
Abstract
A novel and simple in situ synthetic strategy was used to fabricate CdS/g-C3N4 hybrid nanocomposite catalysts with visible-light-driven photocatalytic activity from cadmium-containing carbon nitride compounds. X-ray diffraction measurements, high-resolution transmission electron microscopy images, and Fourier transform infrared spectra showed heterojunctions with a close interface between the g-C3N4 and the CdS nanoparticles and nanorods in the composite. Ultraviolet visible diffuse reflectance spectra exhibited a red shift that further presented the CdS in the polymer g-C3N4 skeleton, which allowed the efficient utilization of the solar spectrum for creating photogenerated electrons and holes. The photoluminescence spectra of the nanocomposites suggested charge transfer from g-C3N4 to CdS. The photocurrent intensity of hybrid nanocomposites was 2.3 times than that of pure g-C3N4 sample, and photocatalytic activity for the photodegradation of methyl orange was 2.5 times, and hydrogen evolution reaction was 2.8 times. Enhanced photocatalytic activity and photocurrent for the CdS/g-C3N4 hybrid nanocomposites were achieved.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Serious environmental pollution and global energy shortage have recently led to increasing interest in studying the application of photocatalyst technology on water pollution treatment and hydrogen evolution [1–3]. In 1972, Fujishima [4] reported TiO2 as the electrode for water in photolysis, and several researchers have exerted great efforts in searching for suitable semiconductor materials to apply as photocatalysts since then [5, 6]. A typical semiconductor material CdS has a wide range of application in photocatalysis and electronic devices [7]. CdS has shown promise in enhancing photodegradation because it can efficiently separate photogenerated charge carriers [8]. However, CdS is unstable under photo-corrosion because the photogenerated holes self-oxidize during the photocatalytic process.
Wang [9] has recently produced a polymeric carbon nitride material (C3N4), which has been used as a metal-free photocatalyst for water splitting produce hydrogen application. Carbon nitride (g-C3N4), which is essentially composed of covalent bonds, is the most promising candidate to complement carbon in phocatalytic applications. Carbon nitride posses a high thermal and chemical stability and has attracted attention because of its outstanding mechanical, electrical, thermal, and optical properties. Nevertheless, the low efficiency caused by the high recombination rate of photogenerated charges limits the practical application of the current material. Several methods, including fabrication porous structure doped with metal or nonmetal elements, have been developed to enhance the phtocatalytic capability of carbon nitride [10–20]. Furthermore, Bai et al. fabricated a g-C3N4 from nanoplates to nanorods, which enhanced the photocatalytic activity and photocurrent intensity about 1.5 and 2.0 times those of g-C3N4 nanoplates, respectively [21]. Combining C3N4 with other semiconductors to form heterostructures particularly provides a feasible route to inhibit the recombination of photogenerated electron-hole pairs. Efforts are exerted to design organic and inorganic hybrid structures that exhibit improved selectivity and efficiency toward light energy conversion [22].
The energy levels of C3N4 and CdS are compared and their well-matched overlapping band-structures are suitable to construct heterostructures that can effectively separate and transfer photogenerated charges. Therefore, combining C3N4 and CdS overcomes the disadvantages that exist in C3N4 and CdS simultaneously [23]. Feng et al. recently prepared CdS/g-C3N4 composites through chemical impregnation and subsequent calcination of separately prepared CdS particles and g-C3N4 [24]. Cao et al. reported an in situ growth CdS/g-C3N4 composites using a solvothermal method, the combination of as-prepared g-C3N4 and Cd resource in dimethyl sulfoxide as the S resource solution [25]. Fu et al. prepared CdS/g-C3N4 composites through an in situ precipitation–deposition method; the composites of the Cd and S resources were first prepared then combined with as-prepared g-C3N4 [26]. Jiang et al. fabricated g-C3N4-CdS composite through a precipitation method; the as-prepared g-C3N4 was first combined with the Cd resource and then combined with the S resource [22]. Lu et al. have also reported that the as-prepared ultrathin g-C3N4 nanosheets–CdS nanocomposite exhibited significantly enhanced photocatalytic activity for methyl orange (MO) degradation under visible light irradiation [27]. Significant advances have recently been made to the design and concern loaded type CdS-C3N4 composite particle. However, the phtocatalytic activity of the loaded type CdS-C3N4 needs further improvement because of the fast recombination of photogenerated charge carriers.
Thus, we report a novel and simple in situ synthetic approach toward CdS/g-C3N4 hybrid nanocomposites, which are based on the growth of CdS nanoparticles and nanorods within the g-C3N4 polymers from metal-containing carbon nitride compounds, similar to the synthesis of Fe-g-C3N4 [28] and ZnO-g-C3N4 [29]. CdS/g-C3N4 hybrid nanocomposites are synthesized using low-cost melamine and Cd(CH3COO)2·2H2O as precursors to obtain the Cd-g-C3N4, which are combined through a polycondensation process at elevated temperatures, and combined with sulfur. This process assures the two active components are efficiently in contact with each other. The synergistic effects of the two components significantly enhance the photocatalytic performance.
Experimental section
Chemicals
All reagents were analytical grade without any further purification before the experiment.
Synthesis of Cd/g-C3N4 compounds
First, Cd/g-C3N4 compounds were synthesized through a hydrothermal method. An amount of 4 g of melamine mixed with 100 mL of deionized water and 1 g of Cd(CH3COO)2·2H2O was heated and stirred at 80 °C for 40 min to create Cd/g-C3N4. The mixture was transferred into a Teflon-lined stainless autoclave (50 mL), maintained at 150 °C for 4 h, and then naturally cooled at room temperature. The precipitation was washed several times with deionized water to remove residual reactants and then dried in a vacuum oven at 60 °C for 3 h. The product was ground into powder and heated in a covered crucible under nitrogen atmosphere for 4 h to reach 550 °C. The Cd/g-C3N4 precursors were obtained by naturally cooling down to room temperature under nitrogen gas.
Synthesis of CdS/g-C3N4 hybrid materials
The obtained light yellow bulk was ground into powder and dispersed in saturated thiourea solution (35 mL) through ultrasonic treatment for 10 min. The mixed suspension was transferred into Teflon-lined stainless autoclave, kept in an oven at 150 °C for 4 h, and then naturally cooled to room temperature. Finally, the product was collected, washed with distilled water, vacuum dried, grounded, and stored for characterization.
The pure g-C3N4 was obtained through the same conditions but without adding the Cd(CH3COO)2·2H2O powder.
Characterization
The structures and compositions of the products were analyzed using X-ray diffraction (XRD) on a BRUKER D8 ADVANCE X-ray powder diffractometer with Cu Kα 1 radiation (λ = 1.5406 Å) within the 2θ range of 5°–80°. The accelerating voltage and emission current were 40 kV and 40 mA, respectively. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images were measured on FEI Tecnai G2 (F30 S–TWIN/X-MAX80) at 200 kV. Ultraviolet visible (UV–vis) diffuse reflectance spectra were measured using a UV–vis spectrophotometer (Agilent Cary 5000, USA). A Hitachi F-2500 spectroscope recorded the products of the photoluminescence (PL) spectra and is equipped with an integrated sphere attachment. Fourier transform infrared (FT-IR) spectra were recorded using Spectrum100 (PerkinElmer, USA). FT-IR spectrometry within the wavenumber range of 600–4000 cm−1 was applied using the KBr pellet technique. The Brunauer–Emmett–Teller (BET) surface area was determined by a multipoint BET method using Gemini VII 2390. Electrochemical measurements were performed with a CHI 660E electrochemical workstation (Chenhua Instrument, Shanghai, China) in a standard three-electrode system. The visible irradiation was obtained from a 500 W Xe lamp (CHF-MX, Beijing).
Photocatalytic activity for pollutant degradation
Photoelectrochemical measurements
Photocurrent measurements were performed on a CHI 660E electrochemical workstation in a conventional three-electrode configuration using a Pt foil and Ag/AgCl (saturated KCl) as counter and reference electrodes, respectively. A 500 W Xe arc lamp served as the light source. A 0.5 M Na2SO4 aqueous solution was used as the electrolyte. The working electrodes were prepared as follows: 10 mg of the prepared product was ground with 20 μL of a PEDOT-PSS (Sigma-Aldrich, 1.3–1.7 %) aqueous solution and 100 μL of distilled water to create a slurry. The slurry was then spread on a 2 × 1.5 cm2 FTO glass substrate, with an active area of approximately 0.5 cm2, using the doctor-blade method; adhesive tape was used as the space. The film was air dried and annealed in a vacuum oven at 150 °C for 30 min. The photoresponses of the samples as the light was turned on and off were measured at 0.2 V.
Measurement of photocatalytic activity
Degradation of methyl orange
The photocatalytic activities were evaluated through the decomposition of methyl orange (MO) under visible light irradiation (λ = 400 nm), which was obtained from a 300 W mercury lamp (XGY-1 Nanjing University) with cut-off filter of 400 nm. An amount of 0.1 g of the photocatalyst was dispersed in 200 mL of MO solution (the initial concentration was 10 mg L−1) in the dark and stirred for 30 min to reach the adsorption equilibrium before the photocatalytic reaction. Approximately 10 mL of the mixture was sampled at selected times during the photoreaction process, and the suspension was then centrifuged and filtered to separate the photocatalyst particles. The temperature of reaction solutions was sustained at 25 °C with cooling water circulating during the reaction. The concentration of MO was determined by the UV–vis spectrophotometer using a Shimadzu UV-2450 spectrophotometer at 464 nm.
Evolution of hydrogen
The photocatalytic hydrogen evolution was conducted in a closed gas circulation system, and a top window Pyrex cell was used as the reactor. The irradiation light source was a 300 W Xe lamp with a cut-off filter (λ > 420 nm), which was positioned 20 cm away from the reactor. In a typical photocatalytic experiment, the photocatalyst g-C3N4 (0.1 g) reaction system contained 10 ml of triethanolamine (TEOA) [30] aqueous solution, and the reaction system of CdS/g-C3N4 (0.1 g) was the aqueous solution with 1.0 M Na2SO3 and 0.35 M Na2S [31]. TEOA, Na2SO3, and Na2S were used as sacrificial reagents for photocatalytic hydrogen generation. The Pt cocatalyst (2 wt%) was photodeposited on the photocatalyst particles by directly dissolving H2PtCl6·6H2O into the reactant suspension. The temperature of the reaction solutions was sustained in an ambient temperature because of the flow of cooling water during the reaction. The other test conditions were equivalent. The evolved gas was determined by the online gas chromatography (Shimazu GC-14, with nitrogen as a carrier gas).
Results and discussion
Hybrid structure and optical properties
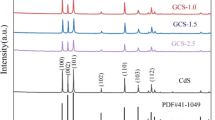
Figure 1(left) shows the XRD patterns of g-C3N4-CdS composite photocatalysts with different ratios of CdS/g-C3N4, pure g-C3N4, and Cd/g-C3N4. The XRD patterns reveal graphitic stacking of C3N4 layers in pure g-C3N4, Cd/g-C3N4, and CdS/g-C3N4. The peaks of 13.1° and 27.77° were the two characteristic diffractions of the g-C3N4 sample. The peak at 13.1° corresponded to the in-plane structural packing motif of the tristriazine unit and is indexed as the (100) peak [9]. The strong diffraction peak at 27.77° indicated the graphite-like stacking of the conjugated aromatic units of CN, which was indexed to the (002) plane of the hexagonal g-C3N4 (JCPDS# 87-1526). Both g-C3N4-CdS samples exhibited diffraction peaks corresponding to C3N4 and hexagonal wurtzite-structured CdS (JCPDS# 65-3414). No other impure peaks were observed, which suggests a two-phase composition of g-C3N4 and CdS in these composites. The two peaks of 13.1° and 27.77° gradually decreased in intensity when CdS was introduced, which is similar to the report of Fu Jie [26]. The XRD pattern of 1:4 CdS/g-C3N4 composites revealed distinct reflections at 24.925°, 26.450°, 28.217°, 36.424°, and 43.858° that corresponded to the (100), (002), (101), (102), and (110) planes of CdS, respectively.

XRD patterns of (a) pure g-C3N4, (b) Cd/g-C3N4, (c) 1/10CdS/g-C3N4, (d) 1/4CdS/g-C3N4 (left) and EDS spectrum of 1/4CdS/g-C3N4 (right)
Figure 1(right) shows the typical EDS spectrum of the CdS/g-C3N4 composites. Peaks associated with Cd, S, C, and N were observed in the EDS spectrum. The C/N molar ratio is nearly 0.75, and the S/Cd molar ratio is nearly 1.1. Both of them are close to the theoretically predicted of the C3N4 and CdS empirical stoichiometry, respectively. Similar observations were also reported for CdS nanostructured materials synthesized by solvothermal and hydrothermal methods [32]. The BET surface areas of g-C3N4, CdS, and CdS/g-C3N4 (1:4) photocatalysts were shown in Table 1. The BET surface areas of the pristine g-C3N4 and CdS were about 21.1 m2 g−1 and 12.8 m2 g−1, respectively. After CdS was doped, the BET of CdS/g-C3N4 (1:4) was 20.8 m2 g−1. The change in BET may be related to the hybrid of CdS and g-C3N4. Accordingly, the pore volume of the g-C3N4–CdS (1:3) is 0.13 cm3/g, between that of g-C3N4 or CdS.
Figure 2a shows the TEM image of pure g-C3N4. The pure g-C3N4 had an obviously thin laminar structure. Figure 2b–d shows the lower and higher magnification TEM images of the obtained CdS/g-C3N4 (1:4) composites. The obtained CdS/g-C3N4 nanocomposites were composed of two phases: hexagonal CdS and g-C3N4 polymer. The CdS/g-C3N4 hybrid nanocomposites were randomly distributed on the surface of g-C3N4 sheet (Fig. 2b). Several nanoparticles and nanorods were embedded in the obtained CdS/g-C3N4 nanocomposites. The particles were 3–5 nm in size and the rods had diameters of 4–5 nm and the length of 25–30 nm were gathered and intertwined to form sphere-like nanocomposites (Fig. 2c, d). These hybrid nanocomposites had a size of 65 nm (Fig. 2c). Similar observations were also reported for CdS microspheres prepared through a self-assembly method [32–35]. The HRTEM image (Fig. 2c–d) revealed the exact structure of the obtained CdS/g-C3N4 hybrid nanocomposites: the single crystalline CdS nanoparticles were embedded into the g-C3N4 polymers and nanorods located at the nanosphere border. The lattice part was CdS nanocrystals and the amorphous part was g-C3N4 polymers. The HRTEM image clearly revealed a close interface between the g-C3N4 and the CdS nanoparticles and nanorods in the composite, which formed heterojunctions. Consequently, the charge transfer between the two semiconductors was spatially smooth, which was fundamental to improving photocatalytic activity. The CdS crystallite in the hybrid nanocomposites exhibited three lattice planes with spacings of 0.357, 0.336, and 0.181 nm, which corresponded to the (100), (002), and (103) lattice planes of the hexagonal structured CdS nanocrystal, respectively. The lattice spacings of 0.336 and 0.181 nm observed in Fig. 2d also agreed well with the interplanar distances of the (002) and (103) direction parallel in the wurtzine phase of CdS.

TEM images of a pure g-C3N4 and b, c, and d CdS/g-C3N4 (1:4)
Figure 3 shows the FT-IR spectra of three kinds of samples. Several strong bands were found in the 1200–1650 cm−1 region, which corresponded to the typical stretching modes of the aromatic CN heterocycles as reported [2, 36]. The peaks were approximately at 1236, 1312, 1397, and 1636 cm−1. Moreover, the characteristic breathing mode of the triazine units was observed at 802 cm−1 [2, 37]. The broad bands at approximately 3000 cm−1 corresponded to the stretching mode of the terminal NH2 or NH groups at the defect sites of the aromatic ring [38]. The bands at 1533, 1457, 1129, and 711 cm−1 were attributed to the Cd–S bond [26, 29]. The FT-IR spectrum further confirmed the formation of composites that contained the two phases of CdS and g-C3N4.

FT-IR spectrum of (a) g-C3N4, (b) 1:10CdS/g-C3N4, and (c) 1:4CdS/g-C3N4
Figure 4 shows the UV–Vis spectrum of the pure g-C3N4, 1:4 CdS/g-C3N4, and 1:10 CdS/g-C3N4 composites. The pure g-C3N4 exhibited an absorption edge at 457 nm, which corresponded to the band gap of 2.71 eV. The samples of CdS/g-C3N4 with mass ratio of 1:10 and 1:4 had absorption edges at 490 and 497 nm, which corresponded to the band gaps of 2.53 and 2.49 eV, respectively. The absorption value and band gap of the pure CdS sample was 514 nm, which corresponded to the band gap of 2.41 eV according to the report of Cao [24]. Thus, the CdS/g-C3N4 composites exhibited hybrid absorption features of g-C3N4 and CdS, which allowed for efficiently utilizing the solar spectrum to create photogenerated electrons and holes. Figure 4 shows a red shift at −40 nm of the absorption edge (from pure g-C3N4 to CdS/g-C3N4) and the enhanced absorption intensity of CdS/g-C3N4 samples, which cause photocatalytic activity [39].

Absorption spectra of (a) pure g-C3N4, (b) 1:10CdS/g-C3N4, and (c) 1:4CdS/g-C3N4
Figure 5 shows the photoluminescence spectra of pure g-C3N4, 1:10 CdS/g-C3N4, and 1:4CdS/g-C3N4 with an excitation wavelength of 275 nm. The pure g-C3N4 sample exhibited a strong emission peak centered at 460 nm. The intensity of the emission spectral bands significantly decreased when CdS was introduced, which indicated an efficient transferring of photoexcited electrons from g-C3N4 to CdS. This spectra were useful in the migration, transfer of charge carriers, and separation and recombination processes of the photogenerated electron-hole pairs as Samanta S. reported [40, 41].

Photoluminescence spectra of (a) pure g-C3N4, (b) 1:10 CdS/g-C3N4, and (c) 1:4 CdS/g-C3N4
Photocatalytic activity and photocurrent response
Figure 6 shows the MO degradation curves of the photocatalysts of g-C3N4, CdS, and CdS/g-C3N4 under light irradiation (λ = 400 nm). Adsorption tests of photocatalysts were performed before running the photocatalytic reaction. All photocatalytic systems completely reached adsorption equilibrium with light irradiation after 30 min. The pure g-C3N4 solution and CdS solution was fully faded at 25 min and 60 min, respectively, but the suspension was fully faded at 15 min when the CdS/g-C3N4 hybrid nanocomposites were under light irradiation. To quantitatively investigate the reaction kinetics of the MB photocatalytic degradation, the photocatalytic data are analyzed using pseudo-first-order kinetics [26], which can be described as follows:

Photocatalytic degradation of MO over (a) g-C3N4, (b) CdS/g-C3N4 and (c) CdS under visible light (λ > 400 nm)
where C 0 is the initial MO concentration after stirring the suspension solution for 30 min, C t is the MO concentration at irradiation time t, and k obs is the observed pseudo-first-order rate constant. The observed first-order rate constant k obs can be obtained from the linear relationship of ln(C 0/C t) versus t. The calculated k obs of g-C3N4–CdS (1:4) is 0.2745 min−1, which is about 2 times as much as that of g-C3N4 (0.1187 min−1), indicative of an enhanced photocatalytic activity of g-C3N4–CdS photocatalysts. This result was due to the significant difference in the interface of the samples; the heterojunction photocatalyst exhibited a closely contacted interface, which is fundamental to improving photocatalytic activity.
The photocatalytic hydrogen evolution activity of the prepared CdS/g-C3N4 composite was evaluated under visible light irradiation (λ > 420 nm) using Pt (2.0 wt%) as the cocatalyst to reduce the overall potential for H2 evolution. The sacrificial reagents of 1.0 M Na2SO3 and 0.35 M Na2S were used to consume the photo-induced holes. In a similar experiment with pure g-C3N4, the triethanolamine was the sacrificial reagent. Other test conditions were equivalent. Figure 7 shows the H2 evolution rate over the sample under visible light irradiation. The catalytic g-C3N4 and CdS/g-C3N4 were uniformly loaded Pt (2.0 wt%). The hydrogen yield performance capacity of CdS/g-C3N4 was obviously higher than the g-C3N4. The maximum hydrogen production rate of CdS/g-C3N4 reached 259 μmol h−1, which was 2.8 times more than that of pure g-C3N4. A photocatalyst must have good stability to convert solar energy. Thus, the catalyst was subjected to continuous illumination experiments to evaluate its stability. A two-cycle experiment was performed. The amount of hydrogen reached approximately 1300 μmol after 6 h in the first cycle, and the two-cycle experiment had a hydrogen yield of 1000 μmol. This result demonstrated that photocatalysis performed better under visible light.

Plots of photocatalytic H2 evolution amount vs. irradiation (λ > 420 nm) times for (a) g-C3N4 and (b) CdS/g-C3N4
Figure 8 shows the results of the transient photocurrent response obtained from the pure g-C3N4 and CdS/g-C3N4 samples. The photocurrent intensity remained at a constant value when the light was on and rapidly decreased when the light was turned off. The photocurrent over CdS/g-C3N4 greatly improved, which was 2.3 times better than that of pure g-C3N4.

Transient photocurrent responses for (a) pure g-C3N4 and (b) CdS/g-C3N4
Figure 9 shows the schematic of the photogenerated charge-transfer process between g-C3N4 and CdS in photocatalytic hydrogen under visible light. Both g-C3N4 and CdS were excited by the visible light and produced photogenerated electrons and holes. The conduction and valence band edge potentials of g-C3N4 were located at −1.12 and 1.57 eV [42] and CdS was located at −0.5 and 1.9 eV. Under light irradiation, the photo-induced electrons on the conduction band (CB) of g-C3N4 directly transferred to the CB of CdS. These electrons, together with the electrons excited from the valent band (VB) of CdS, accumulated on the Pt deposited on CdS nanoparticle surfaces. Pt can effectively capture electrons. Meanwhile, holes on the VB of CdS spontaneously transferred to the VB of g-C3N4. Therefore, an effective charge separation was achieved, which resulted in a prolonged lifetime for the photogenerated electrons and holes to enhance photocatalytic activity. Consequently, the photo-induced holes oxidized SO3 2− and S2− on the g-C3N4 surface. Meanwhile, the separated electrons had ample time to reduce H2O to H2 on the Pt particle surface. However, CdS is unstable in aqueous solutions under irradiation [10]. Thus, CdS/g-C3N4 heterojunctions can effectively promote the charge separation and transfer corrosive holes from CdS to g-C3N4. This phenomenon makes the charge separation efficient, reduces the probability of charge recombination, and prevents the oxidation of CdS by holes, which results in enhanced photocatalytic activity.

Schematic of the photogenerated charge-transfer processes between g-C3N4 and CdS in photocatalytic hydrogen under visible light
Conclusions
Heterojunctions between polymers g-C3N4 and semiconductors CdS were successfully fabricated using a simple in situ growth method. Heterojunctions with close interfaces between the g-C3N4 and the CdS nanoparticles and nanorods in the composite were confirmed by the XRD, TEM, and FT-IR measurements. The UV–vis diffuse reflectance spectra exhibited a red shift that showed the CdS was introduced into the polymer g-C3N4, which allowed for the efficient utilization of the solar spectrum to create photogenerated electrons and holes. The PL spectra of the nanocomposites indicated a charge transfer from g-C3N4 to CdS. The photocatalytic activity and photocurrent for the CdS/g-C3N4 hybrid nanocomposites were significantly enhanced, which was due to an increased interfacial contact between the CdS and g-C3N4 polymer, fundamental to improving photocatalytic activity.
References
Hoffmann MR, Martin ST, Yong CW, Bahnemann DW (1995) Environmental applications of semiconductor photocatalysis. Chem Rev 95:69–96
Yan SC, Li ZS, Zou ZG (2009) Photodegradation performance of g-C3N4 fabricated by directly heating melamine. Langmuir 25:10397–10401
Li FB, Li XZ (2002) Photocatalytic properties of gold/gold ion-modified titanium dioxide for wastewater treatment. Appl Catal A: Gen 228:15–27
Fujishima A, Honda K (1972) Electrochemical photolysis of water at a semiconductor electrode. Nature 238:37–38
Cho IS, Chen Z, Forman AJ, Kim DR, Rao PM, Jaramillo TF, Zheng X (2011) Branched TiO2 nanorods for photoelectrochemical hydrogen production. Nano Lett 11:4978–4984
Li FB, Li XZ (2002) The enhancement of photodegradation effciency using Pt-TiO2 catalyst. Chemoshere 48:1103–1111
Granot E, Patolsky F, Willner I (2004) Electrochemical assembly of a CdS semiconductor nanoparticle monolayer on surfaces: structural properties and photoelectrochemical applications. J Phys Chem B 108:5875–5881
Pawar RC, Khare V, Lee CS (2014) Hybrid photocatalysts using graphitic carbon nitride/cadmium sulfide/reduced graphene oxide (g-CN/CdS/RGO) for superior photodegradation of organic pollutants under UV and visible light. Dalton Trans 43:12514–12527
Wang XC, Maeda K, Thomas A, Takanabe K, Xin G, Carlsson JM, Domen K, Antonietti M (2009) A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat Mater 8:76–80
Cao S, Yu J (2014) g-C3N4-Based photocatalysts for hydrogen generation. J Phys Chem Lett 5:2101–2107
Chen J, Shen SH, Guo PH, Wu P, Guo LJ (2014) Spatial engineering of photo-active sites on g-C3N4 for efficient solar hydrogen generation. J Mater Chem A 2:4605–4612
Cui YJ, Ding ZX, Liu P, Antonietti M, Fu XZ, Wang XC (2012) Metal-free activation of H2O2 by g-C3N4 under visible light irradiation for the degradation of organic pollutants. Phys Chem Chem Phys 14:1455–1462
Di Y, Wang XC, Thomas A, Antonietti M Making (2010) Metal carbon nitride heterojunctions for improved photocatalytic hydrogen evolution with visible light. ChemCatChem 2:834–838
Dong F, Sun YJ, Wu LW, Fu M, Wu ZB (2012) Facile transformation of low cost thiourea into nitrogen-rich graphitic carbon nitride nanocatalyst with high visible light photocatalytic performance. Catal Sci Technol 2:1332–1335
Ye S, Qiu LG, Yuan YP, Zhu YJ, Xia J, Zhu JF (2013) Facile fabrication of magnetically separable graphitic carbon nitride photocatalysts with enhanced photocatalytic activity under visible light. J Mater Chem A 1:3008–3015
Wang Y, Wang XC, Antonietti M (2012) Polymeric graphitic carbon nitride as a heterogeneous organocatalyst: from photochemistry to multipurpose catalysis to sustainable chemistry. Angew Chem Int Ed 51:68–89
Sun LM, Qi Y, Jia CJ, Jin Z, Fan WL (2014) Enhanced visible-light photocatalytic activity of g-C3N4/Zn2GeO4 heterojunctions with effective interfaces based on band match. Nanoscale 6:2649–2659
Min YL, Qi XF, Xu QJ, Chen YC (2014) Enhanced reactive oxygen species on a phosphate modified C3N4/graphene photocatalyst for pollutant degradation. CrystEngComm 16:1287–1295
Zhang YJ, Mori T, Ye JH, Antonietti M (2010) Phosphorus-doped carbon nitride solid: enhanced electrical conductivity and photocurrent generation. J Am Chem Soc 132:6294–6295
Yan SC, Lv SB, Li ZS, Zou ZG (2010) Organic-inorganic composite photocatalyst of g-C3N4 and TaON with improved visible light photocatalytic activities. Dalton Trans 39:1488–1491
Bai X, Wang L, Zong R, Zhu Y (2013) Photocatalytic activity enhanced via g-C3N4 nanoplates to nanorods. J Phys Chem C 117:9952–9961
Kamat PV (2008) Quantum dot solar cells: semiconductor nanocrystals as light harvesters. J Phys Chem C 112:18737–18753
Fu J, Chang BB, Tian YL, Xi FN, Dong XP (2013) Novel C3N4–CdS composite photocatalysts with organic–inorganic heterojunctions: in situ synthesis, exceptional activity, high stability and photocatalytic mechanism. J Mater Chem A 1:3083–3090
Cao SW, Yuan YP, Fang J, Shahjamali MM, Boey FY, Barber J, Joachim Loo SC, Xue C (2013) In-situ growth of CdS quantum dots on g-C3N4 nanosheets for highly efficient photocatalytic hydrogen generation under visible light irradiation. Int J Hydrog Energy 38:1258–1266
Ge L, Zuo F, Liu J, Ma Q, Wang C, Sun D, Bartels L, Feng P (2012) Synthesis and efficient visible light photocatalytic hydrogen evolution of polymeric g-C3N4 coupled with CdS quantum dots. J Phys Chem C 116:13708–13714
Jiang F, Yan TT, Chen H, Sun AW, Xu CM, Wang X (2014) A g-C3N4–CdS composite catalyst with high visible-light-driven catalytic activity and photostability for methylene blue degradation. Appl Surf Sci 295:164–172
Lu M, Pei Z, Weng S, Feng W, Fang Z, Zheng Z, Huang M, Liu P (2014) Constructing atomic layer g-C3N4–CdS nanoheterojunctions with efficiently enhanced visible light photocatalytic activity. Phys Chem Chem Phys 16:21280-21288
Chen XF, Zhang JS, Fu XZ, Antonietti M, Wang XC (2009) Fe-g-C3N4-catalyzed oxidation of benzene to phenol using hydrogen peroxide and visible light. J Am Chem Soc 131:11658–11659
Sun JX, Yuan YP, Qiu LG, Jiang X, Xie AJ, Shen YH, Zhu JF (2012) Fabrication of composite photocatalyst g-C3N4-ZnO and enhancement of photocatalytic activity under light. Dalton Trans 41:6756–6763
Wang XC, Maeda K, Chen XF, Takanabe K, Domen K, Hou YD, Fu XZ, Antonietti M (2009) Polymer semiconductors for artificial photosynthesis: hydrogen evolution by mesoporous graphitic carbon nitride with visible light. J Am Chem Soc 131:1680–1681
Bao N, Shen L, Takata T, Domen K (2008) Self-Templated synthesis of nanoporous CdS nanostructures for highly efficient photocatalytic hydrogen production under visible light. Chem Mater 20:110–117
Rengaraj S, Venkataraj S, Jee SH, Kim Y, Tai CW, Repo E, Koistinen A, Ferancova A, Sillanpaa M (2011) Cauliflower-like CdS microspheres composed of nanocrystals and their physicochemical properties. Langmuir 27:352–358
Li G, Jiang L, Peng H, Zhang B (2008) Self-assembled cadmium sulfide microspheres from nanorods and their optical properties. Mater Lett 62:1881–1883
Zhang B, Jian JK, Zheng Y, Sun Y, Chen Y, Cui L (2008) Low temperature hydrothermal synthesis of CdS submicro- and microspheres self-assembled from nanoparticles. Mater Lett 62:1827–1830
Wang Z, Pan L, Wang L, Wang H (2011) Urchin-like CdS microspheres self-assembled from CdS nanorods and their photocatalytic properties. Solid State Sci 13:970–975
Bojdys MJ, Müller JO, Antonietti M, Thomas A (2008) Ionothermal synthesis of crystalline, condensed, graphitic carbon nitride. Chem Eur J 14:8177–8182
Liao G, Chen S, Quan X, Yu H, Zhao H (2012) Graphene oxide modified g-C3N4 hybrid with enhanced photocatalytic capability under visible light irradiation. J Mater Chem 22:2721–2726
Liu L, Ma D, Zheng H, Li X, Cheng M, Bao X (2008) Synthesis and characterization of microporous carbon nitride. Micropor Mesopor Mater 110:216–222
Bai X, Zong R, Li C, Liu D, Liu Y, Zhu Y (2014) Enhancement of visible photocatalytic activity via Ag@C3N4 core–shell plasmonic composite. Appl Catal B Environ 147:82–91
Samanta S, Martha S, Parida K (2014) Facile synthesis of Au/g-C3N4 nanocomposites: an inorganic/organic hybrid plasmonic photocatalyst with enhanced hydrogen gas evolution under visible-light irradiation. ChemCatChem 6:1453–1462
Dai X, Xie M, Meng S, Fu X, Chen S (2014) Coupled systems for selective oxidation of aromatic alcohols to aldehydes and reduction of nitrobenzene into aniline using CdS/g-C3N4 photocatalyst under visible light irradiation. Appl Catal B Environ 158–159:382–390
Martha S, Nashim A, Parida KM (2013) Facile synthesis of highly active g-C3N4 for efficient hydrogen production under visible light. J Mater Chem A 1:7816–7824
Acknowledgements
This research was supported by NSF of China (Grant No. 21363027) and the Scientific Research Foundation of GuangXi University (Grant No. XGZ130765).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fang, Z.L., Rong, H.F., Ya, Z.L. et al. In-situ synthesis of CdS/g-C3N4 hybrid nanocomposites with enhanced visible photocatalytic activity. J Mater Sci 50, 3057–3064 (2015). https://doi.org/10.1007/s10853-015-8865-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-015-8865-8