
Abstract
The fast recombination of photo-generated conduction band electrons (e −cb ) and valance band holes (h +vb ) of TiO2 results in an unsatisfactory photocatalytic performance for organic degradation. To increase the efficiency of charge separation, TiO2 was modified by Cu–Ce co-doping considering the better redox properties of copper–ceria oxide with respect to the single oxide, i.e., an easier electron capturing ability. An optimal Cu–Ce co-doped TiO2 with the initial molar ratio of Cu/Ce at 3:1 was prepared by a hydrothermal method with the aim to greatly promote the charge separation, and characterized by XRD, BET, DRS, PL, HR-TEM, and XPS techniques. Upon ultraviolet light irradiation, it exhibits significantly enhanced photocatalytic activity, about 5.8 times that of Ti–HF. The presence of Cu2+ and Ce3+/Ce4+ benefits electrons captured by molecular oxygen, while an increased hydroxyl groups upon Cu–Ce co-doping consume more holes, resulting in prolonged lifetime of photo-generated carriers. Moreover, it is proved that electron transfers preferably from conduction band (CB) of TiO2 to CB of CuO and then to nearby CeO2.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Semiconductor photocatalysts have gained significant attention and much research fascination due to their promising applications in converting solar energy into chemical energy and removing organic pollutants from water [1–5]. Among various metal oxide semiconductor materials, TiO2 has received considerable interest because of its favorable physical/chemical properties, high stability, low cost, and nontoxicity [6, 7]. However, the fast recombination of photo-generated conduction band electrons (e −cb ) and valance band holes (h +vb ) of TiO2 results in an unsatisfactory photocatalytic performance for organic degradation.
To increase the efficiency of charge separation, one of the strategies is to dope metal ions, which can trap electrons and thus free up the excited holes. Noble metal nanoparticles (e.g., Au, Ag, and Pt) [8] were used to quickly capture electrons from photoexcited TiO2 but with high cost. Hence, surface modification of TiO2 with transition metal ions has been employed, such as Cu2+ [9, 10], which promotes the multielectron reduction reaction of oxygen.
On the other hand, enriching the dissolved O2 onto the oxide surface from aqueous solution ensures the subsequent surface catalytic reaction for molecular oxygen activation and thus high quantum efficiency [11]. Recently, it is proved that ceria used commonly as oxygen storage material in the automotive exhaust purification is also a good oxygen promoter of TiO2 photocatalysis [12].
Considering the better redox properties of copper–ceria oxide with respect to the single oxide [13], i.e., an easier electron capturing ability, we synthesized coupled Cu–Ce–Ti oxide using hydrothermal method and investigated its photocatalytic degradation performance. In that case, photoexcited electrons will be more easily trapped by Cu sites and then transferred to activated oxygen species on Ce sites nearby, resulting in a prolonged charge separation time and thus an effective surface reaction. Since the band edges of TiO2 bracket those of CuO, the doping amount of copper–ceria oxide should be appropriate, which was fixed at 3 wt% in the form of CuO and CeO2 related to TiO2 in this study. Otherwise, photoexcited charge carriers in TiO2 may transfer to and ultimately recombine in CuO [14] or a shielding effect may happen. The present work demonstrates that rational design and construction of Cu–Ce co-doped TiO2 is an effective strategy for modifying TiO2 in photocatalytic performances.
Experimental
Hydrothermal synthesis of Cu–Ce–Ti mixed oxides
In a typical synthesis, 5 mL of Ti(OBu)4 was mixed with 20 mL of absolute ethanol and 0.6 mL of 40 % HF under vigorous stirring. Then, required amounts of Cu(NO3)2·3H2O and Ce(NO3)3·6H2O were added to the above solution. After continuously stirring for 3 h, the resulting solution is transferred to a Teflon lined stainless-steel autoclave and then kept at 160 °C for 24 h. When the autoclave was cooled to room temperature, the precipitate was centrifuged, thoroughly washed with absolute ethanol, and subsequently dried at 70 °C in vacuum overnight. For convenience, the obtained powders are denoted as xCuyCe–Ti–HF, Cu–Ti–HF, and Ce–Ti–HF, respectively, where xCuyCe represents the initial molar ratio of Cu to Ce at 1:3, 1:1, and 3:1 during preparation processes. The actual weight ratios of Cu and Ce elements in xCuyCe–Ti–HF were analyzed by inductively coupled plasma (ICP) performed on a JY Ultima2 spectrometer and listed in Table 1. For comparison, a similar process was adopted to synthesize Ti–HF and Ti–H2O without adding copper and cerium resources, and Ti–H2O means the substitution of HF with 0.6 mL of H2O.
Photocatalytic evaluation
The photocatalytic activity of the samples was evaluated by monitoring the decomposition of methyl orange (MO) under ultraviolet (UV) light at room temperature. The radiation source used for this study was UV-C lamp from Philips (Model TUV PL-S 9W/2P 1CT), and the intensity of the incident light was 10 mW/cm2. Each prepared sample (15 mg) was suspended in 150 mL MO aqueous solution with a concentration of 9.8 mg L−1 in a beaker. The suspension was magnetically stirred for 0.5 h to reach absorption equilibrium before UV-light exposure. After the start of photocatalytic reaction, samples were taken at regular time and the supernatant MO dye concentration was measured at the maximum absorption wavelength (464 nm).
Characterization
X-ray diffraction (XRD) patterns were collected on a MiniFlex600, and the accelerating voltage and the applied current were 40 kV and 40 mA, respectively. The specific surface area, total pore volume, and average pore radius were measured by N2 physisorption at −196 °C (BELSORP-mini II instrument). The UV–Vis diffuse reflectance spectra (DRS) were performed at a Varian Cary 500 apparatus equipped with an integrating sphere, using BaSO4 as a reference. Photoluminescence (PL) spectra were recorded on RF-5301PC spectrophotometer using room temperature photoluminescence with a 280 nm excitation wavelength. The microstructures of the sample were investigated using a high-resolution transmission electron microscope (JEM-2010) with an accelerating voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) measurements were performed on Physical Electronics Quantum 2000, equipped with a monochromatic Al-K α source (K α = 1486.6 eV) and a charge neutralizer. The C 1s peak at 284.6 eV of the adventitious carbon was referenced to rectify the binding energies.
Results and discussion
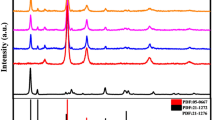
Figure 1 shows the XRD diffractograms of the undoped and Cu–Ce-doped TiO2. For undoped TiO2, the observed peaks at 2θ of 25.3°, 37.8°, 48.0°, 53.8°, 54.9°, and 62.7° are ascribed to the diffraction of the (101), (004), (200), (105), (211), and (204) crystal planes of anatase (JCPDS 21-1272). The characteristic peaks in Ti–HF are more intense than those in Ti–H2O, indicating HF is favorable to the TiO2 crystallization. Moreover, the weak (004) diffraction peak in Ti–HF excludes the morphology-controlling role of HF in favoring the formation of (001) plane in our work [15]. After doping Cu–Ce oxides, the anatase crystallization is further enhanced (see the inset of Fig. 1) and no diffraction peaks about Cu or Ce species can be detected due to their small content below the detection limit of XRD. Furthermore, the weak diffraction peak at 2θ of 27.5° indexed to rutile (110) crystalline plane is found.

XRD patterns of a Ti–H2O, b Ti–HF, c 1Cu3Ce–Ti–HF, d 1Cu1Ce–Ti–HF, and e 3Cu1Ce–Ti–HF
The specific surface area, pore volume, and average pore diameter are summarized in Table 1. The specific surface area of Ti–HF and Ti–H2O is 161 and 227 m2/g, respectively. A drop in S BET is observed over Cu–Ce-modified TiO2. 3Cu1Ce–Ti–HF exhibits an apparent larger surface area than 1Cu3Ce–Ti–HF and 1Cu1Ce–Ti–HF, coinciding with the crystallization degree as indicated in the above XRD analysis.
Photocatalytic performance was evaluated for degrading aqueous solution of MO under UV irradiation. As seen in Fig. 2A, Ti–H2O with the largest surface area shows the lowest UV-light photoactivity. This should be related to its poor anatase crystallinity accelerating charge recombination. Mono-doping samples exhibit inferior performance than co-doping samples, indicating a synergistic effect between Cu and Ce species. Especially, 3Cu1Ce–Ti–HF shows the best degradation efficiency, about 5.8 times that of Ti–HF calculated from the kinetic constants (Fig. 2B). The kinetic linear simulation curves over the five catalysts show that the degradation reactions follow a Langmuir–Hinshelwood apparent first-order kinetic model. Combined with XRD results, it is concluded that the enhanced degree of anatase crystallization is one factor improving photoactivity for 3Cu1Ce–Ti–HF with respect to Ti–HF.

A Degradation curves and B kinetic curves of MO catalyzed by samples: (a) Ti–H2O, (b) Ti–HF, (c) 1Cu3Ce–Ti–HF, (d) 1Cu1Ce–Ti–HF, (e) 3Cu1Ce–Ti–HF, (f) Cu–Ti–HF, (g) Ce–Ti–HF
The optical properties of TiO2-based samples were examined using UV–Vis spectroscopy, which are illustrated in Fig. 3A. Ti–H2O and Ti–HF present typical UV–Vis diffuse reflectance spectra of anatase with a cut-off wavelength at ~400 nm, corresponding to its band gap of ca. 3.2 eV. In the presence of Cu–Ce oxide, no obvious red shift for the cut-off wavelength can be observed, suggesting that Cu–Ce co-doping does not alter the band gap of TiO2. However, the intensity of absorption in the 200–400 nm increases with the raise of Ce component for Cu–Ce-modified TiO2. The observed enhanced UV-light absorbance is expected to increase the utilization of UV-light for photocatalytic reaction. Moreover, we employ photoluminescence spectra (Fig. 3B) to characterize the recombination probability of photoexcited carriers in TiO2-based samples.

A UV–Vis diffuse reflectance spetra; B PL spectra of (a) Ti–H2O, (b) Ti–HF, (c) 1Cu3Ce–Ti–HF, (d) 1Cu1Ce–Ti–HF, (e) 3Cu1Ce–Ti–HF; C HR-TEM image of 3Cu1Ce–Ti–HF; and D effects of a series of scavengers on the degradation of MO over 3Cu1Ce–Ti–HF
A high PL spectra intensity of bare TiO2 shown in Fig. 3B implies a high recombination tendency of the photo-generated charged carriers [16], while the addition of Cu–Ce oxide results in an apparent lower PL emission peak. Thus, the efficiency of charge separation in TiO2 has been significantly promoted by Cu–Ce doping, and a best photocatalytic performance is obtained over 3Cu1Ce–Ti–HF. It is speculated that there is a tendency of electron transfer from the conduction band (CB) of TiO2 to the CB of CuO rather than Cu2O due to the potential difference [14, 17], and then to CeO2 nearby that adsorbs O2 from aqueous solution, consequently ensuring effective electron transfer and surface oxygen activation. The successful construction of 3Cu1Ce–Ti–HF is further demonstrated by the HR-TEM images in Fig. 3C. The lattice d-spacing of observed particles are measured to be 0.353, 0.234, and 0.316 nm, matching with the lattice spacing of TiO2 (101), CuO (111), and CeO2 (111), respectively.
The main reactive species that trigger the oxidation are disclosed by adding scavengers of KBrO3, ammonium oxalate (AO), benzoquinone (BQ), and isopropanol (IPA), which are used to quench CB electrons, h+, ·O2 −, and ·OH, respectively. As shown in Fig. 3D, after adding AO and IPA, a bit more decreased efficiency is found for AO, indicating ·OH is one main reactive species since ·OH can be converted from h+. If h+ is the main reactive species, the addition of IPA will show little suppression effect. However, KBrO3 scavenger shows a positive effect on the photocatalytic oxidation of MO, indicating e− is not involved in the phototransformation of MO. As for BQ scavenger, it shows an obvious suppression effect, demonstrating ·O2 − is another main reactive species.
To verify the assumption of the electron transfer pathway, XPS studies are conducted and XPS spectra of Ti 2p, O 1s, Cu 2p3/2, and Ce 3d core level profiles are shown in parts a–d of Fig. 4. The Ti 2p3/2 and Ti 2p1/2 are located near binding energies of 458.9 and 464.7 eV, respectively, agreeing with the values of Ti4+ in the TiO2 lattice [18]. After adding Cu–Ce oxide, the Ti 2p3/2 and Ti 2p1/2 binding energy values are slightly shifted to higher binding energy regions, implying the formation of Ti–O–Cu bonds based on the changes in electron densities [19]. That is, the increasing binding energies of Ti 2p could be explained by considering the electrons transfer from TiO2 to Cu species.

XPS of samples: A Ti 2p, B O 1s, C Cu 2p3/2, and D Ce 3d
The O 1s peak at 530.0 eV (denoted as OI) could be attributed to lattice oxygen of TiO2, while the peak at 530.6 eV (denoted as OII) is ascribed to adsorbed OH− on the surface of TiO2 [20]. As listed in Table 1, the amount of hydroxyl groups is in the following order: 3Cu1Ce–Ti–HF > 1Cu3Ce–Ti–HF > 1Cu1Ce–Ti–HF. It is considered that the hydroxyl radicals can be formed by the reaction of photo-generated holes with the hydroxyl groups, thus consuming photoexcited holes and promoting charge separation. Cu 2p3/2 characteristic peaks for Cu+ and Cu2+ appear at 932.6 and 933.6 eV, respectively [21]. Meanwhile, there is no evident satellite at 940–950 eV belonging to Cu2+ probably due to the low content of Cu element [14]. According to Table 1, the content of Cu2+ is calculated to be the highest for 3Cu1Ce–Ti–HF, agreeing well with its best performance and reinforcing the idea of the electron transfer pathway from the conduction CB of TiO2 to the CB of CuO. Regarding Ce 3d, due to the low content of Ce and self-complicated cures of Ce 3d, the spectra are not fitted and the binding energies of Ce 3d3/2 peak at 917.5 eV and Ce 3d5/2 peak at 885.2 eV are distinguished as a judge for the existence of Ce4+ and Ce3+, respectively [22]. The redox pair of cerium could not only act as electron scavenger but also increase oxygen reserve, which retards the recombination of electron–hole pair and greatly promotes the catalytic oxidation activity.
Conclusion
Cu–Ce-modified TiO2 were synthesized through a hydrothermal method, which shows better anatase crystallization than Ti–HF. A decreased surface area is found over Cu–Ce co-doped TiO2. Cu–Ce co-doping influences the band gap little, but greatly promotes lifetime of photo-generated carriers. As a result, Cu–Ce co-doped TiO2 exhibits higher degradation efficiency than bare TiO2, and 3Cu1Ce–Ti–HF possesses the best performance. Moreover, it is proposed that the higher content of Cu2+ and existence of Ce4+/Ce3+ benefits the electron transfer and accelerates electron capture on surface molecular oxygen. HR-TEM provides a visual existence of Ti–Cu–Ce construction, while XPS indicates the formation of Ti–O–Cu bonds. Additionally, Cu–Ce co-doping results in more hydroxyl groups which can be attacked by photoinduced holes. All in all, the charge separation in TiO2 can be promoted by the synergistic effects of Cu and Ce co-doping.
References
Chen X, Mao SS (2007) Titanium dioxide nanomaterials: synthesis, properties, modifications, and applications. Chem Rev 107:2891–2959
Baransi K, Dubowski Y, Sabbah I (2012) Synergetic effect between photocatalytic degradation and adsorption processes on the removal of phenolic compounds from olive mill wastewater. Water Res 46:789–798
Wei H, Wu Y, Lun N, Zhao F (2004) Preparation and photocatalysis of TiO2 nanoparticles co-doped with nitrogen and lanthanum. J Mater Sci 39:1305–1308. doi:10.1023/B:JMSC.0000013889.63705.f3
Ni M, Leung MKH, Leung DYC, Sumathy K (2007) A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renew Sustain Energy Rev 11:401–425
Nagaraju G, Manjunath K, Ravishankar TN, Ravikumar BS, Nagabhushan H, Ebeling G, Dupont J (2013) Ionic liquid-assisted hydrothermal synthesis of TiO2 nanoparticles and its application in photocatalysis. J Mater Sci 48:8420–8426. doi:10.1007/s10853-013-7654-5
Zhang J, Xu Q, Feng Z, Li M, Li C (2008) Importance of the relationship between surface phases and photocatalytic activity of TiO2. Angew Chem Int Ed 47:1766–1769
Liu S, Guo E, Yin L (2012) Tailored visible-light driven anatase TiO2 photocatalysts based on controllable metal ion doping and ordered mesoporous structure. J Mater Chem 22:5031–5041. doi:10.1039/C2JM15965A
Jang JS, Kim HG, Lee JS (2012) Heterojunction semiconductors: a strategy to develop efficient photocatalytic materials for visible light water splitting. Catal Today 185:270–277
Zhang F, Cheng Z, Kang L, Cui L, Liu W, Hou G, Yang H, Xu X (2014) 3D controllable preparation of composite CuO/TiO2 nanofibers. RSC Adv 4:63520–63525
Zhang H, Guo L, Wang D, Zhao L, Wan B (2015) Light-induced efficient molecular oxygen activation on a Cu (II)-grafted TiO2/graphene photocatalyst for phenol degradation. ACS Appl Mater Interfaces 7:1816–1823
Peirco AM, Colombo C, Doyle G, Nelson J, Mills A, Durrant JR (2006) Photochemical reduction of oxygen adsorbed to nanocrystalline TiO2 films: a transient absorption and oxygen scavenging study of different TiO2 preparations. J Phys Chem B 110:23255–23263
Li Z, Sheng J, Zhang Y, Li X, Xu Y (2015) Role of CeO2 as oxygen promoter in the accelerated photocatalytic degradation of phenol over rutile TiO2. Appl Catal B 166–167:313–319
Luo Y, Wang K, Xu Y, Wang X, Qian Q, Chen Q (2015) The role of Cu species in electrospun CuO-CeO2 nanofibers for total benzene oxidation. New J Chem 39:1001–1005
Li G, Dimitrijevic NM, Chen L, Rajh T, Gray KA (2008) Role of surface/interfacial Cu2+ sites in the photocatalytic activity of coupled CuO–TiO2 nanocomposites. J Phys Chem C 112:19040–19044
Zheng J, Bao S, Guo Y, Jin P (2014) Anatase TiO2 films with dominant 001 facets Fabricated by direct-current reactive magnetron sputtering at room temperature: oxygen defects and enhanced visible-light photocatalytic behaviors. ACS Appl Mater Interfaces 6:5940–5946
Muñoz-Batista MJ, Fernández-García M, Kubacka A (2015) Promotion of CeO2–TiO2 photoactivity by gC3N4: ultraviolet and visible light elimination of toluene. Appl Catal B 164:261–270
Huang L, Peng F, Wang H, Yu H, Li Z (2009) Preparation and characterization of Cu2O/TiO2 nano-nano heterostructure photocatalysts. Catal Commun 10:1839–1843
Erdem B, Hunsicker RA, Simmons GW, Sudol ED, Dimonie VL, El-Aasser MS (2001) XPS and FTIR surface characterization of TiO2 particles used in polymer encapsulation. Langmuir 17:2664–2669
Lin C, Yang W (2014) Ordered mesostructured Cu-doped TiO2 spheres as active visible-light-driven photocatalysts for degradation of paracetamol. Chem Eng J 237:131–137
Toma F, Bertrand G, Begin S, Meunier C, Barres O, Klein D, Coddet C (2006) Microstructure and environmental functionalities of TiO2-supported photocatalysts obtained by suspension plasma spraying. Appl Catal B 68:74–84
Moulder JF, Stickle WF, Sobol PE, Bomben KD (1995) Handbook of X-ray photoelectron spectroscopy: a reference book of standard spectra for identification and interpretation of xps data. Perkin-Elmer Corporation, Waltham
Wang S, Zhang J, Jiang J, Liu R, Zhu B, Xu M, Wang Y, Cao J, Li M, Yuan Z, Zhang S, Huang W, Wu S (2009) Porous ceria hollow microspheres: synthesis and characterization. Micropor Mesopor Mater 1–3:349–353
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21407025, 21307012), National Science Foundation of Fujian Province (2016J01047, 2014J01035), National Program for the Fujian Provincial Science and Technology and the Provincial Financial Department (2013H6007), Educational Commission of Fujian Province (JA14090), and Outstanding Youth Research Training Program of University of Fujian Province.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Luo, Y., Xu, Y., Liu, X. et al. Design of Cu–Ce co-doped TiO2 for improved photocatalysis. J Mater Sci 52, 1265–1271 (2017). https://doi.org/10.1007/s10853-016-0421-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-016-0421-7