
Abstract
Li3V2(PO4)3/C (LVP/C) composite materials have been successfully synthesized via a sol–gel method with oxalic acid as chelating agent and polyethylene glycol (PEG) as the supplementary carbon source, in which oxalic acid and PEG serve as double carbon sources. The X-ray diffraction patterns indicate that all of the samples are well crystallized. Transmission electron microscopy images reveal that the LVP/C sample prepared with 10 wt% PEG is uniformly coated by carbon layer with an appropriate thickness of 10–16 nm, resulting in a high electrical conductivity and a fast kinetics. The Li+ diffusion coefficient in the LVP/C sample prepared with PEG is 3.482 × 10−13 cm2 s−1, which is larger than that of the LVP/C sample prepared without PEG. In the range of 3.0–4.3 V, the LVP/C-10 electrodes exhibit good rate capability and excellent cyclic performance, which discharge capacities are 131.9 mAh g−1 at 0.1 C and 105.3 mAh g−1 at 5 C. The present work provides a valuable route for preparing lithium metal phosphates with double carbon sources to improve the conductivity and hence the electrochemical performance.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Due to their relatively high energy density and power density and their environmental friendliness, lithium–ion batteries are used in laptop computers, cell phones, renewable energy storage devices, electric vehicles and hybrid electric vehicles. Monoclinic Li3V2(PO4)3 (LVP) has attracted much attention as a potential cathode material for lithium–ion batteries, due to its fast lithium ion migration resulting from its three-dimensional framework, which shows good ion mobility [1–4], high theoretical capacity and high operating voltage [3, 5].
For the structure of Li3V2(PO4)3, a slightly distorted VO6 octahedron and a PO4 tetrahedron share an oxygen atom to form a (V–O–P–O)n bond, each VO6 octahedron surrounding six PO4 tetrahedrons and each PO4 tetrahedron surrounding four VO6 octahedrons to form a three-dimensional network structure, where the lithium atoms are in the structure of the hole clearance [1]. VO6 octahedron contains two V positions (i.e., V (1) and V (2)), and the average length of V–O bond are 2.003 and 2.006 Å [3]. Similarly, Li has three crystallography positions, where Li (1) occupies the tetrahedron and Li (2) and Li (3) occupy a quasi-tetrahedron, which has a slightly longer Li–O bond [3].
The charge and discharge complex mechanism of the Li3V2(PO4)3 have been studied. At a voltage window of 3.0–4.3 V, in the process of charging, the emergence of Li+ occurs step by step in the two phase transition mechanism which involves three plateaus. Among these plateaus, the emergence of the first lithium in two steps, which occurs at voltage plateaus of 3.60 and 3.68 V is due to the presence of an orderly lithium phase (Li2.5V2(PO4)3). In this phase structure, Li (1) and Li (2) are completely filled, and Li (3) is half filled because the position has the highest energy. The V (1)–O bond relative to the initial phase (Li3V2(PO4)3) becomes shorter, which suggests a change in the vanadium that is confirmed by the paramagnetic resonance spectrum indicating the presence of V4+ [6]. The V (2)–O bond remains unchanged. When the first lithium is completely removed, the Li (3) site is vacant, and Li (2) converts into a tetrahedron similar to the Li (1) position. The V (1)–O bond corresponds to a V4+–O bond length of 1.91 Å, and the V (2)–O bond corresponds to a V3+–O bond length of 1.99 Å. Therefore, Li2V2(PO4)3 contains V3+/V4+ mixed oxidation states [3]. The removal of the second lithium corresponds to a 4.10 V voltage plateau where Li (1) is empty and Li (2) converts into a five heavy position. The length of V (2)–O bond shortens to 1.907 Å, which is similar to the V (1)–O bond where the V4+ oxidation state results in the formation of Li1V2(PO4)3. At 3.0–4.3 V, the charging and discharging processes follow the two phase transfer mechanism, and there is no solid solution behavior. Nearly all of the lithium equivalents (i.e., 2 mol/FU) have been extracted for a theoretical value of 132 mAh g−1.
However, Li3V2(PO4)3 exhibits low electronic conductivity (i.e., approximately 2.3 × 10−8 S cm−1) at 300 K due to the polarization of the V–O bond [6], which substantially restricts its practical application in lithium–ion batteries. Therefore, to overcome this disadvantage, several methods have been developed, such as coating with carbon or metal [1, 7–13], doping with metal cations [14–16], and decreasing the particle size [17, 18]. Carbon coating is a simple and effective method for improving the electronic conductivity of the Li3V2(PO4)3 materials. Not only Carbon coating can substantially improve the electronic conductivity of the LVP but also prevent direct contact between the LVP particles and the electrolyte, which is helpful in alleviating dissolution of vanadium ions in the electrolyte [19]. Recent results have demonstrated that the carbon-coating is primarily dependent on the carbon source. Therefore, various carbon sources have been employed for carbon-coated LVP, such as glucose [9, 20–24], acetylene black [25], sucrose [26], maltose [27], citric acid [28], starch [21], polyvinyl alcohol [29, 30], oxalic acid [31] and polyethylene glycol (PEG) [32].
To the best of our knowledge, this is the first report on the synthesis of LVP/C using oxalic acid and PEG as double carbon sources. The PEG with an ordered and uniform chain structure is easily adsorbed onto the surface of particles [33] and can effectively form a carbon layer on the surface of the particles after carbonization at high temperature. In addition, PEG has been extensively used to synthesize LiFePO4 and Li3V2(PO4)3 cathode materials to improve their electronic conductivity. Double carbon sources have been recently used to improve the quality of the carbon coating in LiFePO4 by Sung Woo Oh etc. [34]. The double carbon-coated LiFePO4 electrode has high tap density, 1.5 g cm−3. This value is much greater than those obtained for common LiFePO4 electrode materials, which are limited to a maximum of 1.0 g cm−3. As well known, a high tap density is a very welcome property for electrode materials in lithium batteries, since it reflects high volumetric energy density. Indeed, on the basis of the cathode tap density and the charge–discharge response above discussed, researchers estimated for Li/LiFePO4 cell a volumetric energy density of 820 Wh m−3. The results of this work clearly demonstrate that the double carbon-coated LiFePO4 electrode has highly desirable properties: a specific capacity (close to the theoretical value), stable cycle life, exceptional rate capability, and a high tap density. The researchers believe that their double carbon-coated LiFePO4 electrode is a serious candidate for the development of high performance, advanced lithium batteries directed to the hybrid vehicle and electric vehicle markets. The uniformity of the carbon coating is crucial to achieving high electronic conductivity and Li+ transfer. The sol–gel method is commonly employed to obtain homogeneous morphology and uniform carbon layered electrode materials.
In this study, we have successfully synthesized Li3V2(PO4)3/C composite materials via an oxalic acid-based sol–gel route using PEG. It is should be noted that PEG is simultaneously used as a supplementary carbon source and a surface active agent for the LVP/C composites, which is prepared with the sol–gel method and ensure the quality of the carbon coating layer. The Li3V2(PO4)3/C composite materials are characterized using X-ray diffraction (XRD), scanning electron microscopy (SEM) and thermogravimetric/differential scanning calorimetry (TG/DSC). The electrochemical performance of the Li3V2(PO4)3/C/Li cells are examined using a galvanostatic charge–discharge method, cyclic voltammetry and electrochemical impedance spectroscopy (EIS) method. The results indicate that the use of double carbon sources in the formation of a high quality carbon coated layer that ensure excellent electrochemical performance, which may be beneficial for the preparation of lithium metal phosphate ions.
2 Experimental
2.1 Synthesis of Li3V2(PO4)3/C composite cathode materials
The Li3V2(PO4)3/C composites were synthesized via an oxalic acid-based sol–gel route using V2O5 (A.R.), LiOH·H2O (A.R.), NH4H2PO4 (A.R.), C2H2O4·2H2O (oxalic acid, A.R.) and polyethylene glycol (PEG, A.R.). In the first step, V2O5 and oxalic acid in a stoichiometric molar ratio of 1:3 were dissolved with stirring in deionized water at 80 °C to form a clear blue solution. Second, a stoichiometric mixture of LiOH·H2O and NH4H2PO4 were added to the solution in turn. The mixture was heated to 80 °C until a green gel formed. Finally, the gel was dried at 80 °C overnight in vacuum to obtain the dry gel. The dry gel was pre-heated at 350 °C for 6 h in a N2 atmosphere and then cooled to room temperature to obtain the precursor powder. Various amounts of PEG (i.e., 0, 5, 10, 15, 20 wt% of the precursor powder) were added, and milled with the precursor powder in ethanol for 30 min followed by drying at 65 °C for 8 h in an oven. This mixture was sintered at 700 °C for 10 h in a N2 atmosphere to obtain the LVP/C composite material. The overall synthesis process is schematically shown in Fig. 1. The samples prepared with 0, 5, 10, 15 and 20 wt% of PEG were denoted LVP/C-0, LVP/C-5, LVP/C-10, LVP/C-15 and LVP/C-20, respectively.

Schematic representation of the synthesis process for the LVP/C samples
2.2 Material characterization
The LVP/C samples were characterized by X-ray diffraction (Bruker/D8-Focus, Germany) with a CuKα radiation source. The morphological observations of LVP/C were performed using scanning electron microscopy (SEM, Hitachi/SU8081, Japan). Carbon coating of the LVP/C powders were evaluated by transmission electron microscopy (TEM, */JEM2100F, Japan). Thermogravimetric/differential scanning calorimetry (TG/DSC) curves were obtained using an STA 449F3 analyzer (NETZSCH) at 30–900 °C in an alumina crucible under an air atmosphere with a heating rate of 10 °C min−1.
2.3 Electrochemical property measurements
The electrochemical performance of the Li/Li3V2(PO4)3 was tested by simulating battery. The working electrodes were prepared by mixing the active Li3V2(PO4)3/C materials, acetylene black and polyvinylidene fluoride binder in a weight ratio of 75:15:10 in N-methyl pyrrolidine solvent. The slurry of the mixture was pasted onto aluminum foil and dried in an oven at 80 °C for 10 h. The resulting film was punched into discs (a diameter of Ø 15 mm). Metallic lithium foil was employed as the anode. The electrolyte was 1 M LiPF6 in ethylene carbonate and dimethyl carbonate (1:1 in volume), and the separator was Celgard 2300. The Li3V2(PO4)3/C composite batteries were tested in a voltage range of 3.0–4.3 V (vs. Li/Li+). Cyclic voltammetry (CV) and EIS were conducted on a Vmp3 (Biologic) electrochemical workstation. CV tests were performed at a scan rate of 0.5 mV s−1. The EIS spectra were obtained over a frequency range of 100 kHz to 10 mHz. The Li3V2(PO4)3/C composite batteries were charged with a constant current and constant-voltage protocol and discharged in a constant-current mode at varied C rates by using a battery tester (Arbin BT2000). All of the tests were performed at room temperature.
3 Results and discussion
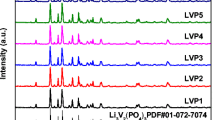
The XRD patterns of the prepared Li3V2(PO4)3/C are shown in Fig. 2. The diffraction peaks for all samples are well indexed as monoclinic Li3V2(PO4)3 with a space group of P21/n (JCPDS No. 80-1515). Table 1 shows the crystal cell parameters of various LVP/C materials with the standard atlas (JCPDS No. 80-1515) of the crystal cell parameters. The residual carbon source did not affect the LVP/C material crystal structure. The carbon is not observed on the XRD diffraction pattern, which indicates that the residual carbon is an amorphous structure or that the thickness of the carbon layer on the LVP is too thin to be detected [35]. Table 1 shows the crystal cell parameters of various LVP/C materials, after coated carbon cell volume decreases; relative to the not coating LVP, LVP/C composite materials main diffraction peaks of the half peak width basically increase, and the diffraction peak intensity gradually weaken. According to the Scherrer’s formula, width of the peak shows that the decrease of the grain size, so the carbon coating can reduces grain size, achieve the goal of refining grain. With the addition PEG, the value of a, b and c slightly decrease. The decrease of the diffusion path is advantageous for the extraction/insertion of the lithium ions, which improves the material performance.

XRD patterns for the as-prepared LVP/C samples
To determine the carbon content in the end products, a TGA (thermal gravimetric analysis) measurement is performed employing a CR-G type high-temperatureμ-DTA apparatus. As shown in Fig. 3 thermogravimetric analysis (TGA) is adopted to estimate the amorphous carbon content in the LVP/C-10. The individual causes of the total mass changes (△m) can be determined from room temperature to 900 °C: evaporation of moisture (m w ), gasification of carbon (m c ), and oxidation of Li3V2(PO4)3/C (m o ), namely:
where m w = ~2.65 %, △m = ~2.52 %, and the oxidation coefficient (k = 0.0784) is obtained from the Kuang et al. [36]. Results on Li3V2(PO4)3/C oxidation over the temperature range of 50–900 °C. Based on Eqs. (1) and (2), m c = 2.31 %.

TGA curve for Li3V2(PO4)3/C-10
The amount of other composites residual carbon is also calculated by this method. The carbon content of the LVP/C samples is listed in Table 2. The residual carbon can be used as a conductive agent, improves the LVP electronic conductivity, and prevents LVP particles from coming into direct contact with the electrolyte, which inhibit the dissolution of V in the electrolyte [19]. Obviously, the content of residual carbon in LVP/C-0 prepared without additional PEG is the lowest (only 0.12 %), resulting in few carbon coating and incomplete conversion of V5+–V3+. Thereby, it is necessary to provide additional carbon source to cover the shortage of residual carbon in the product obtained from stoichiometric oxalic acid and V2O5.
SEM images of the LVP/C composites are shown in Fig. 4. The surface of the five composite materials is rough, and some small particles are attached on the surface of LVP, which correspond to scattered carbon particles. The stabilization of V3+ is favored, and the formation of composite materials with superior conductivity (Table 2; pure LVP, approximately 2.3 × 10−8 S cm−1) and Li+ ion diffusion is favored due to the residual carbon. In addition, notable agglomeration (LVP/C particles) is not observed for the LVP/C-0, LVP/C-5, and LVP/C-10 samples, which may be related to the decomposition of oxalate to produce a substantial amount of gas and the existence of the carbon layer. With the PEG content increasing, the reunion phenomenon strengthens because the residual carbon in the sample acts as a bonding agent. For LVP/C-15 and LVP/C-20, although the residual carbon content is relatively high, the reunion phenomenon is the most serious, which reduces the tap density of the material. However, the LVP/C-10 particles morphology present spherical.

SEM images of LVP/C-0 (a, b), LVP/C-5 (c, d), LVP/C-10 (e, f), LVP/C-15 (g, h), and LVP/C-20 (i, j)
TEM images are further used to investigate the influence of the additional PEG on microstructure for the LVP/C samples, as shown in Fig. 5. The thickness of the carbon coating agrees well with the content of residual carbon in Table 2. The more the residual content, the thicker the carbon coating is. For LVP/C-0 (Fig. 5a), no obvious carbon coating is found on the particle surface, but adhesion phenomenon is observed. For LVP/C-20 (Fig. 5e), multilayer carbon coating with irregular shape tightly wraps around the LVP particles. Typical core–shell structures are observed in the images of LVP/C-5, LVP/C-10 and LVP/C15 (Fig. 5b, c and d). The large space (SEM) between the LVP/C-0 and LVP/C-5 particles increases the electrochemical reaction resistance. For LVP/C-0 and LVP/C-5 materials, due to low carbon content, lead to not form a complete carbon coating or the thickness of carbon coating is too thin. Due to not form a complete conductive network, so the abundance of grain boundaries leads to poor connection between the particles resulting in the high contact resistance. The high electrochemical reaction resistance and contact resistance may be responsible for the low initial discharge specific capacities of LVP/C-0 and LVP/C-5. Figure 5c shows uniform carbon coating on the surface particles, which can results in full absorption of the electrolyte and good contact between the LVP and the electrolyte. Further clarification that layer of carbon coat on the spherical LVP/C-10 particles surface (Fig. 4f). Meanwhile, the 10–16 nm thickness of carbon coating on the surface of LVP/C-10 is more appropriate for forming an ideal conducting network to enhance conductivity and alleviate vanadium dissolution, resulting in a faster lithium ion transfer and better electrochemical performance.

TEM images of the LVP/C-0 (a), LVP/C-5 (b), LVP/C-10 (c), LVP/C-15 (d), and LVP/C-20 (e)
The conductivity values (σ) are calculated from Eq. (3) [37]:
where R ct is the charge-transfer resistances, t is the thickness of the cathode, and A is the area of the electrode surface. Based on Eq. (3), the σ values of LVP/C-0, LVP/C-5, LVP/C-10, LVP/C-15 and LVP/C-20 are calculated to be 1.577 × 10−4, 1.792 × 10−4, 2.736 × 10−4, 2.326 × 10−4 and 2.424 × 10−4 S cm−1, respectively (Table 2).
The addition of a small amount of the PEG (i.e., less carbon residue, for LVP/C-0 and LVP/C-5) does not improve the electronic conductivity of the LVP. However, a product with larger particles is formed. Therefore, the lithium ion migration distance to the center of the particles of the active material is long, which is likely to lead to the active material not being completely used. The addition of a large amount of the PEG (i.e., more carbon residue, for LVP/C-15 and LVP/C-20) substantially improve the LVP electronic conductivity but the surface of the LVP with a thick carbon layer, which does not favor lithium ion transport. Based on these two results, there must be an optimal PEG addition amount. LVP/C-10 exhibits the largest conductivity values (σ), which improves the electrochemical performance of the battery.
Figure 6a shows the initial charge and discharge curves for the LVP/C cathode material in the range of 3.0–4.3 V. At the 0.1 C charge curve, three plateaus are observed at 3.61, 3.70 and 4.10 V, which correspond to the extraction of two lithium ions and the phase transition of LixV2(PO4)3 with x = 3.0 to 2.5, 2.0 and 1.0, respectively. The first lithium ion is extracted in two steps due to the existence of the ordered phase Li2.5V2(PO4)3. With the PEG content increasing, charge plateaus become wider, and the discharge plateaus shift upward and become wider. The observation of voltage polarization is based on the difference between the charge voltage and the discharge voltage. The difference becomes small indicating that the polarization between charge and discharge is suppressed due to the addition of PEG, which improves the electrical conductivity of the materials. Because the role of carbon layer is to improve the electrical conductivity of materials, the extraction/insertion of the lithium ions occurs more quickly. A reduction of the electrode polarization reduces the capacity loss. In addition, the length of the charging and discharging platform significantly increases, which increases the discharge specific capacity of the materials. The results indicate that LVP/C-10 has the longest discharge plateau comparing to the other samples, and its initial discharge capacity is 131.9 mAh g−1 at 0.1 C, which is in good agreement with the SEM results.

Initial charge/discharge curves (a) and cycle performance (b) of LVP/C at 0.1 C
The cyclic performance of the five samples at 0.1 C for 200 cycles is shown in Fig. 6b. The capacity retention of all of the LVP/C samples with additional PEG is higher than that of LVP/C-0. Visible carbon coating improves the stability of the LVP structure of the material on certain degree. The discharge capacity profiles are substantially improved after addition of PEG. LVP/C-0 only delivers a discharge capacity of 70.9 mAh g−1 with a coulombic efficiency of 71.36 % at 0.1 C, and the discharge capacity decreases to 50.6 mAh g−1 after 200 cycles. LVP/C-10 delivers the highest capacity of 131.9 mAh g−1 with a coulombic efficiency of 83.47 % at 0.1 C and maintains a discharge capacity of 110.1 mAh g−1 even after 200 cycles.
The low residual carbon content of the sample resulting in a lower initial capacity fades rapidly. This result is primarily due to the low electronic conductivity of the samples that has low carbon content, and the larger crystal grains mean that the active material cannot be fully utilized. These factors lead to non-ideal electrochemical properties. The carbon content of LVP/C-10, LVP/C-15 and LVP/C-20 deliver a good capacity retention rate, which indicate the good conductivity of the material. However, the discharge capacity of LVP/C-15 and LVP/C-20 are lower than that of LVP/C-10, which are primarily due to the high carbon content in the sample resulting in a relatively low content of active material. The residual carbon content of LVP/C-10 is 3.21 %, which is adequate for the formation of an effective carbon coating that can provides a good conduction network for the kinetic extraction/insertion reaction of Li+.
Figure 7 shows the rate capability performance of the LVP/C composites based on various amounts of PEG under different discharge rates (i.e., 0.1, 0.5, 1, 2, 5 and 0.1 C). The initial discharge at 0.1 C, the LVP/C-10 sample exhibits a high capacity of 130.1 mAh g−1 and remains nearly unchanged at the 10th cycling test. At a discharge rate of 0.5 C, capacity approximately maintains 128.3 mAh g−1 after ten rounds, which demonstrates its high reversible performance. At a high discharge rate of 1, 2 and 5 C, the capacity approximately maintains 118.2 (1 C), 114.9 (2 C) and 101.7 mAh g−1 (5 C) after ten rounds, respectively. Therefore, the LVP/C-10 sample exhibits the best rate performance due to the additional 10 wt% PEG, which is more appropriate for boosting the electronic conductivity from the layer of coated carbon.

Rate capabilities of LVP/C under different discharge rates (i.e., 0.1, 0.5, 1, 2, 5 and 0.1 C)
This result may be due to the low carbon content of LVP/C-0 and LVP/C-5 where the electronic conductive performance is poor. In addition, the larger particles of the material result in a poor rate performance. However, the higher carbon content of LVP/C-15 and LVP/C-20 materials results in a rate performance that is not as good as the LVP/C-10 materials because the carbon coating on LVP/C-15 and LVP/C-20 materials is too thick, which reduces the diffusion rate of the lithium ions. However, the small particle size and appropriate carbon layer thickness of LVP/C-10 resulting in small electrochemical polarization and good reversibility during charging and discharging, which produce excellent rate properties.
As can be seen, LVP/C-10 exhibits the best rate capability and excellent cyclic performance with a discharge capacity as high as 131.9 mAh g−1 with a coulombic efficiency of 83.47 % at 0.1 C. In addition, the sample maintains a discharge capacity of 110.1 mAh g−1 even after 200 cycles. Comparing with conventional synthetic methods (single carbon source), carbon content of this synthesis material is relatively low, to ensure a sufficient tap density. As well known, a high tap density is a very welcome property for electrode materials in lithium batteries, since it reflects high volumetric energy density. Thus relatively high tap density can ensure excellent discharge capacity, which also greatly improves the conductivity of the material of the LVP, exhibiting excellent electrochemical characteristics and cycle stability.
Figure 8a shows the cyclic voltammetry curves for the LVP/C electrodes at a scanning rate of 0.5 mV s−1 between 3.0 and 4.3 V. With the additional PEG, the redox peaks become sharp well-shaped. Three anodic peaks are located at 3.65, 3.75, and 4.15 V, and three cathodic peaks are observed at 3.50, 3.60, and 4.09 V, which agrees well with the charge–discharge curve in Fig. 4a. With additional PEG, the difference in the three pairs of redox peaks corresponding to the peak potential is smaller than those for the material without PEG, which indicates smaller polarization and better electrochemical performance. With the CV peak current increasing, the Li+ diffusion coefficient increases. The relationship between the peak current and Li+ diffusion can be explained by the Randles–Sevcik equation [38]:
where I p is the CV peak current (A), A is the electrode area (cm2), D is the Li+ diffusion coefficient (cm2 s−1), v is the potential scan rate (V s−1), C is the concentration of lithium ions in the cathode (mol cm−3), and n is the number of electrons transferred per molecule during the intercalation (n is 2 for Li3V2(PO4)3). Based on Eq. (4), the Li+ diffusion coefficients of the LVP/C-0, LVP/C-5, LVP/C-10, LVP/C-15 and LVP/C-20 samples are calculated to be 4.127 × 10−14, 3.991 × 10−14, 3.482 × 10−13, 2.095 × 10−13 and 2.455 × 10−13 cm2 s−1, respectively (Table 2). LVP/C-0 exhibits the lowest Li+ diffusion coefficient, and LVP/C-10 exhibits the highest Li+ diffusion coefficient, which is indicative of an enhancement in reversibility of the electrode reaction due to the formation of the carbon layer with the appropriate amount of PEG.

CV curves (a) and EIS spectra (b) for the LVP/C composites
Nyquist plots of the LVP/C samples are shown in Fig. 8b. Based on the formation of stable SEI (solid electrolyte interphase) films on the surface of the electrode [39], the second cycle is chosen for the EIS measurement. The EIS spectra of the five LVP/C samples are similar, which consist of a small intercept at high frequency, a semicircle at high to medium frequency and an inclined line at low frequency. The inset in Fig. 8b shows the equivalent circuit. R C represents the bulk resistance of the cell, which corresponds to the small intercept. R ct and C dl are the charge-transfer resistances and the double-layer capacitance, respectively, which correspond to the depressed semicircle. Z w is the Warburg impedance, which corresponds to the inclined line at low frequency. A lower charge transfer resistance (R ct ) is more conducive to electrochemical reaction kinetics. In Table 2, the R ct values of LVP/C samples with additional PEG are lower than that of LVP/C-0, which indicate the formation of a more conductive network resulting in transfer of lithium ions during solid phase migration and improvement in the charge transfer between the particles in the environment. It should be noted out that with the amount of PEG increasing, the R ct values first decrease, but these values increase when the PEG amount is higher than 10 wt%. Therefore, excessive residual carbon in the sample resulting in the formation of a thick carbon coated layer that inhibits charge migration. The LVP/C-10 samples possess minimum charge transfer resistance and effectively overcome the dynamics of the process limits, which result in improving dynamic performance and a high charge and discharge performance. This result is consistent with the cyclic voltammetry and high-rate performance test results.
The values of the exchange current density (i o) are calculated using Eq. (5) [37]:
where R is the gas constant (8.314 J mol−1 K−1), T is the temperature (298.5 K), and F is Faraday’s constant (96,500 C mol−1). The exchange current density is a parameter that indicates the reversibility of the electrode. The largest i o value is observed for the LVP/C-10 sample (Table 2), which indicates its superior reversibility comparing to the other samples.
Figure 9a shows charge and discharge curves for the LVP/C-10 cathode material at 0.1 C in the range of 3.0–4.3 V at the first five cycles. The results indicate that with the cycling time increasing, the charge plateaus of LVP/C-10 become shorter, less flat and shift upward. Figure 9b shows charge and discharge curves for the LVP/C-10 cathode material at 0.5 C at the first five cycles. The results indicate that with the cycling time increasing, the charge plateaus of LVP/C-10 become wider, more flat and shift downward. Based on a comparing these two results, a good trend develop at 0.1 C but not at 0.5 C. In particular, the corresponding to the 4.1 V plateau (i.e., the second lithium ion deintercalation) of the material after coating with a carbon layer is due to an increasing in its electrical conductivity. Therefore, at large charge and discharge rates, the material performance is relatively better. For the LVP/C materials, the high rate performance is superior to the low rate performance.

LVP/C-10 samples for the first five cycles at 0.1 C (a) and 0.5 C (b)
4 Conclusions
The carbon-coated Li3V2(PO4)3 cathode materials have been successfully synthesized via an oxalic acid-based sol–gel route using PEG, in which the oxalic acid and PEG serve as double carbon sources. The electrochemical performance of the LVP/C composites substantially improves with the addition PEG. Our results indicate that the addition of PEG as a second carbon source gives rise to uniform carbon coating, which results in high electrical conductivity and a large Li+ diffusion coefficient. The optimized LVP/C sample with 10 % PEG, which the 10–16 nm thickness of carbon coating on the surface of LVP/C-10 is more appropriate for forming an ideal conducting network to enhance conductivity and alleviate vanadium dissolution, resulting in a faster lithium ion transfer and better electrochemical performance. Therefore, for preparation of the cathode materials with poor intrinsic electronic transport such as lithium metal phosphates, it is helpful to use double carbon sources to form high quality carbon layer to achieve superior electrochemical performance.
References
Huang H, Yin SC, Kerr T, Taylor N, Nazar LF (2002) Adv Mater 14:1525–1528
Saıdi M, Barker J, Huang H, Swoyer J, Adamson G (2002) Electrochem Solid-State Lett 5:A149–A151
Yin S-C, Grondey H, Strobel P, Anne M, Nazar L (2003) J Am Chem Soc 125:10402–10411
Zhang LL, Zhang X, Sun YM, Luo W, Hu XL, Wu XJ, Huang YH (2011) J Electrochem Soc 158:A924–A929
Huang H, Faulkner T, Barker J, Saidi M (2009) J Power Sources 189:748–751
Yin SC, Strobel P, Grondey H, Nazar L (2004) Chem Mater 16:1456–1465
Fu P, Zhao Y, Dong Y, An X, Shen G (2006) J Power Sources 162:651–657
Li Y, Zhou Z, Ren M, Gao X, Yan J (2006) Electrochim Acta 51:6498–6502
Chang C, Xiang J, Shi X, Han X, Yuan L, Sun J (2008) Electrochim Acta 54:623–627
Pan A, Liu J, Zhang JG, Xu W, Cao G, Nie Z, Arey BW, Liang S (2010) Electrochem Commun 12:1674–1677
Qiao Y, Wang X, Zhou Y, Xiang J, Zhang D, Shi S, Tu J (2010) Electrochim Acta 56:510–516
Zhang L, Wang X, Xiang J, Zhou Y, Shi S, Tu J (2010) J Power Sources 195:5057–5061
Rui X, Jin Y, Feng X, Zhang L, Chen C (2011) J Power Sources 196:2109–2114
Ren M, Zhou Z, Li Y, Gao X, Yan J (2006) J Power Sources 162:1357–1362
Chen Y, Zhao Y, An X, Liu J, Dong Y, Chen L (2009) Electrochim Acta 54:5844–5850
Jiang T, Wei Y, Pan W, Li Z, Ming X, Chen G, Wang C (2009) J Alloys Compd 488:L26–L29
Kim DK, Muralidharan P, Lee H-W, Ruffo R, Yang Y, Chan CK, Peng H, Huggins RA, Cui Y (2008) Nano Lett 8:3948–3952
Teng F, Hu ZH, Ma XH, Zhang LC, Ding CX, Yu Y, Chen CH (2013) Electrochim Acta 91:43–49
Patoux S, Wurm C, Morcrette M, Rousse G, Masquelier C (2003) J Power Sources 119:278–284
Ren M, Zhou Z, Gao X, Peng W, Wei J (2008) J Phys Chem C 112:5689–5693
Rui X, Li C, Chen C (2009) Electrochim Acta 54:3374–3380
Zhou X, Liu Y, Guo Y (2009) Electrochim Acta 54:2253–2258
Wang L, Zhang LC, Lieberwirth I, Xu HW, Chen C-H (2010) Electrochem Commun 12:52–55
Wang L, Zhou X, Guo Y (2010) J Power Sources 195:2844–2850
Liu M, Guo X (2008) Rare Met 27:571–574
Ying JR (2006) GAO J, Jiang CY, Li W, Tang CP. J Inorg Mater 21:1097–1102
Rui X, Li C, Liu J, Cheng T, Chen C (2010) Electrochim Acta 55:6761–6767
Li Y, Zhou Z, Gao X, Yan J (2007) Electrochim Acta 52:4922–4926
Jiang T, Pan W, Wang J, Bie X, Du F, Wei Y, Wang C, Chen G (2010) Electrochim Acta 55:3864–3869
Wang J, Zhang X, Liu J, Yang G, Ge Y, Yu Z, Wang R, Pan X (2010) Electrochim Acta 55:6879–6884
Zheng JC, Li XH, Wang ZX, Niu SS, Liu DR, Wu L, Li LJ, Li JH, Guo HJ (2010) J Power Sources 195:2935–2938
Wang J, Liu J, Yang G, Zhang X, Yan X, Pan X, Wang R (2009) Electrochim Acta 54:6451–6454
Dobryszycki J, Biallozor S (2001) Corros Sci 43:1309–1319
Sung WO, Seung TM, Seung MO, Kyu HO, Khalil A, Bruno S, Yang KS (2010) Adv Mater 22:4842–4845
Cui Y, Zhao X, Guo R (2010) Electrochim Acta 55:922–926
Kuang Q, Zhao YM, An XN, Liu JM, Dong YZ, Chen L (2010) Electrochim Acta 55:1575–1581
Shin HC, Chob WI, Jang H (2006) J Power Sources 159:1383–1388
Dahn J, Jiang J, Moshurchak L, Fleischauer M, Buhrmester C, Krause L (2005) J Electrochem Soc 152:A1283–A1289
Reddy M, Pecquenard B, Vinatier P, Levasseur A (2007) Electrochem Commun 9:409–415
Acknowledgments
This work was supported by the National Natural Science Foundation of China (21173198) and the Science and Technology Support Program of Hubei Province, China (2013BHE014).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tao, D., Wang, S. Synthesis and performance of carbon-coated Li3V2(PO4)3 cathode materials via an oxalic acid-based sol–gel route using PEG. J Sol-Gel Sci Technol 74, 121–131 (2015). https://doi.org/10.1007/s10971-014-3585-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-014-3585-0