its non-toxic, environmental friendliness, stability in nature, inexpensive, more abundant, as well as more appropriate working voltage of negative potential (− 1.0–0 V) and higher theoretical specific capacitance (3265 F g−1) [11, 12]. However, as a pseudocapacitive material for supercapacitor application, α-Fe2O3 still has some limitations in low electrical conductivity (10−14 Scm-1) and poor ion diffusion rate, which cause the practical capacitance far below theoretical specific capacitance [13, 14].
It is well known that high capacitance is almost ascribed to high electrochemical activity, suitable structure, and large specific surface area of the working electrode materials, which lead to a fast electron/ion transfer rate as well as large electrochemical active surface area. Therefore, a great many researchers and scientists focus on tailoring Fe2O3 materials with various morphologies including nanoparticles nanorods, nanowires, nanocubes, and mesoporous structures to improve the charge storage capacity, which could shorten transmission path and accelerate ion and electron transfer effectively [15,16,17,18]. However, developing α-Fe2O3 electrode materials with high capacitance performance and good cycle stability still remains a challenge.
Herein, we demonstrate a facile solution method to prepare one-dimensional (1D) FeC2O4·2H2O nanowires under the assistance of ethylene glycol (EG). The as-prepared FeC2O4·2H2O nanowires subsequently convert to mesoporous α-Fe2O3 nanowires after calcinations in air, while their original shapes are maintained well during the conversion. The electrochemical performance of the as-prepared mesoporous α-Fe2O3 nanowires has been analyzed through cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS) measurements. Interestingly, the obtained mesoporous α-Fe2O3 nanowires display excellent pseudocapacitive performance containing high specific capacitance and long cycle stability indicating that mesoporous α-Fe2O3 nanowires have a great application prospect as electrode materials for SCs.
2 Experimental section
2.1 Preparation of mesoporous α-Fe2O3 nanowires
The mesoporous α-Fe2O3 nanowires were synthesized via solid-state thermal conversion of FeC2O4·2H2O precursors. In a typical procedure, 0.199 g FeCl2·4H2O was added in a solution mixture containing 10 ml distilled water and 20 ml EG by magnetic stirring to form a transparent solution (A) and then 0.5 g H2C2O4 was added in another solution mixture containing 10 ml distilled water and 20 ml EG to form a transparent solution (B). Then, solutions A and B were mixed together and stirred vigorously at room temperature for 20 min. After that, the mixture should be kept in 50 °C water bath for 4 h under static conditions. The yellow ferrous oxalate dehydrate precursors were collected via centrifugation, washed with distilled water and absolute ethanol for three times, respectively, followed by drying in a vacuum oven at 60 °C for 5 h. Finally, the as-prepared mesoporous α-Fe2O3 nanowires were obtained by calcining the precursor in air at 450 °C for 2 h. The chemical reactions involved in the formation of the mesoporous α-Fe2O3 nanowires have been described as follows:
3 Structural and morphology characterization
The crystallographic structure and phase purity of the products were investigated by X-ray diffraction (XRD) utilizing a D/Max-IIIA X-ray diffractometer equipped with Cu Kα radiation. X-ray photoelectron spectroscopy (XPS) spectrum was measured on a thermo ESCALAB 250 electron spectrometer. The morphology and microstructure of the samples were examined by means of field emission scanning electron microscopy (FESEM, Hitachi S4800) and transmission electron microscopy (TEM, Philips Tecnai G2 F20). Thermogravimetry and differential thermal analysis (TG–DTA) tests were characterized by NETZSCH-STA 449 F3 simultaneous thermal analyzer under N2 atmosphere. Fourier transform infrared (FT-IR) spectrum was determined by TENSOR 27 spectrometer. N2 adsorption–desorption isotherm was measured by surface area analyzer (NOVOE 4000). The specific surface area and average pore size distribution were studied through Brunauer–Emmett–Teller (BET) principle and Barrett–Joyner–Halenda (BJH) analysis, respectively.
4 Electrode preparation and electrochemical characterization
The mesoporous α-Fe2O3 nanowires as electrode material applied for electrochemical characterization was prepared through mixing the active material, polyvinylidene fluoride (PVDF), and acetylene black as binder with 1-methyl-2-pyrrolidone (NMP) in the proportions of 75:15:10. Then, the mixture was mixed homogeneously, coated on a piece of Ni foam (1 cm × 1 cm), and then vacuum dried at 80 °C for 20 h. The mesoporous α-Fe2O3 nanowires electrode contained active material of about 3 mg cm−2. All the electrochemical characterizations were tested in a three-electrode system at room temperature composed of mesoporous α-Fe2O3 nanowires electrode as working electrode, Pt foil (1 cm × 1 cm) as counter electrode, as well as saturated calomel electrode (SCE, Hg/HgO) for reference electrode. Electrochemical measurements consisting of cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) were analyzed in 1-mol L−1 KOH aqueous solution by CHI 760e electrochemical workstation. Electrochemical impedance spectroscopy (EIS) was obtained between the frequency range of 10−2 and 105 Hz at open-circuit voltage with an amplitude of 5 mV.
5 Results and discussion
The crystallographic structure of the ferrous oxalate precursor prepared in water bath at 50 °C for 4 h is displayed in Fig. 1. All the diffraction peaks can be well indexed to pure FeC2O4·2H2O with lattice parameters of a = 12.06 Å, b = 5.55 Å, and c = 9.804 Å, which are in good correspondence with the standard card (JCPDS Card 72-1305). The characteristic diffraction peaks of ferrous oxalate with 2θ values at 18.36°, 22.93°, 29.28°, 34.25°, 37.78°, 39.95°, 42.67°, 45.88°, 48.14°, 50.21°, 57.20°, and 61.30° are in good agreement with the crystal planes of (200), (002), (311), (021), (−404), (022), (202), (221), (113), (−131), (114), and (223), respectively. Evidently, there are no diffraction peaks from impurities produced in the sample, indicating the high purity of the ferrous oxalate precursors.

XRD pattern of the FeC2O4·2H2O precursor
The decomposition of ferrous oxalate dihydrate precursor was studied by TG–DSC analysis with a heating rate of 2 °C min−1 from 50 to 800 °C in flowing N2 atmosphere as shown in Fig. 2. From the TG curve, it is observed two weight loss steps in the range 130–200 °C and 300–440 °C, respectively. The first step in the thermal decomposition of ferrous oxalate dihydrate is a dehydration process, which is represented by a remarkable exothermic peak at 170 °C in the DSC curve. And about 19.74% weight loss is obtained, which is in agreement with the expectation of the complete dehydration of two H2O molecules [19, 20]. The anhydrous FeC2O4 is decomposed in the second step, which is represented by a remarkable exothermic peak at 386 °C in the DSC curve. The decomposition weight loss of about 49.68% is in a good agreement with the formation of α-Fe2O3 in the N2 flow, which is further verified by the XRD pattern coming from the final sample of the decomposition.

TG–DSC graph for the decomposition of FeC2O4·2H2O precursor
The chemical bonding of the obtained FeC2O4·2H2O was examined by FT-IR spectra, as exhibited in Fig. 3. It is noted that a strong absorption peak centered at 1627 cm−1 is assigned to C=O stretching vibration. And the peaks at 1363 and 1317 cm−1 are related to C–O asymmetric and symmetric vibrations, respectively, suggesting the existence of bridging oxalates in association with all four oxygen atoms coordinated to the metal atoms. The asymmetric δ (O–C–O) absorption band locates at 822 cm−1, while the broad peak at 495 cm−1 is ascribed to Fe–O vibration [21]. Additionally, the presence of the broad band at 3356 cm−1 is ascribed to the stretching vibration of the O–H group in H2O [22, 23]. These results further verify the formation of FeC2O4·2H2O, which is in correspondence with the XRD and TG–DSC results.

FT-IR spectra of the FeC2O4·2H2O precursor
The morphologies and microstructures of FeC2O4·2H2O precursor were observed by FESEM and TEM tests. Figure 4a exhibits a representative low magnification FESEM image of the as-prepared product, which clearly reveals that the FeC2O4·2H2O precursor is composed of uniform nanowires. Figure 4b displays a high-magnification FESEM image of the FeC2O4·2H2O nanowires, which clearly demonstrates that the nanowires have a diameter of 150–450 nm with a length of several micrometers. Figure 4c and d displays the TEM images of FeC2O4·2H2O nanowires. It is obvious that the sample exhibits nanowire morphology, which is in correspondence with the SEM results.

a and b SEM images of the as-prepared FeC2O4·2H2O nanowires; c and d TEM images of FeC2O4·2H2O nanowires
In this study, solvents play a key role in controlling the morphology of FeC2O4·2H2O precursor. When no EG was added, the product was composed of irregular rod-like structures with obvious cracks (Fig. 5a). When isometric 1,2-propylene glycol replaced EG, a large number of rods with the diameter of 300–1000 nm and length of 5–8 μm were produced (Fig. 5b). Generally, 1,2-propylene glycol has higher viscosity than EG (1,2-propylene glycol 60.5 cP; EG 19.9 cP), which reduced the diffusion of FeC2O4·2H2O nuclei and growth of nanocrystals, thus resulting in the generation of rods. Evidently, the mixture solution of EG and deionized water favored the reaction rate and the growth of 1D nanowires.

FESEM images of the FeC2O4·2H2O precursor obtained in water bath at 50 °C for 4 h: a in the absence of EG and b in the solution mixture of H2O and 1,2-propylene glycol; the evolution process is monitored by FESEM test in the solution mixture of H2O and EG: c 1 h and d 2 h; e schematic diagram illustrating the growth process of FeC2O4·2H2O nanowires
In order to investigate the formation process of FeC2O4·2H2O nanowires, the morphology of the intermediate samples obtained at different reaction stages were observed through SEM analysis. When H2C2O4 solution was added to EG/water solution mixture of FeCl2·4H2O, a yellow turbid suspension generated immediately. 1 h later, the product consisted of short rod-like structures with the diameter of 500–1000 nm and length of 5–10 μm (Fig. 5c). After 2 h of reaction, it can be seen that that most of the rod-like structures began to evolve into wires (Fig. 5d). When the reaction time was 4 h, uniform FeC2O4·2H2O nanowires finally formed (Fig. 4a).
According to the above experimental results, a plausible growth mechanism for FeC2O4·2H2O nanowires is proposed. Figure 5e exhibits the schematic illustration for the formation process of FeC2O4·2H2O nanowires. The whole evolution could be separated into three stages: (1) nucleation and successive formation of rod-like structures: First, based on Eq. (1), H2C2O4 was dissociated into C2O42− oxalate anions. Subsequently, Fe2+ reacted with C2O42− in the EG/water solution mixture generating FeC2O4·2H2O molecular, which formed FeC2O4·2H2O nuclei quickly in the supersaturated solution. Whereas, the H2O molecules coordinated FeC2O4 since they have the high ligand field strength [24]. Then, the FeC2O4·2H2O nuclei began to grow up and formed rod-like structures. As explained in the previous literature [25], the C2O42− ions in aqueous solution may act as a bidentate ligand to generate the stable complex. In the complex, the central Fe atom was bonded by two C2O42− ions to produce a planar molecule. And perpendicular to the molecular plane there were two coordinated water molecules. As a result, the complex was further constructed into rod-like structure. (2) Crystal growth: When the reaction time was proceeded, 1D FeC2O4·2H2O nanowires were finally formed. The growth of nanowires is presumably facilitated by the stabilization effect of EG molecules, which will go to be along the sides of the linear structure of a single chain of FeC2O4·2H2O. Such formation mechanism is analogous to the growth process of the reported literature [26].
Thermal decomposition of the precursor is a common and facile route to prepare metal oxides. According to the TG result, the calcination temperature of the FeC2O4·2H2O precursor was set as 450 °C for 2 h to guarantee complete decomposition of FeC2O4·2H2O precursor to α-Fe2O3.
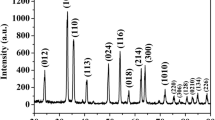
The XRD pattern of the as-prepared mesoporous α-Fe2O3 nanowires is depicted in Fig. 6a. All the diffraction peaks could be in agreement with the hexagonal phase α-Fe2O3 (hematite, JCPDS No. 33-0664) with lattice constants of a = b = 5.04 Å as well as c = 13.75 Å. The three strong peaks with 2θ values of 33.15°, 35.58°, and 54.09° correspond to the crystal planes of (104), (110), and (116) of the hexagonal phase α-Fe2O3, respectively. No impurities phase was detected.

XRD pattern of the as-prepared mesoporous α-Fe2O3 nanowires from FeC2O4·2H2O precursor. XPS spectra of as-prepared mesoporous α-Fe2O3 nanowires: a survey; b Fe 2p; and c O 1s
The composition and purity of the mesoporous α-Fe2O3 nanowires were researched by XPS test. Figure 6b displays XPS survey spectrum of as-obtained α-Fe2O3. According to the binding energies of the peaks, the elements in the compound have been established, such as 284 eV for C 1s, 528 eV for O 1s, as well as 709 eV for Fe 2p. The spectrum of Fe 2p (Fig. 6c) exhibits two distinct peaks for Fe 2p3/2 and Fe 2p1/2 binding energies corresponding to 710.7 and 724.4 eV, respectively. It can be found that an energy distance between the peaks of Fe 2p3/2 and Fe 2p1/2 is about to be 13.7 eV, which is in accordance with the reported value of Fe2O3 [27]. And the other two peaks at 716.4 and 731.6 eV correspond to the shake-up satellite peaks of Fe 2p3/2 and Fe 2p1/2 binding energies, respectively, indicating the existence of Fe3+ in Fe2O3 [28]. Such result is also consistent with the XRD characterization [29]. The spectrum of O 1s (Fig. 6d) exhibits two distinct peaks at the binding energies of 529.4 and 531 eV, which can be ascribed to the lattice oxygen (Olatt) and hydroxyl oxygen (Ohyd), respectively [30].
The morphology and microstructure of the mesoporous α-Fe2O3 nanowires were observed via SEM and TEM tests, as depicted in Fig. 7. It is noted that the as-prepared α-Fe2O3 retained the morphology of FeC2O4·2H2O precursor and exhibited 1D wire-like characteristic (Fig. 7a). Figure 7b displays high-magnification SEM images, which further reveal the α-Fe2O3 nanowires with a width of 100–200 nm as well as length up to several micrometers. Moreover, by careful observation from the SEM image, it is obvious that the α-Fe2O3 nanowires possess unique mesoporous loose structures, which are assembled through many interconnected small nanoparticles, due to the removal of H2O and CO2 during the pyrolysis process.

a, b FESEM images of mesoporous α-Fe2O3 nanowires; c TEM image of α-Fe2O3 nanowires; d TEM image of a single α-Fe2O3 nanowire. The inset is the corresponding electron diffraction pattern; e SAED pattern obtained from a nanoparticle in an individual α-Fe2O3 nanowire; f high-resolution TEM image of an individual α-Fe2O3 nanowire
Figure 7c exhibits the representative TEM image of α-Fe2O3 nanowires. Figure 7d clearly demonstrates that a single α-Fe2O3 nanowire possesses a diameter of 170 nm with small intercrystallite pore and coarse surface, which consists with the SEM observations. The inset in Fig. 7d exhibits the corresponding selected area electron diffraction (SAED) pattern, suggesting the polycrystalline nature of the as-obtained mesoporous α-Fe2O3 nanowires. The diffraction rings in the SAED pattern can be indexed as (012), (104), (024), (214), and (300) reflections, respectively, implying the formation of hexagonal phase α-Fe2O3, in accordance with the XRD analysis. Figure 7e clearly shows the corresponding SAED pattern of a nanoparticle in the α-Fe2O3 nanowire, which indicates its single-crystalline structure and also presents intense reflection spots of hexagonal phase α-Fe2O3. Figure 7f is the high-resolution TEM (HRTEM) image, and the observed lattice spacing of 0.27 nm is well matched with the (104) diffraction of hexagonal phase α-Fe2O3.
Figure 8a displays a type IV characteristic of N2 adsorption–desorption isotherms for the α-Fe2O3 nanowires performed at 77 K, suggesting the presence of pores on the surface of the α-Fe2O3 nanowires. The specific surface area of the mesoporous α-Fe2O3 nanowires is measured to be 70.6 m2 g−1, which is much higher than the Fe2O3 nanosheets (13.1 m2 g−1), pure α-Fe2O3 powder (14.3 m2 g−1), and α-Fe2O3/rGO (18.3 m2 g−1) reported in the previous literatures [31, 32]. The average pore size distribution of the α-Fe2O3 nanowires shown in Fig. 8b is calculated to be 2.5 nm with the total pore volume of 0.111 cm3 g−1, which is the optimal pore size for ions diffusion and electrons transition within active electrode materials [33]. Apparently, high surface area, ample pore structure, as well as proper pore volume could be expected to form more active sites and quicken the interface of active electrode materials with the electrolyte solution, thus causing a significant enhancement of the electrochemical performance [34, 35]. Such characteristics of the mesoporous α-Fe2O3 nanowires are critical for the redox reaction, which enable easy access of electrons and ions between the electrode/electrolyte interface.

a N2 adsorption–desorption isotherm of mesoporous α-Fe2O3 nanowires; b the pore size distribution curve of mesoporous α-Fe2O3 nanowires
The electrochemical performance of the as-obtained α-Fe2O3 nanowires as electrode materials for SCs was examined by means of CV, GCD, and EIS measurements. CV test is a common technique favored to research the capacitive characteristic of any electrode materials. Figure 9a represents a series of CV curves at different scan rates of 5, 10, 20, 50, 75, and 100 mV s−1 in the potential range of − 1.2 to – 0.2 V. Notably, two typical redox peaks were observed clearly in all the CV curves, implying that the capacitance behavior of the mesoporous α-Fe2O3 nanowires electrode are mainly dominated by Faradaic redox reactions, probably corresponding to the reversible conversion between Fe3+/Fe2+ [36]. The shape of the CV curve changes at higher scan rate, suggesting the slower reaction kinetics at relatively high scan rate. The electrode reaction of as-prepared α-Fe2O3 nanowires is represented as follows:

a CV curves of the mesoporous α-Fe2O3 nanowires at different scan rates. b Average specific capacitances at different scan rates. c Galvanostatic discharge curves at various discharge current densities. d Corresponding specific capacitances at different discharge current densities. e Cycling performance at a current density of 2 A g−1 after 2000 cycles. The inset is the corresponding charge/discharge curves for the first 8 cycles. f Nyquist plots of the mesoporous α-Fe2O3 nanowires
The specific capacitance (C, F g−1) from the CV curves can be calculated by the following equation:
where Q (C) represents the average charge during anodic and cathodic scan, m (g) designates the mass of the active material, and △V (V) indicates the potential window. The specific capacitance at different scan rates of 5, 10, 20, 50, 75, and 100 mV s−1 from CV curves can be calculated to be about 459.3, 354.6, 242, 142.7, 124.6, and 104.5 F g−1, respectively (Fig. 9b). It is found that the specific capacitance decreases with the increase of the scan rate, which is a typical characteristic of the electrochemical system resulting from the diffusion limit of the electrolyte ions. At lower scan rates, the electrolyte ions have enough time to diffuse and could attain access to the maximum surface area of the electroactive material, which deliver a higher specific capacitance [37]. At higher scan rates, the effective interaction between electrolyte electrode and ions is restricted with increasing the scan rates, which results in a decrease of the specific capacitance [38].
Figure 9c demonstrates the GCD tests of the as-prepared α-Fe2O3 nanowires electrode at different current densities from 1 to 10 A g−1 with the potential range of − 1.0 to − 0.2 V. The specific capacitance of the mesoporous α-Fe2O3 nanowires can be computed according to the following equation:
where C (F g−1) indicates specific capacitance, I (mA) stands for charge–discharge current, △t (s) refers to the discharge time, △V (V) represents the potential change in the galvanostatic discharge process, and m (mg) means the mass loading of active material on the working electrode. So, the specific capacitance at current densities of 1, 2, 3, 4, 5, 8 and 10 A g−1 from each discharge curve was about 330, 267.5, 210, 170, 137.5, 110, and 99.6 F g−1, respectively (Fig. 9d). It can be seen that the specific capacitance of the mesoporous α-Fe2O3 nanowires electrode gradually decreases along with the increase in the current density, which can be owing to the truth that some active surface of the active material grows inaccessible for charge storage at higher current density [39]. Comparison of the specific capacitances between the present work and other α-Fe2O3 electrodes reported in earlier literatures is represented in Table 1 [32, 40,41,42,43,44,45,46,47,48,49,50,51,52,53]. The specific capacitance of the mesoporous α-Fe2O3 nanowires is verified to be among the best, indicating that the design of the mesoporous α-Fe2O3 nanowires electrode with a mesoporous structure is highly efficient for the applications of SCs.
Figure 9e represents the cycling performance of the mesoporous α-Fe2O3 nanowires carried out through continuous GCD tests at 2 A g−1 for 2000 cycles with the potential range of −1.0 to −0.2 V. It is noted that the specific capacitance is gradually reduced from 267.5 to 233.5 F g−1 after 2000 cycles, which maintains 87% of the initial capacitance, suggesting excellent long-term electrochemical cyclability of the obtained α-Fe2O3 nanowires. The cycling stability of the mesoporous α-Fe2O3 nanowires is much better than porous flower-like α-Fe2O3 nanostructures obtained by Munichandraiah [49], which presents the cycling stability of 79.5% capacitance retention after 1000 cycles at 1 A g−1. It could be owing to the unique mesoporous 1D structure and high surface area of the α-Fe2O3 nanowires, which can afford a short diffusion path for electrons/ions transport between the electrode/electrolyte interface to improve the redox kinetics.
EIS measurements were performed further to investigate the electrochemical behaviors of the mesoporous α-Fe2O3 nanowires. Figure 9f displays the Nyquist plot of the mesoporous α-Fe2O3 nanowires in the frequency range of 10−2–105 Hz measured at an open-circuit voltage state. The EIS data were well fitted with Zview software utilizing equivalent circuit model as exhibited in the inset of Fig. 9f, which is in accordance with the measured one. Noticeably, the impedance spectrum consists of a semicircle in high-frequency region and a straight line in low-frequency region, indicating a typical capacitor behavior. In the high-frequency region, the intercept on the real axis reflects the resistance of the electrolyte (Rs), while the diameter of the small semicircle represents the charge–transfer resistance (Rct). The Rs and Rct of the as-prepared mesoporous α-Fe2O3 nanowires were measured to be 1.5 and 1.4 Ω, respectively, which are much lower than other morphologies of α-Fe2O3 reported in previous literatures [54,55,56], such as hollow microboxes, nanoplates, and nanorods., indicating that the mesoporous α-Fe2O3 nanowires electrode has good intrinsic electrical conductivity for SCs. Additionally, the mesoporous α-Fe2O3 nanowires exhibit a vertical curve in the low-frequency region, implying the rapid ion diffusion in electrolyte as well as adsorption on the surface of the electrode. It facilitates the effective access to the electrode for electrolyte ions, which can help to achieve a high pseudocapacitance.
6 Conclusion
In summary, mesoporous α-Fe2O3 nanowires were successfully prepared in high yield through solid-state thermal conversion of ferrous oxalate dihydrate precursor for supercapacitor application. Electrochemical performance indicates that the mesoporous α-Fe2O3 nanowires exhibits a high specific capacitance up to 267.5 F g−1 at 2 A g−1 with a good capacitance retention of 87% after 2000 cycles, which could have great potential applications for SCs. Moreover, the facile and environmentally friendly methodology to synthesize mesoporous α-Fe2O3 nanowires can be well extended to the fabrication of other transitional metal oxides.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
M.F. EL-Kady, V. Strong, S. Dubin, R.B. Kaner, Laser scribing of high-performance and flexible graphene-based electrochemical capacitors. Science 335(6074), 1326–1330 (2012). https://doi.org/10.1126/science.1216744
Z. Yu, L. Tetard, L. Zhai, J. Thomas, Supercapacitor electrode materials: nanostructures from 0 to 3 dimensions. Energy Environ. Sci. 8(3), 702–730 (2015). https://doi.org/10.1039/C4EE03229B
X. Ren, M.G. Li, L.Y. Qiu, X. Guo, F.Y. Tian, G.H. Han, W.W. Yang, Y.S. Yu, Cationic vacancies and interface engineering on crystalline–amorphous gamma-phase Ni–Co oxyhydroxides achieve ultrahigh mass/areal/volumetric energy density flexible all-solid-state asymmetric supercapacitor. J. Mater. Chem. A 11, 5754–5765 (2023). https://doi.org/10.1039/d2ta09035j
X. Guo, M.G. Li, L.Y. Qiu, F.Y. Tian, L. He, S. Geng, Y.Q. Liu, Y. Song, W.W. Yang, Y.S. Yu, Engineering electron redistribution of bimetallic phosphates with CeO2 enables high-performance overall water splitting. Chem. Eng. J. 453, 139796 (2023). https://doi.org/10.1016/j.cej.2022.139796
M.Y. Gao, F.Y. Tian, Z. Guo, X. Zhang, Z.J. Li, J. Zhou, X. Zhou, Y.S. Yu, W.W. Yang, Mutual-modification effect in adjacent pt nanoparticles and single atoms with sub-nanometer inter-site distances to boost photocatalytic hydrogen evolution. Chem. Eng. J. 446, 137127 (2022). https://doi.org/10.1016/j.cej.2022.137127
X. Zhang, M.Y. Gao, L.Y. Qiu, J. Sheng, W.W. Yang, Y.S. Yu, Sulfur vacancies-induced ‘‘Electron Bridge” in Ni4Mo/Sv–ZnxCd1−xS regulates electron transfer for efficient H2-releasing photocatalysis. J. Energy Chem. 79, 64–71 (2023). https://doi.org/10.1016/j.jechem.2023.01.001
X. Zhang, C.X. Zhu, L.Y. Qiu, M.Y. Gao, F.Y. Tian, Y.Q. Liu, W.W. Yang, Y.S. Yu, Concentrating photoelectrons on sulfur sites of ZnxCd1–xS to active H–OH bond of absorbed water boosts photocatalytic hydrogen generation. Surf. Interfaces 34, 102312 (2022). https://doi.org/10.1016/j.surfin.2022.102312
G.R. Li, H. Xu, X.F. Lu, J.X. Feng, Y.X. Tong, C.Y. Su, Electrochemical synthesis of nanostructured materials for electrochemical energy conversion and storage. Nanoscale 5(10), 4056–4069 (2013). https://doi.org/10.1039/C3NR00607G
J. Xu, Q. Wang, X. Wang, Q. Xiang, B. Hang, D. Chen, G. Shen, Flexible asymmetric supercapacitors based upon Co9S8 nanorod Co3O4@RuO2 nanosheet arrays on carbon cloth. ACS Nano 7(6), 5453–5462 (2013). https://doi.org/10.1021/nn401450s
H. Wang, H. Yi, X. Chen, X. Wang, One-step strategy to three-dimensional graphene/VO2 nanobelt composite hydrogels for high performance supercapacitors. J. Mater. Chem. A 2(4), 1165–1173 (2014). https://doi.org/10.1039/C3TA13932H
Y. Li, J. Xu, T. Feng, Q.F. Yao, J.P. Xie, H. Xia, Fe2O3 nanoneedles on ultrafine nickel nanotube arrays as efficient anode for high-performance asymmetric supercapacitors. Adv. Funct. Mater. 27(14), 1606728 (2017). https://doi.org/10.1002/adfm.201606728
L. Liu, J. Lang, P. Zhang, B. Hu, X. Yan, Facile synthesis of Fe2O3 nano-dots@nitrogen-doped graphene for supercapacitor electrode with ultralong cycle life in KOH electrolyte. ACS Appl. Mater. Interfaces 8(14), 9335–9344 (2016). https://doi.org/10.1021/acsami.6b00225
Z. Wang, C.J. Liu, Preparation and application of iron oxide/graphene based composites for electrochemical energy storage and energy conversion devices: current status and perspective. Nano Energy 11, 277–293 (2015). https://doi.org/10.1016/j.nanoen.2014.10.022
D. Sarkar, G.G. Khan, A.K. Singh, K. Mandal, High-performance pseudocapacitor electrodes based on α-Fe2O3/MnO2 core-shell nanowire heterostructure arrays. J. Phys. Chem. C 117(30), 15523–15531 (2013). https://doi.org/10.1021/jp4039573
P. Zhao, W. Li, G. Wang, B. Yu, X. Li, J. Bai, Z. Ren, Facile hydrothermal fabrication of nitrogen-doped graphene/Fe2O3 composites as high performance electrode materials for supercapacitor. J. Alloys Compd. 604, 87–93 (2014). https://doi.org/10.1021/j.jallcom.2014.03.106
Z. Ma, X. Huang, S. Dou, J. Wu, S. Wang, One-pot synthesis of Fe2O3 nanoparticles on nitrogen-doped graphene as advanced supercapacitor electrode materials. J. Phys. Chem. C 118(31), 17231–17239 (2014). https://doi.org/10.1021/jp502226j
L. Xu, J. Xia, H. Xu, S. Yin, K. Wang, L. Huang, L. Wang, H. Li, Reactable ionic liquid assisted solvothermal synthesis of graphite-like C3N4 hybridized α-Fe2O3 hollow microspheres with enhanced supercapacitive performance. J. Power Sources 245, 866–874 (2014). https://doi.org/10.1016/j.jpowsour.2013.07.014
Q.X. Low, G.W. Ho, Facile structural tuning and compositing of iron oxide-graphene anode towards enhanced supacapacitive performance. Nano Energy 5, 28–35 (2014). https://doi.org/10.1016/j.nanoen.2014.01.002
D. Dollimore, D.L. Griffiths, D. Nicholson, The thermal decomposition of oxalates. Part II. Thermogravimetric analysis of various oxalates in air and in nitrogen. J. Chem. Soc. 488, 2617–2623 (1963). https://doi.org/10.1016/j.nanoen.2014.01.002
E.D. Macklen, Influence of atmosphere on the thermal decomposition of ferrous oxalate dehydrate. J. Inorg. Nucl. Chem. 29, 1229–1234 (1967). https://doi.org/10.1016/0022-1902(67)80362-2
Y.M. Zhao, Y.H. Li, R.Z. Ma, M.J. Roe, D.G. McCartney, Y.Q. Zhu, Growth and characterization of iron oxide nanorods/nanobelts prepared by a simple iron-water reaction. Small 2(3), 422–427 (2006). https://doi.org/10.1002/smll.200500347
H.L. Du, J.Z. Wang, B. Wang, D.Q. Cang, Preparation of cobalt oxalate powders with the presence of a pulsed electromagnetic field. Powder Technol. 199(2), 149–153 (2010). https://doi.org/10.1016/j.powtec.2009.12.015
N. Du, Y.F. Xu, H. Zhang, C.X. Zhai, D.R. Yang, Selective synthesis of Fe2O3 and Fe3O4 nanowires via a single precursor: a general method for metal oxide nanowires. Nanoscale Res. Lett. 5(8), 1295–1300 (2010). https://doi.org/10.1007/s11671-010-9641-y
C.E. Houseroft, Inorganic Chemistry (Pearson Education; Edinburgh Gate, London, 2005)
X.L. Li, J.F. Liu, Y.D. Li, Low-temperature conversion synthesis of M(OH)2 (M=Ni, Co, Fe) nanoflakes and nanorods. Mater. Chem. Phys. 80(1), 222–227 (2003). https://doi.org/10.1016/S0254-0584(02)00488-1
Y.D. Li, H.W. Liao, Y. Ding, Y. Fan, Y. Zhang, Y.T. Qian, Solvothermal elemental direct reaction to CdE (E=S, Se, Te) semiconductor nanorod. Inorg. Chem. 38(7), 1382–1387 (1999). https://doi.org/10.1021/ic980878f
P.H. Zhao, W.L. Li, G. Wang, B.Z. Yu, X.J. Li, J.T. Bai, Z.Y. Ren, Facile hydrothermal fabrication of nitrogen-doped graphene/Fe2O3 composites as high performance electrode materials for supercapacitor. J. Alloy Compd. 604, 87–93 (2014). https://doi.org/10.1016/j.jallcom.2014.03.106
J. Zhao, Z.J. Li, X.C. Yuan, Z. Yang, M. Zhang, A. Meng, Q.D. Li, A high-energy density asymmetric supercapacitor based on Fe2O3 nanoneedle arrays and NiCo2O4/Ni(OH)2 hybrid nanosheet arrays grown on SiC nanowire networks as free-standing advanced electrodes. Adv. Energy Mater. 8, 1702787 (2018). https://doi.org/10.1002/aenm.201702787
Q. Tang, W. Wang, G. Wang, The perfect matching between the low-cost Fe2O3 nanowire anode and the NiO nanoflake cathode significantly enhances the energy density of asymmetric supercapacitors. J. Mater. Chem. A 3(12), 6662–6670 (2015). https://doi.org/10.1039/CSTA00328H
X.J. Yang, H.M. Sun, L.S. Zhang, L.J. Zhao, J.S. Lian, Q. Jiang, High efficient photo-fenton catalyst of α-Fe2O3/MoS2 hierarchical nanoheterostructures: reutilization for supercapacitors. Sci. Rep. 6, 31591 (2016). https://doi.org/10.1038/srep31591
J.C. Huang, S.N. Yang, Y. Xu, X.B. Zhou, X. Jiang, N.N. Shi, D.X. Cao, J.L. Yin, G.L. Wang, Fe2O3 sheets grown on nickel foam as electrode material for electrochemical capacitors. J. Electroanal. Chem. 713, 98–102 (2014). https://doi.org/10.1016/j.jelechem.2013.12.009
Y.D. Dong, L. Xing, F. Hu, A. Umar, X. Wu, α-Fe2O3/rGO nanospindles as electrode materials for supercapacitors with long cycle life. Mater. Res. Bull. 107, 391–396 (2018). https://doi.org/10.1016/j.materresbull.2018.07.038
S. Yang, X. Song, P. Zhang, J. Sun, L. Gao, Self-assembled α-Fe2O3 mesocrystals-graphene nanohybrid for enhanced electrochemical capacitors. Small 10(11), 2270–2279 (2014). https://doi.org/10.1002/smll.201303922
K. Deori, S.K. Ujjain, R.K. Sharma, S. Deka, Morphology controlled synthesis of nanoporous Co3O4 nanostructures and their charge storage characteristics in supercapacitors. ACS Appl. Mater. Interfaces 5(21), 10665–10672 (2013). https://doi.org/10.1021/am4027482
Y. Wang, M.M. Zhang, D.H. Pan, Y. Li, T.J. Ma, J.M. Xie, Nitrogen/sulfur co-doped grapheme networks uniformly coupled N-Fe2O3 nanoparticles achieving enhanced supercapacitor. Electrochim. Acta 266, 242–253 (2018). https://doi.org/10.1016/j.electacta.2018.02.040
C.L. Long, T. Wei, J. Yan, L.L. Jiang, Z.J. Fan, Supercapacitors based on graphene-supported iron nanosheets as negative electrode materials. ACS Nano 7(12), 11325–11332 (2013). https://doi.org/10.1021/nn405192s
U.M. Patil, K.V. Gurav, V.J. Fulari, C.D. Lokhande, O.S. Joo, Characterization of honeycomb-like “β-Ni(OH)2” thin films synthesized by chemical bath deposition method and their supercapacitor application. J. Power Sources 188(1), 338–342 (2009). https://doi.org/10.1016/j.jpowsour.2008.11.136
N.K. Chaudhari, Cube-like α-Fe2O3 supported on ordered multimodal porous carbon as high performance electrode material for supercapacitors. ChemSusChem 7(11), 3102–3111 (2014). https://doi.org/10.1002/cssc.201402526
B.J. Lokhande, R.C. Ambare, R.S. Mane, S.R. Bharadwaj, Concentration-dependent electrochemical supercapacitive performance of Fe2O3. Curr. Appl. Phys. 13(6), 985–989 (2013). https://doi.org/10.1016/j.cap.2013.01.047
K.Y. Xie, J. Li, Y.Q. Lai, W. Lu, Z. Zhang, Y.X. Liu, L.M. Zhou, H.T. Huang, Highly ordered iron oxide nanotube arrays as electrodes for electrochemical energy storage. Electrochem. Commun. 13(6), 657–660 (2011). https://doi.org/10.1016/j.elecom.2011.03.040
K.K. Lee, S. Deng, H.M. Fan, S. Mhaisalkar, H.R. Tan, E.S. Tok, K.P. Loh, W.S. Chin, Sow, α-Fe2O3 nanotubes-reduced graphene oxide composites as synergistic electrochemical capacitor materials. Nanoscale 4(9), 2958–2961 (2012). https://doi.org/10.1039/C2NR11902A
S. Sun, J. Lang, R. Wang, L. Kong, X. Li, X. Yan, Identifying pseudocapacitance of Fe2O3 in an ionic liquid and its application in asymmetric supercapacitors. J. Mater. Chem. A 2(35), 14550–14556 (2014). https://doi.org/10.1039/C4TA02026J
P.M. Padwal, S.L. Kadam, S.M. Mane, S.B. Kulkarni, Enhanced specific capacitance and supercapacitive properties of polyaniline–iron oxide (PANI–Fe2O3) composite electrode material. J. Mater. Sci. 51(23), 10499–10505 (2016). https://doi.org/10.1007/s10853-016-0270-4
X. Zheng, X.Q. Yan, Y.H. Sun, Y.S. Yu, G.J. Zhang, Y.W. Shen, Q.J. Liang, Q.L. Liao, Y. Zhang, Temperature-dependent electrochemical capacitive performance of the α-Fe2O3 hollow nanoshuttles as supercapacitor electrodes. J. Colloid Interface Sci. 466, 291–296 (2016). https://doi.org/10.1016/j.jcis.2015.12.024
S. Lee, H. Kim, H.M. Jung, Interfacial generation of plates assembled with α-Fe2O3 nano-flakes for electrochemical capacitors. J. Electroanal. Chem. 770, 44–49 (2016). https://doi.org/10.1016/j.jelechem.2016.03.035
L.C. Yue, S.G. Zhang, H.Q. Zhao, M. Wang, D.F. Wang, J. Mi, Microwave-assisted one-pot synthesis of Fe2O3/CNTs composite as supercapacitor electrode materials. J. Alloy Compd. 765, 1263–1266 (2018). https://doi.org/10.1016/j.jallcom.2018.06.283
M.Y. Zhu, J.R. Kan, J.M. Pan, W.J. Tong, Q. Chen, J.C. Wang, One-pot hydrothermal fabrication of α-Fe2O3@C nanocomposites for electrochemical energy storage. J. Energy Chem. 28, 1–8 (2019). https://doi.org/10.1016/j.jechem.2017.09.021
S. Shivakumara, T.R. Penki, N. Munichandraiah, Preparation and electrochemical performance of porous hematite (α-Fe2O3) nanostructures as supercapacitor electrode material. J. Solid State Electrochem. 18(4), 1057–1066 (2014). https://doi.org/10.1007/s10008-013-2355-1
S. Shivakumara, T.R. Penki, N. Munichandraiah, Synthesis and characterization of porous flowerlike α-Fe2O3 nanostructures for supercapacitor application. ECS Electrochem. Lett. 2(7), A60–A62 (2013). https://doi.org/10.1149/2.002307eel
B.P. Prasanna, D.N. Avadhani, M.S. Raghu, K.K. Yogesh, Synthesis of polyaniline/α-Fe2O3 nanocomposite electrode material for supercapacitor applications. Mater. Today Commun. 12, 782–787 (2017). https://doi.org/10.1016/j.mtcomm.2017.07.002
D. Wang, Q. Wang, T. Wang, Controlled synthesis of mesoporous hematite nanostructures and their application as electrochemical capacitor electrodes. Nanotechnology 22(13), 135604 (2011). https://doi.org/10.1088/0957-4484/22/13/135604
T.Z. Shi, Y.L. Feng, T. Peng, B.G. Yuan, Sea urchin-shaped Fe2O3 coupled with 2D MXene nanosheets as negative electrode for high-performance asymmetric supercapacitors. Electrochim. Acta 381, 138245 (2021). https://doi.org/10.1016/j.electacta.2021.138245
S.B. Tian, B.L. Zhang, D. Han, Z.Q. Gong, X.Y. Li, Fe2O3/porous carbon composite derived from oily sludge waste as an advanced anode material for supercapacitor application. Nanomaterials 12, 3819 (2022). https://doi.org/10.3390/nano12213819
Z.Y. Yu, X.Y. Zhang, L. Wei, X. Guo, MOF-derived porous hollow α-Fe2O3 microboxes modified by silver nanoclusters for enhanced pseudocapacitive storage. Appl. Surf. Sci. 463, 616–625 (2019). https://doi.org/10.1016/j.apsusc.2018.08.262
H.Y. Quan, B.C. Cheng, Y.H. Xiao, S.J. Lei, One-pot synthesis of α-Fe2O3 nanoplates-reduced graphene oxide composites for supercapacitor application. Chem. Eng. J. 286, 165–173 (2016). https://doi.org/10.1016/j.CEJ.2015.10.068
S.W. Zahng, B.S. Yin, Z.B. Wang, F. Peter, Super long-life all solid-state asymmetric supercapacitor based on NiO nanosheets and α-Fe2O3 nanorods. Chem. Eng. J. 306, 193–203 (2016). https://doi.org/10.1016/j.cej.2016.07.057
Funding
This work was supported by the Science and Technology Research Program of Chongqing Municipal Education Commission (Grant Nos. HZ2021013, KJQN202001341, KJQN202001304, and KJZD-K202001305) and the Natural Science Foundation of Chongqing (cstc2019jcyj-msxmX0670 and cstc2020jcyj-msxmX0103).
Author information
Authors and Affiliations
Contributions
HW participated in the investigation, data curation, and writing of the original draft, YL participated in the conceptualization, writing, reviewing, & editing of the manuscript, and supervision. WX participated in the investigation. LT participated in the methodology. JS participated in the validation.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, H., Li, Y., Xiao, W. et al. Preparation and electrochemical properties of mesoporous α-Fe2O3 nanowires for supercapacitor application. J Mater Sci: Mater Electron 34, 1098 (2023). https://doi.org/10.1007/s10854-023-10456-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10854-023-10456-0