
Abstract
Herein, a series of phosphorus-doped CoMoO4 rod bundles electrocatalysts on 3D nickel foams have been successfully synthesized and exploited as efficient hydrogen evolution reaction (HER) electrocatalyst in 1 M KOH. The incorporation of phosphorus into CoMoO4 can significantly promote the HER activity, and the CoMoO4 bundles phosphorized at 350 °C exhibited the optimum activity, which only required low overpotentials of 56 and 148 mV to deliver cathodic current densities of 10 mA cm−2 and 100 mA cm−2, respectively. This electrode also presented considerable long-term electrochemical stability with negligible delay after 30 h operation and 3000 accelerated cyclic voltammetry cycles. Our study provides a superior earth-abundant catalyst for efficient HER in alkaline media, which also suggests that P doping engineering is an effective way to boost the HER activity of transition metal-based oxides.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Water electrolysis driven by renewable energy sources is widely considered as a clean technique to produce pure hydrogen [1,2,3]. Generally, electrochemical water splitting proceeds through two half-reactions, namely hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) [4]. As for HER, by far, the benchmark electrocatalysts are still limited to Pt-based precious metal catalysts [5, 6]. Their large-scale industrial application, however, is largely restricted by the scarcity and high cost. Consequently, the development of effective HER catalysts using earth-abundant materials is urgently needed [1, 2].
Recent years, transition metals oxides, phosphides, and sulfides have shown a great promise [7,8,9,10,11,12,13,14,15,16,17,18,19]. Among them, Co- and Mo-based oxides have been attracting increasing attention for designing low-cost electrocatalysts, due to the unique electron configurations [10]. Particularly, bimetallic CoMoO4 composite oxide exhibits promising electrochemical activity because of the synergistic interactions between different elements [15,16,17,18,19,20,21,22,23,24]. However, for pure CoMoO4, large overpotentials (> 200 mV) are usually required to deliver a current density of − 10 mA cm−2 [16,17,18].
To boost the HER activity, there are some strategies like designing special microstructure and constructing heterostructures. For example, CoMoO4/Co9S8 arrays and heterostructured CoMoO4–Co2Mo3O8 composite have been constructed for HER catalysts with enhanced performance [15, 22]. Further, elemental doping with a variety of element choice is proven to be an efficient way to modulate the physicochemical properties of parent oxides. For example, Li et al. [21]reported that phosphorus-doped β-CoMoO4 nanoplates on Ti foil show greatly enhanced HER activity, which only requires an overpotential of 138 mV at − 10 mA cm−2 in 1 M KOH.
Encouraged by this work, herein, we fabricated and evaluated 3D free-standing P-doped CoMoO4 rod bundles anchored on NF as HER electrocatalysts. Phosphorus heterodoping was achieved through phosphorization with NaH2PO2 at higher temperatures. Electrochemical measurement confirmed that phosphorus incorporation can dramatically enhance the electrocatalytic activity toward HER in alkaline media. Among treated samples, the CoMoO4 bundles phosphorized at 350 °C exhibited the highest activity with a low overpotential of 56 mV @ − 10 mA cm−2 and a small Tafel slope of 78 mV dec−1. High HER activity with excellent stability makes it a very promising Pt-free catalyst for hydrogen evolving application.
Experimental section
Chemicals and materials
Analytical grade of (NH4)6Mo7O24·4H2O, Co(NO3)2·6H2O, CO(NH2)2, NH4F, NaH2PO2·H2O, KOH were all purchased from Aladdin Ltd. Nickel foam (NF, 1 mm in thickness) was obtained from Heze Tianyu Technology Co. Ltd. Deionized (DI) water from a Millipore system was used throughout the experiment.
Catalyst synthesis
Co(OH)F precursors on NFs were first synthesized via a facile hydrothermal method. Nickel foams (2 × 4 cm2) with a thickness of 1 mm were cleaned with 6 M HCl, acetone, ethanol and DI water for 10 min each in an ultrasound bath consecutively. Then, 2 mmol Co(NO3)2·6H2O, 8 mmol NH4F, and 10 mmol CO(NH2)2 were slowly dropped into 36 ml DI water to form a pink solution. NFs and the solution were transferred to a 50-ml Teflon-lined autoclave and kept at 100 °C for 6 h. After cooling, the NFs were dried at 60 °C for 6 h to obtain a Co(OH)F coated on NF. After that, 0.48 mmol of (NH4)6Mo7O24·4H2O was dissolved in 40 mL water and the solution was then transferred to a 50-mL Teflon-lined autoclave with Co(OH)F on NF. The autoclaves were sealed and heated at 140 °C for 4 h. After cooling naturally, NFs coated with CoMoO4 were washed by DI water for 3 times and dried at 60 °C overnight. Finally, the foam specimens (at downstream) and NaH2PO2 (1 g, at upstream) were placed in 2 separate quartz boats in a tube furnace, which were calcined at 300 °C, 350 °C and 400 °C for 2 h using a ramping rate of 2 °C min−1 in N2 flow. As-obtained samples were taken out after cooling and named accordingly as P-CoMoO4-300, P-CoMoO4-350 and P-CoMoO4-400. The loadings of CoMoO4-based catalysts on NFs were estimated by a Sartorius BP211D high-precision balance (about 9–11 mg cm−2).
Characterization
The crystal phases were examined by a Rigaku D/max-2600 X-ray diffraction diffractometer with Cu-Kα radiation. Raman measurement was carried out using a Hobriba J-Y LabRAM spectroscope using 532 nm radiation. The sample morphologies were recorded by using a Hitachi SU-70 field-emission scanning electron microscopy (FE-SEM) and an FEI Tecai G2 F20 transmission electron microscopy (TEM). Further, the chemical states in catalysts were determined by a Thermo-fisher K-Alpha X-ray photoelectron spectroscopy (XPS) with Al Kα radiation, and all the binding energies were referenced using the adventurous carbon (C1s) peak at 284.8 eV.
Electrochemical evaluation
A typical three-electrode configuration consisting of a foam electrocatalyst as working electrode, a graphite rod and a saturated calomel electrode (SCE) as counter electrode and reference electrodes were used. Electrochemical tests were carried out with a Chenhua CHI 660E electrochemical workstation in 1 M KOH electrolyte. Linear sweep voltammetry (LSV) curves were collected at a slow scan rate of 2 mV s−1. All LSV curves were iR-corrected unless noted, and all potentials were converted to reversible hydrogen electrode (RHE) scale using the equation of RHE = E versus SCE + 0.059 × pH + 0.2415(V). Electrochemical impedance spectroscopy (EIS) measurements were recorded within a frequency range of 100 kHz to 0.01 Hz at an AC amplitude of 5 mV. Cyclic voltammetry (CV) test was carried out at non-faradaic region to probe the electrochemical double-layer capacitance (Cdl) of various samples. A series of CV sweeps were performed at various scan rates (20, 40, 60, 80, 100 and 120 mV s−1) in the region of − 0.45 to − 0.35 V versus SCE. By plotting the difference of current density (ja − jc) between the anodic and cathodic sweeps at − 0.4 V versus SCE against the scan rate, a linear trend was observed. The slope of the fitting line is equal to twice of the geometric Cdl, which is proportional to the effective electrode surface area (ECSA) of the catalysts. Therefore, the ECSAs of different samples can be compared based on their Cdl values. Preliminary electrochemical stability of the P-CoMoO4-350 catalyst was investigated by galvanostatically at a current density of − 10 mA cm−2 for 30 h. Accelerated degradation test was performed using CV scans within 0 to − 0.2 V versus RHE at 100 mV s−1 for 3000 continuous scans.
Results and discussion
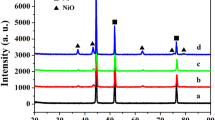
A synthesis procedure of P-doped CoMoO4 bundles on NF is schematically shown in Fig. 1. The crystal phases of obtained catalysts on NF foams were determined by XRD (Fig. 2a). After the first hydrothermal process, the color of NF changes to reddish-brown (Fig. 1) and its diffraction peaks for the precursor could be perfectly indexed as Co(OH)F (JCPDS No. 50-0827) except for the peaks of NF support. The second hydrothermal process changed the sample color to purple, and all the other peaks revealed a mixture of two phases, which can be indexed as the orthorhombic CoMoO4 (JCPDS No. 15-0439) and CoMoO4·0.9H2O (JCPDS No. 14-0086) [25, 26]. Further phosphorization with NaH2PO2 at higher temperatures resulted in black foams and the corresponding XRD patterns suggested the samples treated at 350 and 400 °C transformed to monoclinic CoMoO4 (β-phase, JCPDS No. 21-0868). No additional peaks from phosphides like CoP and MoP are detected, indicating no secondary phases were yielded during phosphorization treatment. XRD patterns of P-CoMoO4-350 and pure CoMoO4 annealed at 350 °C in the air (named as CoMoO4-350-air) were also compared as shown in Fig. 2b. Phosphorization treatment did not change the diffraction peaks obviously. A closer look at the main peak of CoMoO4 at ≈ 26.5° shows a positive shift of 0.2°, suggesting the lattice contraction due to the incorporation of smaller P ions into CoMoO4 lattice [27]. Raman spectra were further probed, and the results are shown in Fig. 2c. The Raman spectrum of pristine CoMoO4 exhibits five Raman bands. The intense doublet at ~ 938 cm−1 could be assigned to Mo=O bond, and the bands at 817 and 857 cm−1 are associated with O–Mo–O bond. Moreover, the peaks at 334 and 358 cm−1 can be assimilated to Co–O–Mo bond [6, 28]. No obvious new peak is observed in phosphorized and air-annealed CoMoO4 bundles. Compared with CoMoO4-350-air sample, the peaks between 800 and 1000 cm−1 (P-CoMoO4-350) show slight blueshift by about 3 cm−1 that could originate from P doping in CoMoO4 lattice. These further confirm the formation of CoMoO4 phase and P doping did not change the crystal structure, consistent with the XRD analysis.

A diagram for the fabrication process of P-CoMoO4 rods on Ni foam (NF). First, reddish-brown Co(OH)F nanosaws were hydrothermally grown on Ni foam, which were then hydrothermally transferred to purple CoMoO4 rods. Finally, black P-CoMoO4 rods were obtained via phosphorization treatment

a XRD patterns of Co(OH)F- and CoMoO4-based materials obtained at different conditions, b XRD patterns of CoMoO4 treated at 350 °C in air (CoMoO4-350-air) and P-CoMoO4-350, c typical Raman spectra of CoMoO4, P-CoMoO4-350 and CoMoO4-350-air
The morphology and microstructure evolution of catalysts were investigated by SEM and TEM techniques. As shown in Fig. 3a, b, the Co(OH)F precursor clearly shows nanosaw feature which uniformly covers on NF surface. After the introduction of Mo ions in hydrothermal condition, the precursor transformed into bundles, as revealed in Fig. 3c, d. These bundles consist of several microrods with a square-like top of about 2 μm in width and 10–15 μm in length. The bundle surface is relatively smooth. Further phosphating treatment retains the bundle character, and a closer look at enlarged image of P-CoMoO4-350 reveals that some particles are formed (Fig. 3e, f). With the increasing of phosphorization temperature, the bundle surface appears to be more coarse (Figure S1), probably due to the hydrate dissociation [21]. The granule size also increases for samples at higher temperatures. In Fig. 3g, EDS mapping images from selected area clearly show even distribution of Mo, Co, O and P elements across the bundles, which also confirms the successful doping of P into CoMoO4 lattice [29], and P doping content is estimated to be 5.6% in P-CoMoO4-350 catalyst (Figure S2).

SEM images of a, b Co(OH)F, c, d CoMoO4, e, f P-CoMoO4-350 and g EDS elemental mapping images of Co, Mo, O, and P in P-CoMoO4-350
The microrod character with some nanoparticles on the surface is also confirmed by TEM analysis (Fig. 4a). Figure 4b presents a high-resolution TEM (HR-TEM) image of P-CoMoO4-350. Lattice fringes with spacings of 0.69 nm and 0.21 nm can be ascribed to the (001) and (222) planes of CoMoO4, respectively, which are in good agreement with XRD analysis [30, 31]. It is anticipated that the unique bundle morphology constructed with many microrods could facilitate both mass transfer and diffusion of gaseous products and provide a high concentration of active sites that are very beneficial to catalytic activity enhancement.

TEM (a) and HR-TEM (b) images of P-CoMoO4-350
The chemical states and surface electronic structure of the CoMoO4 and P-CoMoO4-350 samples were examined by XPS. The survey spectrum of fresh CoMoO4 in Figure S3 clearly verifies the presence of Co, Mo, O and Ni elements. For P-CoMoO4-350 catalyst, two new weak peaks at ~ 134.1 eV and 191.7 eV can be ascribed to P 2p and P 2s signals, respectively. High-resolution P 2p spectrum in Fig. 5a demonstrates a strong peak at 134.1 eV in treated CoMoO4, which is associated with surface oxidized P–O species [32]. This is another solid evidence of P doping, in accordance with EDS analysis. In the Co 2p region (Fig. 5b), two peaks at ~ 781.1 and 797.5 eV are attributed to the Co 2p3/2 and 2p1/2 levels, respectively. [19, 33] After phosphating, the Co 2p3/2 peak of P-CoMoO4-350 shifts to higher energy, which implies that the electronic structure of Co centers has been modified through P doping [34]. The Mo 3d spectrum of bare sample (Fig. 5c) exhibits two main peaks. The one at low binding energy can be specified as Mo 3d5/2, while the other one is Mo 3d3/2. The splitting width of two peaks is 3.1 eV, signifying a majority of Mo6+ oxidation state [35]. Those peaks shift positively by about 0.2 eV in P-doped CoMoO4, and more importantly, two new peaks located at 230.4 eV and 233.6 eV due to Mo5+ species can be deconvoluted. In O 1s spectra (Fig. 5d), two fitted peaks at 530.5 and 532.1 eV can be assigned to the typical metal–oxygen bond (Mo–O–Co) and surface absorbed hydroxyls, respectively [22, 36]. Whereas a new peak at the high energy of 531.4 eV appeared after phosphating which is attributed to P–O [37].

High-resolution XPS spectra of CoMoO4 and P-CoMoO4-350, a P 2p, b Co 2p, c Mo 3d and d O 1s
The HER catalytic performance of pristine CoMoO4 and P-CoMoO4 samples treated at different temperatures were evaluated and compared. The NF support, commercial 20% Pt/C (Johnson Matthey), Co(OH)F precursor and CoMoO4-350-air were also examined in the same condition for reference. LSV polarization curves after iR drop compensation are plotted in Fig. 6a and Figure S4. It can be found that benchmark Pt/C exhibits superior activity with zero onset overpotential, while the NF and Co(OH)F precursor show very poor HER performance. Apparently, the phosphated CoMoO4 bundles display much better activity toward HER than undoped one. Among CoMoO4-based catalysts, the P-CoMoO4-350 demonstrates the highest activity (Table S1) with the lowest overpotential of 56 mV at a cathodic current density of − 10 mA cm−2. For comparison, the required overpotentials to supply the same current density are 169 mV, 73 mV and 63 mV for pure CoMoO4, P-CoMoO4-300 and P-CoMoO4-400, respectively. To afford a higher current density of − 100 mA cm−2, the P-CoMoO4-350 requires an overpotential of 148 mV, still much lower than other tested catalysts except Pt/C (Table S1). Such superior HER electrocatalytic activity of P-CoMoO4-350 electrode outperforms other reported Mo-based oxides electrocatalysts, for example, CoMoO4–Co2Mo3O8 (58 mV@ − 10 mA cm−2) [18], P-doped-β-CoMoO4 nanoplates (138 mV@ − 10 mA cm−2) [21] and CoMoO4 nanowires (121 mV@ − 10 mA cm−2) [23], (see Table S2 for details).

a LSV polarization curves, b Tafel plots c Nyquist plots and d estimated Cdl of CoMoO4 based catalysts
The HER kinetics of these electrodes is analyzed by corresponding Tafel plots. The smallest Tafel slope of 20% Pt/C (38 mV dec−1) is in agreement with a previous report [38], which therefore confirms the reliability of our electrochemical test. As expected, P-CoMoO4-350 exhibits a Tafel slope of 78 mV dec−1, which is smaller than that of P-CoMoO4-300 (108 mV dec−1), P-CoMoO4-400 (90 mV dec−1), pure-CoMoO4 (112 mV dec−1). A smaller Tafel slope implies the higher HER catalyst activity, [39] suggesting the higher intrinsic activity of P-CoMoO4-350. Meanwhile, the Tafel slope is usually used to analysis the HER mechanism and rate-determining step. In this work, the experimentally obtained Tafel slope of P-CoMoO4-350 implies that the HER reaction takes place via a Volmer–Heyrovsky mechanism where Heyrovsky process is considered as the rate-limiting step.
To further study the electron transfer kinetics at catalyst–electrolyte interface, impedance spectra were collected and the Nyquist plots are given in Fig. 6c. By fitting the impedance spectra with a simplified Randles equivalent circuit (insert in Fig. 6c), the charge transfer resistances (Rct) were obtained and listed in Table S1. It is obvious that P-CoMoO4-350 shows a much smaller Rct value (2.5 Ω) than that of P-CoMoO4-300 (6.3 Ω), P-CoMoO4-400 (2.9 Ω), bare CoMoO4 (30.1 Ω) and CoMoO4-350-air (29.0 Ω), indicating the fastest charge transfer process among the CoMoO4-based catalysts, confirming the abovementioned rapid HER kinetics in P-CoMoO4-350 catalyst. To redouble study the intrinsic catalytic activity of P-CoMoO4-350, the turnover frequencies (TOFs) that reflect all the active sites involved in the HER reaction were measured according to references [40, 41]. As shown in Figure S5, the TOF value of P-CoMoO4-350 catalyst is 0.59 s−1 at an overpotential of 100 mV, much greater than that of CoMoO4 (0.092 s−1). The high TOF value of P-CoMoO4-350 indicates its high intrinsic activity due to the successful P doping, which can improve the electrical conductivity and reduce the hydrogen adsorption free energy, as supported by theoretical calculations by Li et al. in P-doped CoMoO4 nanoplates [42]. These are also evidenced by impedance spectra and electrochemical performance in P-doped CoMoO4 rod bundles in our study.
In order to measure the density of active sites, the electrochemically active surface areas (ECSAs) of the foam catalysts were determined by using electrochemical double-layer capacitance (Cdl), which were calculated using CV scans from − 0.35 to − 0.45 V versus SCE at scan rates range of 20–120 mV s−1 (Figure S6). As depicted in Fig. 6d, P-CoMoO4-350 exhibits the highest Cdl value (4.03 mF cm−2) among tested samples, which is about 4.3 and 70 times higher than that of pristine CoMoO4 (0.94 mF cm−2) and Co(OH)F (0.06 mF cm−2). The dramatically increased ECSA of P-CoMoO4-350 reflects the more accessible active sites, due to the P uniformly dope in rods; the rough surface also enables greatly increased exposed surface area [42]. The ECSAs were calculated from Cdl values using the specific capacitance value of a standard 1 cm2 flat surface which is usually between 20 and 60 μF cm−2. An average value of 40 μF cm−2 was used for calculation [17]. Moreover, LSV plots normalized by ECSA and mass of typical electrodes are given in Figure S7a, b, which further confirms the high specific activity of P-CoMoO4-350 catalyst.
In addition to HER activity, considerable stability of a catalyst is also very important for practical application. The durability was first tested by accelerated CV cycling at a scan rate of 100 mV s−1. As compared in Fig. 7a, the polarization curve of P-CoMoO4 approximates to the initial one even after 3000 CV cycles. At the current densities of − 10 and − 100 mA cm−2, the initial overpotentials are 48 and 154 mV, respectively. After test, the corresponding values reached 51 and 157 mV. Furthermore, chronopotentiometric test at − 10 mA cm−2 was carried out (insert in Fig. 7a). Its overpotential shows a slight increase of 0.7 mV after testing for 30 h. Both CV and chronopotentiometric tests prove its strong stability. The XRD peaks turn to be weak after test (Fig. 7b) but a comparison of Raman results (Fig. 7c) for P-CoMoO4-350 catalyst before and after test suggests no detectable phase change occurred. SEM images in Fig. 7d show that the rod bundle feature is maintained, and the enlarged view in the insert reveals a more rough surface with the formation of more particles. The excellent stability of provides more evidence of P-CoMoO4-350 to be a reliable catalyst in alkaline water reduction.

Comparisons of P-CoMoO4-350 catalyst before and after 3000 CV cycles, a polarization curves (insert: chronoamperometric curve without iR correction at the constant overpotential of − 1.13 V, b XRD patterns, c Raman spectra and d SEM images
Based on the discussion above, it is clear that phosphorus doping is an effective strategy in engineering HER activities and the greatly enhanced activity of phosphorized CoMoO4 bundles can be ascribed to the following combined reasons: (1) The unique bundle morphology with rough surface enables the exposure of more active sites with high accessibility and facile diffusion of the electrolyte and gaseous product, (2) direct anchoring of bundles on conductive NF eliminates the use of additional polymer binder and thus enables strong interfaces at the electrocatalyst/electrode, and (3) P doping modulates the electronic structure that enables greatly enhanced HER kinetics.
Conclusion
Phosphorus-doped CoMoO4 rod bundles were successfully prepared by combined hydrothermal and phosphorization processes. The introduction of P ions into CoMoO4 lattice is evidenced by combined XRD, Raman, EDS and XPS techniques. The electrochemical test confirmed that phosphorus incorporation can efficiently promote the electrocatalytic performance. The self-supported CoMoO4 bundles phosphorized at 350 °C exhibited the highest activity in 1 M KOH with low overpotentials of 56 mV @ − 10 mA cm−2 and 148 mV @ − 100 mA cm−2 as well as a low Tafel slope of 78 mV dec−1. The excellent HER activity and high stability enable P-CoMoO4-350 a promising HER electrocatalyst in basic media. Our work suggests that P doping engineering is an effective route to tune the HER performance of transition metal oxides.
References
Roger I, Shipman MA, Symes MD (2017) Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat Rev Chem 1:0003
Zhou HQ, Yu F, Zhu Q, Sun JY, Qin F, Yu L, Bao JM, Yu Y, Chen S, Ren ZF (2018) Water splitting by electrolysis at high current densities under 1.6 volts. Energy Environ Sci 11:2858–2864
Men YN, Li P, Zhou JH, Cheng GZ, Chen SL, Luo W (2019) Tailoring the electronic structure of Co2P by N doping for boosting hydrogen evolution reaction at all pH values. ACS Catal 9:3744–3752
Zhu JQ, Wang ZC, Dai HJ, Wang QQ, Yang R, Yu H, Zhou M, Zhang LJ, Chen W, Wei Z, Li N, Du LJ, Shi DX, Wang WL, Zhang LX, Jiang Y, Zhang GY (2019) Boundary activated hydrogen evolution reaction on monolayer MoS2. Nat Commun 10:1348
Li Y, Guo SW, Jin T, Wang YL, Cheng FY, Jiao LF (2019) Promoted synergy in core-branch CoP@NiFe–OH nanohybrids for efficient electrochemical/photovoltage-driven overall water splitting. Nano Energy 63:103821
Fang LX, Wang F, Zhai TL, Qiu Y, Lan MD, Huang KJ, Jiang QS (2018) Hierarchical CoMoO4 nanoneedle electrodes for advanced supercapacitors and electrocatalytic oxygen evolution. Electrochim Acta 259:552–558
Wu YY, Liu YP, Li GD, Zhou X, Lian XR, Wang DJ, Sun L, Asefa T, Zou XX (2017) Efficient electrocatalysis of overall water splitting by ultrasmall NixCo3−xS4 coupled Ni3S2 nanosheet arrays. Nano Energy 35:161–170
Wang Y, Ma B, Chen Y (2019) Iron phosphides supported on three-dimensional iron foam as an efficient electrocatalyst for water splitting reactions. J Mater Sci 54(24):14872–14883. https://doi.org/10.1007/s10853-019-03985-9
Zhang YY, Zhang X, Wu ZY, Zhang BB, Zhang Y, Jiang WJ, Yang YG, Kong QH, Hu JS (2019) Fe/P dual doping boosts the activity and durability of CoS2 polycrystalline nanowires for hydrogen revolution. J Mater Chem A 7:5195–6200
Tao LM, Li M, Wu SH, Wu SH, Wang QL, Xiao X, Li QW, Wang MK, Fu YQ, Shen Y (2018) Sea coral-like NiCo2O4@(Ni, Co)OOH heterojunctions for enhancing overall water-splitting. Catal Sci Technol 8:4151–4158
Xiang R, Duan YJ, Peng LS, Wang Y, Tong C, Zhang L, Wei ZD (2019) Three-dimensional Core@Shell Co@CoMoO4 nanowire arrays as efficient alkaline hydrogen evolution electro-catalysts. Appl Catal B 246:41–49
Guo JX, Yang DY, Qiu KW, Mu C, Jiao D, Mao J, Wang H, Ling T (2019) High electrocatalytic hydrogen evolution activity on a coupled Ru and CoO hybrid electrocatalyst. J Energy Chem 37:143–147
Guo CY, Sun X, Kuang X, Gao LF, Zhao MZ, Qu L, Zhang Y, Wu D, Ren X, Wei Q (2018) Amorphous Co-doped MoOx nanospheres with a core-shell structure toward an effective oxygen evolution reaction. J Mater Chem A 7:1005–1012
Guo JX, Zhang XQ, Sun YF, Tang L, Zhang X (2017) Self-template synthesis of hierarchical CoMoS3 Nanotubes constructed of ultrathin nanosheets for robust water electrolysis. J Mater Chem A 5:11309
Du X, Huang C, Zhang X (2019) Synthesis of CoMoO4/Co9S8 network arrays on nickel foam as efficient urea oxidation and hydrogen evolution catalyst. Inter J Hydrogen Energy 44:19595–19602
Zhao JX, Ren X, Ma HM, Sun X, Zhang Y, Yan T, Wei Q, Wu D (2017) Synthesis of self-supported amorphous CoMoO4 nanowire array for highly efficient hydrogen evolution reaction. ACS Sustain Chem Eng 5:10093–10098
Jiang D, Xu Y, Yang R, Li D, Meng SC, Chen M (2019) CoP3/comop heterogeneous nanosheet arrays as robust electrocatalyst for pH-universal hydrogen evolution reaction. ACS Sustain Chem Eng 7:9309–9317
Jiang DL, Ma WX, Yang R, Quan B, Li D, Meng SC, Chen M (2020) Nickel–manganese bimetallic phosphides porous nanosheet arrays as highly active bifunctional hydrogen and oxygen evolution electrocatalysts for overall water splitting. Electrochim Acta 329:135121
Yang R, Zhou YM, Xing YY, Li D, Dl Jiang, Chen M, Shi WD, Yuan SQ (2019) Synergistic coupling of CoFe-LDH arrays with NiFe-LDH nanosheet for highly efficient overall water splitting in alkaline media. Appl Catal B Environ 253:131–139
Yan X, Tian L, Atkins S, Liu Y, Murowchick J, Chen XB (2016) Converting CoMoO4 into CoO/MoOx for overall water splitting by hydrogenation. ACS Sustain Chem Eng 4:3743–3749
Li S, Yang N, Liao L, Luo YZ, Wang SY, Gao FF, Zhou W, Huang DK, Chen H (2018) Doping & β-CoMoO4 nanoplates with phosphorus for efficient hydrogen evolution reaction in alkaline media. ACS Appl Mater Interfaces 10:37038–37045
Liu Z, Zhan CH, Peng LK, Gao Y, Chen Y, Ding SJ, Xiao CH, Lai XY, Li JW, Wei SR, Wang JQ, Tu JC (2019) A CoMoO4–Co2Mo3O8 heterostructure with valence-rich molybdenum for a high-performance hydrogen evolution reaction in alkaline solution. J Mater Chem A 7:16761
Liu XH, Wei B, Su R, Zhao CG, Dai DM, Ma X, Xu LL (2019) Mo-doped cobalt phosphide nanosheets for efficient hydrogen generation in alkaline media. Energy Technol 7:1900021
Chi K, Tian X, Wang Q, Zhang Z, Zhang X, Zhang Y, Wang S (2020) Oxygen vacancies engineered CoMoO4 nanosheet arrays as efficient bifunctional electrocatalysts for overall water splitting. J Cata 381:44–52
Wang XH, Rong F, Huang FF, Peng H, Yang Y, Tang JP, Que RH (2019) Facile synthesis of hierarchical CoMoO4@Ni(OH)2 core-shell nanotubes for bifunctional supercapacitors and oxygen electrocatalysts. J Alloys Compds 789:684–692
Ou YQ, Tian WQ, Liu L, Zhang YH, Xiao P (2018) Bimetallic Co2Mo3O8 suboxides coupled with conductive cobalt nanowires for efficient and durable hydrogen evolution in alkaline electrolyte. J Mater Chem A 2:5217–5228
Yang Q, Lin SY (2016) Rationally designed nanosheet-based CoMoO4–NiMoO4 nanotubes for high-performance electrochemical electrodes. RSC Adv 6:10520
Zhang Y, Guo H, Yuan P, Pang K, Cao B, Wu X, Song R (2019) Structural evolution of CoMoO4 to CoOOH by ion electrochemical etching for boosting oxygen evolution reaction. J Power Sour 442:227252
Wei Y, Zhang X, Zhao ZL, Chen HS, Postolek KM, Wang B, Yang P (2019) Controllable synthesis of P-doped MoS2 nanopetals decorated N-doped hollow carbon spheres towards enhanced hydrogen evolution. Electrochimica Acta 297:553–563
Li GC, Yue L, Fan RK, Liu D, Li XB (2017) Synthesis of a Co–Mo sulfide catalyst with a hollow structure for highly efficient hydrodesulfurization of dibenzothiophene. Catal Sci Techol 23:5505–5509
Yin ZX, Chen YJ, Zhao Y, Li CY, Zhu CL, Zhang XT (2015) Hierarchical nanosheet-based CoMoO4–NiMoO4 nanotubes for applications in asymmetric supercapacitors and the oxygen evolution reaction. J Mater Chem A 3:22750
Zhou QW, Shen ZH, Zhu C, Li JC, Ding ZY, Wang P, Pan F, Zhang ZY, Ma HX, Wang SY, Zhang HG (2018) Nitrogen-doped cop electrocatalysts for coupled hydrogen evolution and sulfur generation with low energy consumption. Adv Mater 30:1800140
Sun X, Yang XX, Xiang H, Mi HW, Zhang PX, Ren XZ, Li YL, Li XF (2018) Nitrogen-doped CoOx/carbon nanotubes derived by plasma-enhanced atomic layer deposition: efficient bifunctional electrocatalyst for oxygen reduction and evolution reactions. Electrochim Acta 296:964–971
Yang J, Guo DH, Zhao SL, Lin Y, Yang R, Xu DD, Shi NE, Zhang XH, Lu LZ, Lan YQ, Bao JC, Han M (2019) Cobalt phosphides nanocrystals encapsulated by P-doped carbon and married with P-doped graphene for overall water splitting. Nano Micro Small 28:1804546
Xia ZF, Lei W, Hao QL, Wang WJ, Wang X (2013) One-step synthesis of CoMoO4/graphene composites with enhanced electrochemical properties for supercapacitors. Electrochim Acta 99:253–261
Wang FF, Zhao J, Tian W, Hu ZF, Lv XB, Zhang HL, Yue HR, Zhang YX, Ji JY, Jiang W (2019) Morphology-controlled synthesis of CoMoO4 nanoarchitectures anchored on carbon cloth for high-efficiency oxygen oxidation reaction. RSC Adv 9:1562
Han MM, Hu LL, Zhou YJ, Zhao SQ, Bai L, Sun Y, Huang H, Liu Y, Kang ZH (2018) Z-Scheme in a Co3(PO4)2/α-Fe2O3 photocatalysis system for overall water splitting under visible light. Catal Sci Technol 8:840–846
Ouyang T, Ye YQ, Wu CY, Xiao K, Liu ZQ (2019) Heterostructures composed of N-doped carbon nanotubes encapsulating cobalt and Mo2C nanoparticles as bifunctional electrodes for water splitting. Angew Chem Int Ed 58:4923–4928
Zhang X, Zhang YY, Zhang Y, Jiang WJ, Zhang QH, Yang YG, Gu L, Hu JS, Wan LJ (2019) Phase-controlled synthesis of 1T-MoSe2/NiSe heterostructure nanowire arrays via electronic injection for synergistically enhanced hydrogen evolution. Small Methods 3:1800317
Chen YY, Zhang Y, Zhang X, Tang T, Luo H, Niu S, Dai ZH, Wan LJ, Hu JS (2017) Self-Templated fabrication of MoNi4/MoO3−x nanorod arrays with dual active components for highly efficient hydrogen evolution. Adv Mater 29:1703311
Wu AP, Xie Y, Ma H, Tian CG, Gu Y, Yan HJ, Zhang XM, Yang GY, Fu HG (2018) Integrating the active OER and HER components as the heterostructures for the efficient overall water splitting. Nano Energy 44:356–363
Lamoureux PS, Singh AR, Chan K (2019) pH effects on hydrogen evolution and oxidation over Pt(111): insights from first-principles. ACS Catal 9:6194–6201
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21773048), Heilongjiang Natural Science Foundation (A2015005) and University Nursing Program for Young Scholar with Creative Talent in Heilongjiang Province (UNPYSCT-2015052).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ma, X., Wei, B., Yuan, M. et al. Self-supported phosphorus-doped CoMoO4 rod bundles for efficient hydrogen evolution. J Mater Sci 55, 6502–6512 (2020). https://doi.org/10.1007/s10853-020-04448-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-020-04448-2