
Abstract
An attempt has been made to synthesize the carbon coated Fe-based phospho-olivine nano-crystalline cathode materials using simple binary sources via polyol method. Also, a curious attempt has been made to compare the material with the high temperature methods, viz., hydrothermal and solid state method with the same binary sources as raw materials and oxalic acid as a carbon source. The thermal behaviour of the mixed raw materials of LiFePO4 and thermal stability of the final prepared materials were characterized by TGA. The prepared materials were confirmed as orthorhombic olivine structure with Pnma space group. LiFePO4/C material prepared by Polyol method exhibits an initial reverse capacity of 150 mAh g−1 at 0.1 C rate under room temperature. Also, stable capacity of 143 mAh g−1 has been observed over 50 cycles at 0.1 C rate at room temperature among the other methods studied. It may be due to the coverage of tiny particles by carbon and it leads to provide better electronic conductivity and thereby provides good electrochemical performances.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Nowadays, Lithium batteries are suitable power sources for portable electronics and electric vehicles (EVs), but, further improvements are going to use the hybrid electric vehicles (HEV), plug-in hybrid electric vehicle (PHEV) and space application respectively. In the past decade, most of the researchers are investigating LiFePO4 cathode material, because it exhibits various advantages such as low cost, eco-friendly, low toxicity, high thermal and chemical stability in fully charged state. It can be attained its maximum theoretical specific capacity (170 mAh g−1) and good flat discharge potential at 3.4 V versus Li/Li+ [1–4].
However, the low electronic conductivity and lithium-ion diffusivity significantly restrict the rate performance of LiFePO4. To overcome these problems, many efforts have been taken including reduction of the particle size [5], surface modification using metal oxide, [6] or carbon coating, [7, 8] and lattice modification via doping of other cation [9, 10], etc. For better realization, the synthesis method affects the particle size and morphology. The decrease of particle size will not improve the electronic conductivity, but, can obviously reduce the diffusion distance of lithium ions. This indicates that the particle size and morphology play an important role on electrochemical performances of the material. However, the carbon coating is one of the best method to enhance the electronic conductivity [7]. In this view point, LiFePO4 cathode materials have been prepared via three different methods such as polyol, [11–13] hydrothermal, [14] and solid state reaction methods [15]. Due to its simplicity and controllability, the cost-effective method like polyol process is one of the suitable technique for the production of LiFePO4 in nanoscale without inert atmosphere and post heat treatment.
Kim and Kim [11] introduced the polyol process prepared LiFePO4 using three raw materials with tetra ethylene glycol (TTEG) as a polyol solvent. They controlled particle size using modified reaction time [16]. They obtained an initial capacity of 166 mAh g−1 at a current density of 0.1 mA/cm2 with utilization efficiency of 98 % and 168 mAh g−1 at the 0.1 C rate in the voltage range of 2.5–4 V with well-formed plateau. Munichandraiah et al. [17] reported nanoplate LiFePO4 was synthesized by a polyol route using two reactants, namely, FePO4 and LiOH with tetra ethylene glycol as a solvent. They obtained discharge capacity of 160 and 100 mAh g−1 at 0.15 C and 3.45 C, respectively. Also they attained a stable reversible capacity of 155 mAh g−1 at 0.2 C over 50 cycles. Singh et al. [18] obtained rhombohedral LiFePO4 particles prepared by polyol process using iron nitrate, lithium dihydrogen phosphate and diethylene glycol as a solvent. Dimesso et al. [19] reported 3D carbon nanostructures coated LiFePO4 for the first time, using Pechini-assisted reversed polyol process. Recently, Li et al. [13] reported that the LFP/C composites have been synthesized from a low-cost divalent Fe-containing precursor with tri-ethylene glycol (TEG) as a polyol medium and citric acid as a carbon source. The final LFP/C composites were obtained by heat treatment at 650 °C for 1 h under a flowing nitrogen atmosphere. The optimum electrochemical capacities of these LFP/C composites were 153, 128, 106, and 70 mAh g−1 at 1, 5, 10, and 20 C discharge rates, respectively, with excellent capacity retention. Hence that numerous reports are available based on high temperature and post heat treatment process, to reach the high capacity.
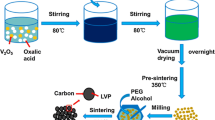
Our intention is to contribute in improving the quality of lithium iron phosphate products via economically low cost and simple binary source materials such as LiH2PO4, FeSO4·7H2O with low boiling point polyol solvent (Diethylene glycol) and low stability constant complexing agent like oxalic acid. These simple binary starting materials could lead to relatively reduce the reaction time and to enhance the cycling stabilities in the resultant LFPs. Polyol solvent acts as a reducing agent and also as a stabilizer. The oxalic acid has been chosen as a complex agent as well as carbon source. According to the crystal field theory, a complexing agent with a low stability constant would be beneficial to easily decompose when heat is applied [20]. In the present work, an attempt has been made to prepare LiFePO4/C by binary resource materials with oxalic acid as a carbon source using polyol technique, a low temperature synthesis. Further attempt has been made to compare the materials with the high temperature methods, viz., hydrothermal method and solid-state reaction. The electrochemical performances of the LiFePO4/C system which are prepared using simple binary materials are the focus of the present investigation to recognize their structure, morphological differences, chemical and magnetic properties.
2 Experimental
2.1 Synthesis of LiFePO4/C
2.1.1 Polyol process
LiFePO4 material was synthesized via low temperature polyol process. Iron (II) sulphate heptahydrate (99.9 % of Alfa-Aesar) (FeSO4·7H2O) and lithium dihydrogen phosphate (LiH2PO4) (99.9 % of Alfa-Aesar) were taken in stoichiometric molar ratio (1:1.05); then oxalic acid was added as a carbon source into the mixed solution, which were dissolved using diethylene glycol (DEG) and heated in a reflux condenser near to the boiling point of the polyol solvent (245 °C) for 18 h. In order to remove the DEG and partial organic compounds, the resulting solution was washed several times with ethanol and acetone. The resulting particles were separated and dried in a vacuum oven at 150 °C for 48 h; finally the carbon coated LiFePO4 fine particles were obtained.
2.1.2 Hydrothermal technique
LiH2PO4 and FeSO4·7H2O were dissolved in 100 ml of deionised water with the molar ratio of 1.05:1. An appropriate amount of oxalic acid has been added to the metal oxide mixed solution. The mixed solution was poured into the air tight Teflon lined 150 ml autoclave and then reacted at 160 °C for 12 h. After the hydrothermal reaction, the reactor was cooled to room temperature. The precipitated powder was filtered and washed sequentially with deionised water, acetone and then with ethanol. The obtained powder was dried at 110 °C for 5 h under vacuum. Finally it was calcined at 500 °C for 6 h under Ar atmosphere.
2.1.3 Solid state method
Starting materials viz, LiH2PO4 and FeSO4·7H2O were taken in the molar ratio of 1.05:1. The precursors with 1 ml of ethanol were initially ball-milled for 3 h in the planetary ball miller (Retzch, Germany). The ball-to-powder weight ratio was taken in 5:1 and with a rotation rate of 250 rpm. For the reaction, the appropriate amount of oxalic acid used as a carbon source. After ball milling, the powders were dried at 150 °C for 6 h in vacuum oven. Then, the dried mixture was preheated at 350 °C for 5 h and followed by calcination at 750 °C for 6 h under Ar atmosphere. The choice of calcination temperature at 750 °C is based on the better electrochemical performance of composite prepared using solid state method with two sources by Drozd et al. [21].
2.2 Characterization of LiFePO4/C
The thermal behaviour of the precursor and prepared cathode materials were examined using a thermo-gravimetric analysis (TGA, Perkin Elmer, TAC 7/DX) under N2 from 30 to 1,000 °C at a heat rate of 10 °C/min. The powder X-ray diffraction (PXRD) analysis of LiFePO4/C samples were performed using a PANalytical X-pert Pro diffractometer with a Cu Kα radiation operated at 40 kV and 30 mA and the radiation of λ = 1.54060 Å in the range of 2θ = 10–70° was used in order to determine the crystal structure of the prepared materials. The functional group vibration was analyzed using Thermo Nicolet 380 FT-IR spectrophotometer using KBr pellets. The presence of carbon structure was identified through Laser Raman Spectrometer (STR 500 Laser Raman spectrometer, SEKI, Japan). The surface and particle morphology of the samples were observed using a Scanning Electron Microscope (SEM) coupled with Energy dispersive X-ray analysis (EDX) and a high-resolution transmission electron microscope with selected area electron diffraction (HRTEM with SAED pattern) (HRTEM, FEI, Tecnai G2, Fischione Instruments, model:3000, USA). Chemical valence states of the elements were analyzed by X-ray photoelectron spectroscopy (XPS, PHI model 5802). Magnetic measurements were performed on the samples at room temperature using a Vibrating Sample Magnetometer Lakeshore 7300.
2.3 Fabrication of coin cell and electrochemical studies
The electrochemical performance of LiFePO4/C powders was analyzed using a CR2032-type coin cell. To fabricate the electrode, a mixture of 80 wt% of each active material, 10 wt% carbon black (Super P) and 10 wt% poly (vinylidene fluoride) as a binder were added and dissolved using N-methyl-2-pyrrolidone (NMP) solvent. This slurry was pasted onto an Al foil and then dried at 120 °C for 6 h in a vacuum oven. The dried paste was roll pressed and then punched as a circular disc. The coin cell was fabricated using the circular disc as cathode and lithium metal as a counter electrode with (electrolyte) 1 M solution of LiPF6 in a mixture 1:1 volume ratio of ethylene carbonate (EC) and diethyl carbonate (DEC). Polypropylene separator (Celgard 2400, Hoechst Celenese Corp) was soaked in the electrolyte for 24 h prior to use. All the weighing procedures and coin cell assembling were performed in the Ar-filled glove box by keeping both the oxygen and moisture levels less than 1 ppm. The galvanostatic charge–discharge analysis was performed by a BTS-55 Neware battery testing system in a potential range of 2.5–4.5 V (vs. Li/Li+) at room temperature with different C-rates.
3 Results and discussion
TGA analysis provides a better platform for estimating the reaction or calcination temperature of the materials in order to obtain the pure phase. Therefore, TGA analysis of the mixed raw materials was performed under N2 atmosphere from room temperature to 1,000 °C and is shown in Fig. 1. From Fig. 1, it is seen that an initial weight loss of 2.96 % at 100 °C has been observed, which represents the physically absorbed moisture during the sample loading. The transition occurred between 100 and 175 °C denotes the 12.76 % weight loss of water molecule of heptahydrate of Iron sulphate [22]. Further, a weight loss of 6.36 % has been obtained between 176 and 263 °C, which represents the decomposition of Lithium dihydrogen phosphate [23]. The phase change around temperature range 264–423 °C denotes the formation of lithium meta phosphate with the weight loss of 7.77 % and decomposition of iron sulphate with weight loss of 7.39 % for the formation of iron oxide. Simultaneously, the formation of LiFePO4 starts about 375 °C is observed with small amount weight loss of organic residuals. When the material heated above 375 °C, it is observed that the weight loss of 9.23 %, which is attributed to the decomposition of oxalic acid. Finally, weight loss of 3.34 % the temperature between 425 and 968 °C is attributed to pyrolysis of carbon content [24, 25].

TGA curve of mixture of LiH2PO4 and FeSO4·7H2O with oxalic acid for the formation of LiFePO4/C under a nitrogen atmosphere with a heating rate of 10 °C/min
The following equation represents the thermal behaviours of metal ion compounds and LiFePO4/C formation.
Oxalic acid decomposition and formation of carbon
Overall reaction for formation of LiFePO4 with carbon content
TGA of LiFePO4/C prepared by polyol (sample A), hydrothermal (sample B) and solid state reactions (sample C) were performed under air and is given in Fig. 2. From the TGA curves, it is clearly observed that there is a weight gain for samples B and C i.e., sample prepared via hydrothermal and solid state reaction methods respectively. This may be due to higher oxidation of LiFePO4. Initially, the sample prepared by polyol method has a weight loss of 1 % is represented the physically absorbed water. Further, a feable weight increment of 0.08 % is due to the oxidation of LiFePO4. Finally, the prepared materials calcined above 900 °C are observed the weight loss of 2.90 % which is attributed to presence of carbon in prepared sample [26]. The higher oxidation is presumably due to larger crystallite size of LiFePO4 and small amount distribution of carbons. Meanwhile, when sample skeleton composed of carbon and LiFePO4 crystallites mixture; the pores are generated inside the skeleton while removing the carbon during pyrolysis. LiFePO4 crystallites in the inner skeletons are easily oxidized owing to the air flow through the pores [27]. This mechanism is happened in sample B and C than in sample A. Similarly, In this case it may be the coating of carbon on the particles is completely surrounded and almost no pores were formed due to the pyrolysis of carbon in polyol process. The polyol solvent DEG itself has acted as a stabilizing agent and surrounded the LiFePO4 particles.

TGA curves of prepared LiFePO4/C polyol process (A), hydrothermal (B) and solid state method (C)
Figure 3 represents the XRD patterns of sample A, B and C respectively. All the diffraction peaks have been indexed to the orthorhombic unit cell olivine type structure with the space group of Pnma and without secondary impurity phase. There are no peaks pertaining to carbon appeared in the pattern; this may be due to the carbon generated from oxalic acid is amorphous natured carbon with low content. The crystal lattice parameters were calculated as a = 10.29 Å, b = 5.99 Å, c = 4.70 Å, V = 289.81 Å3 for sample A, a = 10.31 Å, b = 6.00 Å, c = 4.69 Å, V = 290.23 Å3 for sample B and a = 10.36 Å, b = 6.00 Å, c = 4.71 Å, V = 292.77 Å3 for sample C, respectively. All the calculated lattice constants are in good agreement with JCPDS (83-2092) [28, 29]. Using the Scherrer formula [30], the average crystallite size was estimated respectively as 40, 45 and 49 nm for sample A, B and C.

XRD patterns of (A) polyol process, (B) hydrothermal and (C) solid state method
The local environment of cations against oxygen neighbouring atoms can be estimated by FT-IR, because of the sensitivity of vibration modes of cations. FT-IR spectra of LiFePO4/C prepared via three method samples (A–C) are shown in Fig. 4. From the inset of Fig. 4, it is clearly observed that the vibrations of symmetric and antisymmetric P–O–Fe stretching modes of LiFePO4 are situated at 1,140–950 cm−1 corresponds to the characteristic bond of the [PO4]3−. The bands between 635 and 467 cm−1 are associated with the symmetric and antisymmetric vibrations of O–P–O bending modes of LiFePO4 [31]. The weak peaks in the region of 1,700–1,500 cm−1 are corresponding to the absorbance of the residual carbon [32, 33].

FT-IR spectra of (A) polyol process, (B) hydrothermal and (C) solid state method
Raman spectroscopy is an useful tool for analysing the structure of material. The Raman spectra of LiFePO4/C using different methods are given in Fig. 5. The weak peaks are found in between 995 and 1,061 cm−1, which are assigned to the stretching vibration of PO4 3− in LiFePO4 [34]. The D band corresponds to one of the A1g modes, which is attributed to the sp3 tetrahedral disorder non-conductive form of carbon. The conductive form of carbon located in G band corresponds to one of the E2g modes, which has been assigned as the sp2 graphite-like structure. The ID/IG ratios were determined as 0.967, 0.995, and 1.006, for samples A, B and C respectively (Table 1). Theoretically, higher relative intensity ratios of D/G correspond to a lower degree of ordering in the carbon material. Therefore, the lower ID/IG ratios demonstrated the excellent electrochemical performance [35, 36]. In the present study, the sample A possesses lower intensity ratio of carbon structure when compared to sample B and C. Hence, the Raman spectra reveal the carbon structure on LiFePO4 by polyol process is better than the other methods studied.

Raman spectra of (A) polyol process, (B) hydrothermal and (C) solid state method
The particle size distribution and surface morphology of the LiFePO4/C particles were studied by a scanning electron microscope (SEM) and the images of sample A–C are given in Fig. 6a–c, respectively. The SEM image of sample A has an agglomerated rod shape particles having a size of 100 nm × 1.2 µm (width × length). For sample B, it is observed that there are two kinds of particle morphologies such as primary particles rod-like shape and secondary particles spherical shape; but for sample C, only the spherical particles were observed. Figure 6d–f shows EDX analysis of the prepared samples (A–C), respectively; Also C, O, P and Fe elements were respectively observed with the atomic percentage of 32.29, 59.05, 15.08 and 13.44 % for sample A (Fig. 6d), 23.03, 58.28, 13.06 and 12.84 % for sample B (Fig. 6e) and 19.95, 44.51, 10.01 and 9.42 % for sample C (Fig. 6f). The Fe/P ratio is equal to 1 and O/Fe, O/P equal to 4. The precursor formation of LiFePO4 is in good agreement with the report of Lyczko et al. [37].

a–c SEM images and d–f EDX spectrum of Samples (A–C)
The TEM images of LiFePO4/C prepared by polyol (A), hydrothermal (B) and solid state reaction (C) methods were respectively shown in Fig. 7a–c along with their lattice fringes (Fig. 7d–f). It is observed that from Fig. 7a, the particles are completely covered by carbon only in sample A. From the images Fig. 7b, c it could be observed that the samples are coated with carbon randomly on the surface of the particles. This is may be due to the low stability constant of oxalic acid easily decomposed when heating at higher temperature [20]. Therefore, the oxalic acid would be favourable in uniform coating on LiFePO4 using low temperature process and improving the electrochemical properties. Whereas the higher temperature methods provide random carbon coating on the LFP particles evenif many works were reported on the uniform coating on LFP surfaces via hydrothermal and solid state methods. The polyol method based sample thickness of carbon layer is about 3 nm. This is clearly visualized from the lattice fringes of sample A (Fig. 7d). The width of (3.88 Å) of neighbouring lattice fringes corresponds to (210) plane of LiFePO4. However, the lattice fringes of sample B and C (Fig. 7e, f) were not distinct enough to provide the carbon layer thickness. The selected area electron diffraction (SAED) pattern of sample A represents the highly crystalline nature (Fig. 7d, inset). From the SAED analysis, it is noted that the dark region shows a bright spot pattern, which is typical for crystalline LiFePO4 and diffusion of ring observation indicates the carbon layer of prepared sample [38].

a–c HRTEM image, d–f Lattice image of Samples (A–C) and inset d SAED pattern of sample A
The chemical composition of the elements and valence state of the prepared LiFePO4/C composites were confirmed by XPS analysis and results are shown in Fig. 8a–f. Figure 8a shows the wide range scanning XPS spectrum and it can be seen that all the samples consist of Li, Fe, P, O and C elements on the surface of samples. Figure 8b, illustrates the Li 1 s spectrum, the symmetrical peaks at a binding energy of 55.6 eV for all samples. The Fe 2p peaks (Fig. 8c) are attributed to doublet of binding energy at 711.1 eV and satellite peaks at 724.5 eV, which indicate the spin–orbit splitting of the energy levels of prepared samples. The spin-orbital split energy (Δ) is about 13.4 eV in all samples. These values are in good agreement with Fe2+ ions in all samples [39]. The P 2p and O 1s peaks with binding energies of 133.8 and 531.26 eV corresponding to the tetrahedral PO4 group and oxygen predominant bonding of O2 to Fe ion in the lattice of samples are shown in Fig. 8d, e. The peaks observed at 284.35 and 284. 75 eV (Fig. 8f) are attributed to the C 1s core level that originates from carbon coated on prepared LiFePO4 samples. All the observed binding energy values are in the optimum level and in consistent with literature, which shows the chemical stability of all samples [40, 41].

a Wide XPS spectra b Li 1 s c Fe 2p d P 2p e O 1 s and f C 1 s XPS spectra for sample (A) polyol process, (B) hydrothermal and (C) solid state method
The magnetic behaviour of the prepared samples were analysed by VSM at room temperature and the results are shown in Fig. 9. The M–H curve has been observed as line shape, which corresponds to the spin glass behaviour at room temperature with paramagnetic nature with very small coercivity [42]. This dictates that there are no magnetic impurities such as γ-Fe2O3 and Fe2P phases presented in all samples [43]. Furthermore, this study identified the iron oxidation state is 2+. If the iron is oxidized, the magnetic nature may be observed in ferro/ferric magnetic under the room temperature. According to the M–H curve, when the coercivity increased, the saturation magnetization (Ms) is decreased. In the present study magnetic order of the polyol method based sample is increased among other samples, due to the high crystallinity with small particle size [44, 45].

VSM of (A) polyol process, (B) hydrothermal and (C) solid state method
Figure 10a–c demonstrate the charge/discharge voltage profiles of the cell containing LiFePO4/C electrode at 0.1, 0.5 and 1 C, respectively. The cells provide reverse capacity of 150, 144 and 142 mAh g−1 for sample A, 124, 123 and 119 mAh g−1 for sample B and 106, 97 and 97 mAh g−1 for sample C at 0.1, 0.5 and 1 C rate in room temperature. Sample A has an excellent discharge capacity among the other samples studied. This capacity is comparably higher than the report of Singh et al. [18] for LFP/C prepared via polyol method (91 mAh g−1 at 0.1 C rate @ room temperature and 146 mAh g−1 @ 0.1 C rate @ 40 °C) with binary sources and sucrose as a complexing agent. Figure 10d–f represents the cycle life of samples A–C at 0.1 C at ambient temperature. The maximum discharge capacity has been observed as 143 mAh g−1 over 50 cycles for sample A (Fig. 10d). However, the sample B and C have good stable reverse capacities viz 124 and 110 mAh g−1 over the 50 cycles.

a–c Electrochemical performance of different rate charge discharge profile of Samples (A–C) and d–f Cycle performance of Samples (A–C) at 0.1 C rates under room temperature
The sample A provides very small voltage difference between the charge/discharge plateaus designate excellent kinetics. This may be due to the following reasons.
-
The low temperature synthesis provides the lower particle size when compared to higher temperature methods.
-
The complete coverage of carbon on the prepared materials is appreciable in the low temperature method. According to the crystal field theory, the stability constant of oxalic acid is low (5.2) [20] that lead to the decomposition of complexing agent at high temperature. Therefore, the high temperature methods receive less attention in using oxalic acid as a carbon source.
-
This carbon coverage provides very thin layer which may be formed LiFePO4/C thin plates. On the other hand, higher temperature methods provide thick plates, which induces the polarization between charging and discharging plateaus into larger magnitude. This is due to larger particle and the random coverage of C on the particles [46].
The sample B and C have exhibited low capacity with large difference during the charge/discharge. This may be due to the following reasons are listed hereunder.
-
The sample B and C were prepared at higher temperature which causes the evaporation of some lithium during the heat-treatment process and the addition of oxalic acid was formed randomly distributed carbon on LiFePO4 [47].
-
The oxalic acid has low stability constant; while using the oxalic acid [20] in higher temperature processes (samples B & C), carbon is randomly distributed and increased the crystallite size with inhomogeneity of LiFePO4 particles. In addition, complete coverage of carbon on LiFePO4 particles could be observed in sample A among the other methods studied.
-
The samples B and C cannot be utilized completely due to incomplete coverage of carbon content on LFP (as shown in HRTEM image) which causes the low conductivity and large particle size of the materials. An increment of particles size could provide more space for Lithium intercalation and de-intercalation process [47–50]. During the lithium ion insertion/de-insertion cyclic performance of the samples (B and C) may be attributing to the formation of cracks and subsequent pulverization of the materials because of the volumetric change of the LFP particles [51, 52]. Also, the incomplete dispersion of the electrolyte into the electrode materials at the beginning current rate the intercalation/de-intercalation capacity is more difference obtained in sample B and C.
-
A similar anomaly was observed during the first few cycles which were reported by Rho et al. [53]. An increase of charge capacity at 0.1 C for first few cycles that may be associated with a large polarization and poor reversibility.
In addition, the following reports suggested that when the particle size is increased, the electrochemical performance dramatically decreased, which was reported by Fey et al. [5]. Barker et al. [54] reported electro active materials LiFePO4 and LiFe0.9Mg0.1PO4 via a novel carbothermal reduction (CTR) and suggested that the oxidation of carbon (C + O2 → CO2) has favoured at temperature lower than 650 °C, whereas (2C + O2 → 2CO) has expected to dominate at higher temperature. Zhu et al. [55] reported the LFP via carbothermal reduction method and the results represent increasing of sintering time at 560 °C the capacity would slightly become lower for the increasing of particle size, which would decrease the utilization of the active material.
Based on these above reasons, the electrochemical performance of sample A is better than the other samples studied. By the way, the nature of particle size with carbon content, calcination temperature and time play an important role on electrochemical performances of LFP particles.
4 Conclusion
In summary, LiFePO4/C composite cathode materials were successfully synthesized by low temperature polyol process without any inert atmosphere and post heat treatment. Also, performances were compared with post heat treatment process like hydrothermal and solid state reactions. The phase structure of the prepared material falls under orthorhombic unit cell olivine type structure with the space group of Pnma. There is no impurity detected in the prepared materials that are studied through XRD, FT-IR, XPS and room temperature vibrating sample magnetic analyses. From the TEM analysis, it may be concluded that the particles are completely covered by carbon when the polyol method was adopted. However, the samples are coated by carbon randomly on the surface of the particles that are synthesized via hydrothermal and solid state reaction methods. This may be due to the pyrolysis of coated carbon layer had taken place during calcination. That is, the stability constant of oxalic acid is low (5.2) that lead to the decomposition of oxalic acid at high temperature. It reflects in the electrochemical performances of the prepared samples. All the results denoted the LiFePO4/C sample prepared via polyol process exhibits the excellent thermal, chemical stability and good reverse capacity with cyclic nature at room temperature. A stable reverse capacity of 143 mAh g−1 at 0.1 C rate has been observed over 50 cycles under room temperature. Hence, one can conclude that the polyol process is suitable eco-friendly and easier method to produce nano-sized cathode material for high performance Lithium battery applications.
References
M. Armand, J.M. Tarascon, Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–367 (2001)
J.B. Goodenough, Y. Kim, Challenges for rechargeable Li batteries. Chem. Mater. 22, 587 (2010)
A. Padhi, K. Nanjundaswamy, J.B. Goodenough, Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 144, 1188–1194 (1997)
B.C. Melot, D.O. Scanlon, M. Reynaud, G. Rousse, J.-N. Chotard, M. Henry, J.-M. Tarascon, Chemical and structural indicators for large redox potentials in Fe-based positive electrode materials. ACS Appl. Mater. Interfaces (2014). doi:10.1021/am405579h
G.T.-K. Fey, Y.G. Chen, H.-M. Kao, Electrochemical properties of LiFePO4 prepared via ball-milling. J. Power Sources 189, 169–178 (2009)
J. Wang, X. Sun, Understanding and recent development of carbon coating on LiFePO4 cathode materials for lithium-ion batteries. Energy Environ. Sci. 5, 5163–5185 (2012)
K. Kim, Y.H. Cho, D. Kam, H.S. Kim, J.W. Lee, Effects of organic acids as reducing agents in the synthesis of LiFePO4. J. Alloys Compd. 504, 166–170 (2010)
D. Shao, J. Wang, X. Dong, W. Yu, G. Liu, F. Zhang, L. Wang, Electrospinning fabrication and electrochemical properties of LiFePO4/C composite nanofibers. J. Mater. Sci. Mater. Electron. 24(11), 4263–4269 (2013)
B. Ding, P. Xiao, G. Ji, Y. Ma, L. Lu, J.Y. Lee, High-performance lithium-ion cathode LiMn0.7Fe0.3PO4/C and the mechanism of performance enhancements through Fe substitution. ACS Appl. Mater. Interfaces 5, 12120–12126 (2013)
K.L. Harrison, A. Manthiram, Microwave-assisted solvothermal synthesis and characterization of metastable LiFe1–x(VO)xPO4 cathodes. Inorg. Chem. 50, 3613–3620 (2011)
D.-H. Kim, J.K. Kim, Synthesis of LiFePO4 nanoparticle in polyol medium and their electrochemical properties. Electrochem. Solid State Lett. 9, A439–A442 (2006)
J. Jiang, W. Liu, J.T. Chen, Y.L. Hou, LiFePO4 nanocrystals: liquid-phase reduction synthesis and their electrochemical performance. ACS Appl. Mater. Interfaces 4, 3062–3068 (2012)
S. Li, X. Liu, R. Mi, H. Liu, Y. Li, W-m Lau, J. Mei, A facile route to modify ferrous phosphate and its use as an iron-containing resource for LiFePO4 via a polyol process. ACS Appl. Mater. Interfaces 6, 9449–9457 (2014)
J. Chen, J. Graetz, Study of Antisite Defects in Hydrothermally Prepared LiFePO4 by in Situ X-ray Diffraction. ACS Appl. Mater. Interfaces 3, 1380–1384 (2011)
S.S. Zhang, J.L. Allen, K. Xu, T.R. Jow, Optimization of reaction condition for solid-state synthesis of LiFePO4/C composite cathodes. J. Power Sources 147, 234–240 (2005)
D.-H. Kim, J.K. Kim, Synthesis of LiFePO4 nanoparticles and their electrochemical properties. J. Phys. Chem. Solids 68, 734–737 (2007)
N.N. Sinha, N.J. Munichandraiah, Single-shot preparation of crystalline nanoplate LiFePO4 by a simple polyol route. Electrochem. Soc. 157, A824–A829 (2010)
M. Singh, M. Willert-Porada, Polyol process for the synthesis of LiFePO4 rhombohedral particles. Adv. Powder Technol. 22, 284–289 (2011)
L. Dimesso, C. Spanheimer, S. Jacke, W. Jaegermann, Synthesis and characterization of LiFePO4/3-dimensional carbon nanostructure composites as possible cathode materials for Li-ion batteries. Ionics 17, 429–435 (2011)
R. Yang, E. Kang, B. Jiang, J.-H. Ahn, Effect of complexing agents on the electrochemical performance of LiFePO4/C prepared by sol-gel method. Nanoscale Res. Lett. 40, 1–7 (2012)
V. Drozd, G.Q. Liu, R.S. Liu, H.T. Kuo, C.H. Shen, D.S. Shy, X.K. Xing, Synthesis, electrochemical properties, and characterization of LiFePO4/C composite by a two-source method. J. Alloys Compd. 487, 58–63 (2009)
M.S.R. Swamy, T.P. Prasad, Thermal decomposition of iron (ii) sulphate heptahydrate in the presence of alkali metal carbonates. J. Therm. Anal. 25, 347–354 (1982)
K.-S. Lee, J. Moon, J. Lee, M. Jeon, High-temperature phase transformations in LiH2PO4 and possible solid-state polymerization. Solid State Commun. 147, 74–77 (2008)
S.F. Yang, Y.N. Song, P.Y. Zavalij, M.S. Whittingham, Reactivity, stability and electrochemical behavior of lithium iron phosphates. Electrochem. Commun. 4, 239–244 (2002)
G. Hasegawa, Y. Ishihara, K. Kanamori, K. Miyazaki, Y. Yamada, K. Nakanishi, T. Abe, Facile preparation of monolithic LiFePO4/carbon composites with well-defined macropores for a lithium-ion battery. Chem. Mater. 23, 5208–5216 (2011)
C.-H. Yima, E.A. Baranovab, Y. Abu-Lebdeha, I. Davidsona, Highly ordered LiFePO4 cathode material for Li-ion batteries templated by surfactant-modified polystyrene colloidal crystals. J. Power Sources 205, 414–419 (2012)
J. Lim, V. Mathew, K. Kim, J. Moon, J.K. Kim, One-pot synthesis of multi-morphous LiFePO4 nanoparticles in polyol medium. J. Electrochem. Soc. 158(6), A736–A740 (2011)
A.V. Murugan, T. Muraliganth, P.J. Ferreira, A. Manthiram, Dimensionally modulated, single-crystalline LiMPO4 (M = Mn, Fe Co, and Ni) with Nano-Thumblike shapes for high-power energy storage. Inorg. Chem. 48, 946–952 (2009)
B. D. Cullity, S. R. Stock, Elements of X-Ray Diffraction, 3rd edn., ch. 5.2 (Prentice Hall Publishers, New Jersey, 2001)
K. Zaghib, A. Mauger, F. Gendron, C.M. Julien, Relationship between local structure and electrochemical performance of LiFePO4 in Li-ion batteries. Ionics 14, 271–278 (2008)
K. Vediappan, A. Guerfi, V. Gariépy, G.P. Demopoulos, P. Hovington, J. Trottier, A. Mauger, C.M. Julien, K. Zaghib, Stirring effect in hydrothermal synthesis of nano C-LiFePO4. J. Power Sources 266, 99–106 (2014)
N. Ravet, M. Gauthier, K. Zaghib, J.B. Goodenough, A. Mauger, F. Gendron, C.M. Julien, Mechanism of the Fe3+ reduction at low temperature for LiFePO4 synthesis from a polymeric additive. Chem. Mater. 19(10), 2595–2602 (2007)
H.M. Xie, R.S. Wang, J.R. Ying, L.-Y. Zhang, A.F. Jalbout, H.-Y. Yu, G.-L. Yang, X.-M. Pan, Z.-M. Su, Optimized LiFePO4-polyacene cathode material for lithium-ion batteries. Adv. Mater. 18(19), 2609–2613 (2006)
L. Zhang, R.K. Brow, A Raman study of iron-phosphate crystalline compounds and glasses. J. Am. Ceram. Soc. 94(9), 3123–3130 (2011)
B.-H. Rita, J.-P.P. Ramos, Raman microspectrometry applied to the study of electrode materials for lithium batteries. Chem. Rev. 110, 1278–1319 (2010)
M.M. Doeff, Y.Q. Hu, F. McLarnon, R. Kostecki, Effect of surface carbon structure on the electrochemical performance of LiFePO4. Electrochem. Solid State Lett. 6, A207–A209 (2003)
N. Lyczko, A. Nzihou, P. Sharrock, A. Germeau, C. Toussaint, Characterization of LiFePO4/C cathode for lithium ion batteries. Ind. Eng. Chem. Res. 51(1), 292–300 (2012)
S. Wi, S. Nam, Y. Oh, J. Kim, H. Choi, S. Hong, S. Byun, S. Kang, D.J. Choi, K.-O. Ahn, Y.-H. Kim, B. Park, Facile synthesis of porous-carbon/LiFePO4 nanocomposites. J. Nanopart. Res. 14, 1327 (2012)
Jun Ma, Baohua Li, Du Hongda, Xu Chengjun, Feiyu Kang, Inorganic-based sol–gel synthesis of nano-structured LiFePO4/C composite materials for lithium ion batteries. J. Solid State Electrochem. 16, 1353–1362 (2012)
Z.G. Lu, M.F. Lo, C.Y. Chung, Pulse laser deposition and electrochemical characterization of LiFePO4-C composite thin films. J. Phys. Chem. C 112, 7069–7078 (2008)
R. Dedryvère, M. Maccario, L. Croguennec, F. Le Cras, C. Delmas, D. Gonbeau, X-ray photoelectron spectroscopy investigations of carbon-coated LixFePO4 materials. Chem. Mater. 20, 7164–7170 (2008)
K. Zaghib, A. Mauger, F. Gendron, C.M. Julien, Surface effects on the physical and electrochemical properties of thin LiFePO4 particles. Chem. Mater. 20, 462–469 (2008)
A. Yamada, Y. Takei, H. Koizumi, N. Sonoyama, R. Kanno, K. Itoh, M. Yonemura, T. Kamiyama, Electrochemical, magnetic, and structural investigation of the Li x (Mn y Fe1–y ) PO4 olivine phases. Chem. Mater. 18, 804–813 (2006)
G. Herzer, Nanocrystalline soft magnetic materials. J. Magn. Magn. Mater. 157(158), 133–136 (1996)
K. Zaghib, A. Mauger, J.B. Goodenough, F. Gendron, C.M. Julien, Electronic, optical, and magnetic properties of LiFePO4: small magnetic polaron effects. Chem. Mater. 19, 3740–3747 (2007)
K. Saravanan, M.V. Reddy, P. Balaya, H. Gong, B.V.R. Chowdari, J.J. Vittal, Storage performance of LiFePO4 nanoplates. J. Mater. Chem. 19, 605–610 (2009)
H. Liu, Q. Cao, L.J. Fu, C. Li, Y.P. Wu, H.Q. Wu, Doping effects of zinc on LiFePO4 cathode material for lithium ion batteries. Electrochem. Commun. 8, 1553–1557 (2006)
H. Ning, C.-Y. Wang, X.-Y. Kang, Y.T. Han, Synthesis and electrochemical characterizations of zinc-doped LiFePO4/C by carbothermal reduction. J. Inorg. Mater. 25, 887–892 (2010)
B. Niu, E. Qi, J.-Q. Wang, A simple and facile preparation of LiFePO4 by a one-step microwave hydrothermal method. J. Inorg. Organomet. Polym. 21, 906–912 (2011)
X. Huang, S. Yan, H. Zhao, L. Zhang, R. Guo, C. Chang, X. Kong, H. Han, Electrochemical performance of LiFePO4 nanorods obtained from hydrothermal process. Mater. Charact. 61, 720–725 (2010)
H.-P. Liu, Z.-X. Wang, X.-H. Li, H.-J. Guo, W.-J. Peng, Y.-H. Zhang, Q.-Y. Hu, Synthesis and electrochemical properties of olivine LiFePO4 prepared by a carbothermal reduction method. J. Power Sources 184, 469–472 (2008)
A.S. Andersson, J.O. Thomas, The source of first-cycle capacity loss in LiFePO4. J. Power Sources 97–98, 498–502 (2001)
Y.-H. Rho, L.F. Nazar, L. Perry, D. Ryan, Surface chemistry of LiFePO4 studied by Mössbauer and X-Ray photoelectron spectroscopy and its effect on electrochemical properties. J. Electrochem. Soc. 154, A283–A289 (2007)
J. Barker, M.Y. Saidi, J.L. Swoyer, Lithium iron(II) phospho-olivines prepared by a novel carbothermal reduction method. Electrochem. Solid State lett. 6(3), A53–A55 (2003)
B.Q. Zhu, X.H. Li, Z.X. Wang, H.J. Guo, Novel synthesis of LiFePO4 by aqueous precipitation and carbothermal reduction. Mater. Chem. Phys. 98, 373–376 (2006)
Acknowledgments
The authors M. Sivakumar and R. Muruganantham gratefully acknowledge for the financial support to carry out this work by Department of Science and Technology (DST), New Delhi, Govt. of India under DST-SERC major research project (SR/S2/CMP-0049/2008).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Muruganantham, R., Sivakumar, M., Subadevi, R. et al. A facile synthesis and characterization of LiFePO4/C using simple binary reactants with oxalic acid by polyol technique and other high temperature methods. J Mater Sci: Mater Electron 26, 2095–2106 (2015). https://doi.org/10.1007/s10854-014-2653-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-014-2653-0