
Abstract
Syntheses of Ca(II)Sn(IV)-layered double hydroxides (LDHs) are attempted by the traditional co-precipitation as well as mechanochemical methods. Both the co-precipitation method and the one-step milling operation proved to be unsuccessful; these methods only produced physical mixtures of hydroxides and carbonates of the two metal ions. However, a two-step milling operation (dry milling followed by milling in the presence of minute amount of water) led to successful synthesis, verified by a range of characterisation methods. Surprisingly, it was found that ball-milling was not even necessary; the reaction proceeded on manual grinding of the components in an agate mortar with a pestle. The preparation of nanocomposites through intercalation of the anions of cystine or valine into Ca(II)Sn(IV)-LDH could also be achieved by the two-step milling method verified again by a range of instrumental methods.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Layered double hydroxides (LDHs) are receiving increasing attention in recent years due to their unique structure and wide-ranging technological applications. The structure of LDHs consists of brucite-like layers, in which some of the divalent cations are isomorphously substituted with trivalent cations, thus providing a layer with positive charge. In these layers, the cations are surrounded by hydroxide ions in an octahedral arrangement. To compensate the positive charge of the layers, different types of anions together with water molecules are situated in the interlayer space [1].
The compounds can be described by the general formula of [M(II)1−x M(III) x (OH)2]x+ [(A−) x/n y·H2O]x−, in which M(II) and M(III) are di- and trivalent cations, respectively, and A− is the charge-compensating anion. A large variety of M(II), M(III), and A− can be used to synthesize LDHs. The most frequently used divalent cations are Mg(II), Cu(II), Ca(II), Co(II), Zn(II), etc., and the trivalent ones are Al(III), Fe(III), Cr(III), etc., while the most common anions are CO3 2−, NO3 −, and Cl− [2].
It was proven that LDH structure was formed, even if M(II) was replaced by Li+. Then, the layers have the composition of [LiAl2(OH)6]+; these materials have been studied and characterized thoroughly [3].
Partial or full replacement of the M(III) ions for M(IV) ones is an entirely different matter. The first mention of M(IV)-containing LDHs was as early as 1997 [4]. They reported the successful synthesis of Mg(II)Al(III)Zr(IV)-LDH. Soon after that, they have published a paper, in which they claimed the success of the preparation of Mg(II)Al(III)Sn(IV)-LDH using the simple co-precipitation method [5]. The claim was based on the changes in the lattice parameters due to the presumed incorporation of Sn(IV), obtained from the X-ray diffraction patterns. They also suggested a mechanism for the incorporation of Sn(IV). The theory was that some of the trivalent cations in the LDH layer were substituted by the tetravalent cations. After this, the syntheses of other similar systems (NiAlSn and CoAlSn) were also reported by this group [6, 7].
Soon after that, another group [8, 9] embarked on studying this topic again, and attempted the synthesis of Mg(II)Al(III)Sn(IV)-LDH with the method given earlier [5], and for characterisation, 119Sn Mössbauer and X-ray absorption spectroscopies were applied. It was found that the tetravalent cations were not incorporated in the layers of the LDH, instead, they formed M(IV) oxide-like particles, which were segregated from the layers.
After these findings were published, the scientific community with an interest in M(IV)-type LDHs has been divided into two main groups. Some years later, the first one continued to study M(II)M(III)M(IV) systems [10, 11]. They also found that the Sn(IV) ions were not incorporated in the layers, nevertheless, the segregated Sn(IV) oxide-like particles improved the catalytic properties of these materials. The other one tried a different, and in a way safer approach, and attempted to prepare M(II)Sn(IV)-LDHs [12, 13]. It is safer, since there is no added M(III); thus, there is no competition with the usually more favored M(II)M(III)-LDH formation. Just like in the other cases, solution-phase chemical method (hydrolysis with urea) was applied, and success in the syntheses of Co(II)Sn(IV)-LDH samples were reported.
Obviously, these results are contradicted, and it seemed worth trying to validate them and find a resolution. This is the major objective of this contribution.
The most common technique used for the preparation of LDHs is co-precipitation, a solution-phase chemical method. In this, aqueous solutions of M(II) and M(III) cations are used as precursors. These solutions are mixed together; then, with a base (usually aqueous NaOH or urea solution), the corresponding pH value is set; thus, the metal ions precipitate in the form of LDH [14].
Another not so commonly used preparative technique is the mechanochemical method, which was applied for LDH synthesis only in recent years. A two-step milling operation was developed [15], in which M(II)(OH)2 and M(III)(OH)3 were used as precursors. The first step was dry milling, which was followed by the addition of minute amount of water, and then, milling was continued (wet milling). The addition of water was necessary, because the precursors did not provide enough hydroxide groups to form the LDH. This method has not yet been used for LDHs containing M(IV) cations.
The fact that anions situated (intercalated) between the layers can be replaced with other anions is one of the most important features of LDHs. A great variety of anions can be intercalated to LDHs: simple inorganic anions, oxo- and polyoxometalate anions, anionic complexes of transition metals, organic anions, anionic polymers, etc. Many methods have been developed for the intercalation of the various anions; however, the most frequently applied ones are intercalation using co-precipitation (e.g., [16]), direct anion exchange (e.g., [17]), and the dehydration-rehydration method (e.g., [18]). Intercalation of the anionic form of methotrexatum (an antifolate drug) was intercalated mechanochemically into MgAl-LDH [19], and since this method has not been applied for M(IV)-containing LDH systems yet, to see, whether it works or not for our system was the other objective of our work.
Studying the synthesis possibilities for the Sn(IV)-containing LDHs is important, because they can be used as catalysts in a large variety of reactions like Baeyer–Villiger oxidation of cyclohexanone [20], photocatalysis [21, 22], liquid-phase methanol carbonylation [23], etc.
In this study, our observations collected during the attempted preparation, and intercalation of M(II)M(IV)-LDHs, using the traditional co-precipitation method and the novel mechanochemically assisted route, are described. We are going to show that the solution-phase chemical method does not give M(II)Sn(IV)-LDHs, while the two-step milling as well as the manual grinding methods lead to the formation of the LDH structure, and the synthesis of nanocomposites through the intercalation of amino acid anions can also be performed this way.
Experimental
Materials and methods
Concentrated NaOH (~20 M) stock solution was prepared from MilliQ water (Millipore) and analytical grade solid NaOH (Spectrum 3D), and its carbonate content was minimized. At this base concentration, the solubility of the Na2CO3 precipitate is negligible, and after filtration (polysulfone Nalgene filter) under inert (N2) atmosphere, carbonate-free NaOH solution could be obtained. This stock solution was kept in airtight, caustic-resistant Pyrex bottle, and was then diluted to the desired concentration under inert (N2) atmosphere.
CaCl2, CoCl2 6H2O, Ca(OH)2, l-cystine, dl-valine, and SnCl4 6H2O (Sigma-Aldrich) were of analytical grade and were used as received.
The first technique applied was the co-precipitation method. LDHs were prepared via dropwise addition of the two metal salt solutions (CaCl2 or CoCl2 and SnCl4) in various molar ratios to a vigorously stirred NaOH solution. The pH of the solution containing the two metal ions was set to 1.71 using a 20 % m/m HCl solution (Aldrich), in order to prevent the hydrolysis of the Sn salt. The concentration of NaOH was 1 M, and the quantity was calculated to have a final pH of 9. The mixture of the two solutions was stirred for 24 h. The precipitate was filtered until air dry with the aid of caustic-resistant vacuum filter unit (Nalgene, USA) equipped with an appropriate membrane (Versapor 0.45 μm, Pall Corporation). The solid reaction products were kept at room temperature in desiccators over dry SiO2.
The Ca(II) to Sn(IV) molar ratios were varied in a wide range (from 3:1 to 6:1), while for the Co(II)Sn(IV) samples, it was kept constant at 3:1. In the latter case, the samples were aged for 6 h at a temperature of 70 °C.
The second method, which is a novel technique for these systems, was the mechanochemical route. The mechanochemical syntheses were applied in two ways: by a mixer mill or a simple agate mortar. The mill used was a Retsch MM 400 mixer mill, with two stainless steel grinding jars of 50 cm3 internal volumes each, and stainless steel grinding balls of 20 mm in diameter. The rotation frequency was 11.6 s−1 and was kept constant throughout the syntheses, just like the total weight of the precursors (3 g). Two methods were investigated: the one-step and the two-step millings. In the former one, only dry milling was performed, and the precursors were ground for 3 h. In the latter one, in addition to dry milling, wet milling was also applied. The precursors were ground when dry for 1 h, and then 0.7 ml of water was added, which was followed by a 2-h long wet grinding. After the synthesis, the products were kept at room temperature in a desiccator over dry SiO2.
When using the agate mortar, the results of the above-mentioned methods were meant to be reproduced; thus, the methods applied were analogous to the one mentioned above in both the one-step (dry) and two-step (dry and wet) millings. In this case, the starting materials (3 g altogether) were ground until a uniform, fine powder was obtained, and then, the mixture was divided into two parts. One part was placed in a desiccator and was studied without further treatment. To the other part, 0.35 ml of water was added, and then grinding was continued until a uniform and homogenous mixture was obtained. The product thus received was dried and kept in a desiccator.
The intercalation of various amino acid anions was performed using a method similar to the two-step milling process mentioned above. The first step was dry milling, where the precursors were ground for 1 h; then, a certain amount of 0.1 M NaOH and the amino acid was added, which was followed by 2 h of wet grinding. Preliminary experiments were performed prior to attempt the intercalation, which showed that the ball to sample weight ratio (B/S) had a major effect on the formation of the LDH phase. The results revealed that the B/S = 100 value was optimal for the LDH formation. This value can be achieved by reducing the weight of the samples. In the intercalation experiments, B/S = 100 was used, and therefore, the amount of NaOH had to be reduced. The amount of amino acid was set in the way that the molar ratio of Sn(IV) and the amino acid was 1:1. The addition of the precursors, the amino acids, and the NaOH into the grinding jars, as well as the sealing of the jars were performed under N2 atmosphere in a glove box. In this way, the disturbing CO2 could be excluded. After the synthesis, the products were kept in desiccators, at room temperature.
Apparatus and equipment
Powder X-ray diffraction (XRD) patterns of the dry solid samples were recorded in the 2θ = 3°–60° range (θ is the incidence angle of the X-ray beam) on a Philips PW1710 instrument, using CuKα (λ = 1.5418 Å) radiation.
The morphologies of the substances obtained were studied using a Hitachi S-4700 scanning electron microscope (SEM) at various magnifications. The ratios of the di- and tetravalent ions in the solid samples and elemental mapping were determined using a Röntec QX2 energy dispersive X-ray (EDX) analyzer coupled to the microscope.
The Fourier-transform infrared (FTIR) spectra of the pristine, the organic anion-intercalated LDH nanocomposite, and the intercalated anion were recorded on a BIO-RAD FTS-65A/896 spectrometer equipped with a DTGS detector in diffuse reflectance. Spectral resolution was 4 cm−1, and 256 scans were collected for a spectrum. The spectra were baseline corrected and smoothed using the WIN-IR software package. The samples were finely ground and combined with KBr without pressing into pellets.
Raman spectra were recorded on a BIO-RAD Digilab Division dedicated FT-Raman spectrometer equipped with liquid nitrogen-cooled germanium detector and CaF2 beam-splitter. The excitation line was provided by a Spectra Physics T10-106C Nd:YVO4 laser at 1064 nm. The spectra were recorded in the 3600–100 cm−1 range with 4 cm−1 resolution. 1024 scans were collected for each spectrum. The excitation power was 280 mW at the sample position. The spectrometer was controlled by the BIO-RAD Win-IR 3.3 software. The samples were placed in a standard NMR tube. Spectra were recorded at room temperature. Data were processed by the GRAMS/AI 7.00 software.
Results and discussion
In order to prepare Ca(II)Sn(IV)-LDHs, two preparation methods (co-precipitation and mechanochemistry) were tried. Samples prepared by the co-precipitation method, with their acronyms, molar ratios and synthesis parameters, are presented in Table 1.
The X-ray diffractograms of these samples are presented, without baseline correction or smoothing in Fig. 1.

Powder X-ray diffractograms of Ca(II)Sn(IV) samples prepared by the co-precipitation method: (a) Ca3Sn, (b) Ca4Sn, (c) Ca5Sn, (d) Ca6Sn
The typical diffractogram of an LDH displays three major reflections (003, 006, and 009), from which the 003 is the most significant—it appears around 10° 2θ value. The presence or lack of these reflections was investigated to determine whether the LDH formation was successful or not.
The diffractograms of the Ca(II)Sn(IV) samples do not exhibit drift in the baseline, i.e., the crystallinity is good. The Ca:Sn molar ratio was varied in a wide range. In neither case could the specific reflections of the LDHs be recognised, however. In order to identify the reflections, a randomly chosen diffractogram was analyzed (Fig. 2 ).

Powder X-ray diffractogram of the Ca3Sn sample prepared by the co-precipitation method
The typical reflections of Ca(OH)2 and CaCO3 could be identified (CO2 is most probably airborne). This proves that Ca(OH)2 was precipitated, but the incorporation of the Sn(IV) ions into its layers did not happen. The presence of CaCO3 could be explained by the fact that during the synthesis, the air was not excluded. It can be concluded that using the co-precipitation method, Ca(II)Sn(IV)-LDHs could not be synthesized.
It is worth mentioning that our attempt for the preparation of Co(II)Sn(IV)-LDH (Co(II)/Sn(IV) = 3) by the co-precipitation method was also unsuccessful, in spite of aging the freshly precipitated material for 6 h at 343 K.
After these experiments, we gave up using the solution-phase chemical method and turned to mechanochemistry. Samples prepared using the various mechanochemical and mechanochemically assisted methods, with their designations, molar ratios and synthesis parameters, are presented in Table 2.
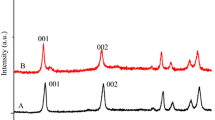
The XRD results for the Ca(II)Sn(IV) samples synthesized via the pure mechanochemical (dry or one-step milling) and the mechanochemically assisted (dry plus wet or two-step milling and wet grinding) methods are presented in Fig. 3.

Powder X-ray diffractogram of Ca(II)Sn(IV) samples prepared via mechanochemical methods (a) Ca4Sn_mm_w, (b) Ca4Sn_mm_d, (c) Ca4Sn_m_w, (d) Ca4Sn_m_d
In the one-step milling operations (B and D curves), there were no LDH formation; the LDH-specific reflections are missing. However, in the two-step milling operations (traces A and C) at 10° 2θ values, the 003 reflection specific to the LDH structure appeared. Thus, it is strongly indicated that the two-step milling method produced Ca(II)Sn(IV)-LDH. The observation that the synthesis was only successful in the two-step milling process is due to the fact that using only dry milling, the crystal water introduced with the precursors could not generate sufficient amounts of hydroxide group necessary for the LDH structure.
Using the Bragg equation, the interlayer spacing of the LDHs could be calculated from the positions of the 003 reflections, and they were 8.38 and 9.05 Å for the Ca4Sn_mm_w and Ca4Sn_m_w samples, respectively.
For providing further proofs of LDH formation, SEM measurements were performed in order to gain information about the morphology of the sample. The hexagonally shaped lamellar structure, the typical morphology of LDHs was sought in the samples. As Figs. 4 and 5 attest the SEM images of Ca(II)Sn(IV), samples prepared with two-step milling or the wet grinding methods show these characteristics.

SEM images of the Ca4Sn_mm_w samples at magnifications of 13000 (a), and 50000 (b)

SEM image of the Ca4Sn_m_w sample at a magnification of 50000
The particle sizes are larger than what one would expect for LDHs; however, it may be assumed that the amorphous phase is due to smaller particles sticking together to form bigger aggregates. The SEM images have confirmed the results of the XRD measurements: LDH was formed indeed, during the two-step mechanochemically assisted syntheses.
Using EDX, the elemental composition could be obtained and projecting it on the corresponding SEM image, the elemental map, and thus the elemental distribution of the samples could be determined. For the Ca4Sn_mm_w sample, the elemental analysis was performed, from which it could be concluded that during the preparation of the samples, iron contamination (iron was the main component of the grinding balls and grinding jars) did not occur.
On the elemental map of both the Ca4Sn_mm_w and Ca4Sn_m_w samples (Figs. 6, 7), it can be observed that the distributions of the Ca and Sn are uniform within the particles, and no aggregation or segregation can be noticed. This is also another piece of evidence supporting that LDH was formed during the two-step milling and also the wet grinding procedures.

Elemental distribution map (a) on the Ca4Sn_mm_w sample (b)

Elemental distribution map (a) on the Ca4Sn_m_w sample (b)
For the preparation of nanocomposites, various organic anions (of cystine and valine) were intercalated in the LDHs formed, using the two-step milling method. The samples prepared in this way, with their designations, molar ratios and synthesis parameters, are presented in Table 3.
The diffractograms of the intercalated samples and that of the pristine LDH is displayed in Fig. 8.

Powder X-ray diffractograms of the amino acid anion–Ca(II)Sn(IV)-LDH samples intercalated by the mechanochemically assisted pathway: (a) Ca3Sn_mm_w, (b) Ca3Sn_mm_w_Val, (c) Ca3Sn_mm_w_Cys (the “original” 003 reflection of the LDH and the 003′ reflection of the intercalated LDH are indicated)
One of the most important proofs that the intercalation of the anions between the layers of the LDH succeeded is the change in the interlayer spacing. This change can be calculated from the position of the 003 reflection, using the Bragg equation. The lower the position of the 003 reflection in terms of 2θ angle, the larger the interlayer distance is. Thus, in the samples prepared, one is looking for the increased interlayer spacing, i.e., the presence of the 003 reflection at lower 2θ values relative to the pristine Ca(II)Sn(IV)-LDH sample. In both cases (using the anions of cystine or valine), this phenomenon could be observed. For the samples containing the anions of valine and cystine, the 003 reflection shifted to 8.8° and 5.6° from 9.8° in the pristine LDH. The calculated interlayer spacings are summarized in Table 4.
Additional support for the success of intercalation is provided by SEM–EDX measurements. Using this method, the elemental distribution maps were taken, shown in Figs. 9 and 10.

Ca–Sn (b) and C–S (c) elemental distribution maps on the Ca3Sn_mm_w_Cys sample (a)

Ca–Sn (b) and N–C (c) elemental distribution maps on the Ca3Sn_mm_w_Val sample (a)
The C–S and N–C elemental maps revealed that in both cases, the distributions of the amino acid anions were uniform. The Ca–Sn elemental maps show that these two elements are also evenly distributed within the particle (which is one of the characteristics of the LDHs).
Other conclusive information could be gained regarding the success of the intercalation by IR spectroscopy. The IR spectra of the intercalated samples were taken and compared to those of the pristine LDH and the amino acids in zwitter ionic forms (Figs. 11, 12).

FTIR spectra of Ca3Sn_mm_w (a), Ca3Sn_mm_w_Val (b), and the zwitter ionic dl-valine (c)

FTIR spectra of Ca3Sn_mm_w (a), Ca3Sn_mm_w_Cys (b), and the zwitter ionic l-cystine (c)
At around 1100 cm−1, the symmetric stretching vibration of the C–N bond is present in the spectra of both the intercalated sample and the zwitter ionic valine, and is not seen in the pristine LDH. This finding verifies that an organic–inorganic composite is in our hands. The presence of the vibrations of the carboxylate ion is additional evidence. Actually, the band corresponding to the asymmetric vibration can only be identified in the composite, since the symmetric vibration coincides with a band of the pristine LDH at 1645 cm−1. Nevertheless, this peak shifted from 1492 to 1520 cm−1 indicating interaction with the layer of the LDH, i.e., the carboxylate ion is situated among the layers.
A similar reasoning may be given for the interaction of the LDH and the anion of cystine, since here, the symmetric carboxylate band can be identified too, and it also shifts in the composite compared to the zwitter ionic form (the shift is smaller though than in the previous case, it is from 1417 to 1428 cm−1). In order to support the conclusion that the anion of cystine is also intercalated, measurements with Raman spectroscopy were also performed Fig. 13.

Raman spectra of Ca3Sn_mm_w (a), Ca3Sn_mm_w_Cys (b), and zwitter ionic l-cystine (c)
The C–S bond stretching vibration at 499 cm−1 appeared in the Raman spectrum of the zwitter ionic l-cystine, and it is seen and shifted to 472 cm−1 in that of the composite sample as well. This confirms the intercalation of the anion of cystine into Ca3Sn-LDH.
Finally, let us say few words about the possible mechanistic steps of the synthesis of the Ca(II)Sn(IV)-LDH in the two-step milling procedure. Direct observation of events occurring in the mill was not possible; therefore, the results of measurements after milling are used for offering a mechanism. It is generally the case in mechanochemistry, however, this may change soon, since very recently, a Raman spectrometer translucent mill setup has been published [24] allowing in situ measurements during milling. Dry milling could not produce the LDH, but it was a necessary prerequisite of the successful synthesis. It provided intimate mixing and activation for the reaction to occur. The second step, wet milling that is, further activated the components, and water was necessary for the formation of the LDH, which possibly was a kind of hydrolysis-co-precipitation.
Conclusions
The synthesis of Ca(II)Sn(IV)-LDH was attempted using the co-precipitation, a pure mechanochemical (one-step milling), mechanochemically assisted methods (two-step milling), and wet grinding. The first two techniques proved to be unsuccessful; however, both the two-step milling and the wet grinding procedures produced Ca(II)Sn(IV)-LDH.
Furthermore, the two-step milling technique performed superbly in the intercalation of the anions of cystine or valine producing organic–inorganic nanocomposites.
To sum up, the combination of dry and wet milling or wet grinding can be viable procedures in the preparation and intercalation of LDHs that cannot be easily approached, if at all, by solution-phase methods.
References
Molls SJ, Christy AG, Génin J-MR, Kameda T, Colombo F (2012) Nomenclature of the hydrotalcite supergroup: natural layered double hydroxides. Miner Mag 76:1289–1336
Evans GD, Slade RCT (2006) Structural aspects of layered double hydroxides. Struct Bond 119:1–87
Besserguenev AV, Fogg AM, Francis RJ, Price SJ, O’Hare D, Isupov VP, Tolochko BP (1997) Synthesis and structure of the gibbsite intercalation compounds [LiAl2(OH)6]X {X = Cl, Br, NO3} and [LiAl2(OH)6]Cl·H2O using synchrotron X-ray and neutron powder diffraction. Chem Mater 9:241–247
Velu S, Ramaswamy V, Ramani A, Chanda BM, Sivasanker S (1997) New hydrotalcite-like anionic clays containing Zr4+ in the layers. Chem Commun 21:2107–2108
Velu S, Suzuki K, Okazaki M, Osaki T, Tomura S, Ohashi F (1999) Synthesis of new Sn-incorporated layered double hydroxides and their thermal evolution to mixed oxides. Chem Mater 11:2163–2172
Velu S, Suzuki K, Kapoor MP, Tomura S, Ohashi F, Osaki T (2000) Effect of Sn incorporation on the thermal transformation and reducibility of M(II)Al-layered double hydroxides [M(II)) Ni or Co]. Chem Mater 12:719–730
Velu S, Suzuki K, Osaki T (2000) A comparative study of reactions of methanol over catalysts derived from NiAl- and CoAl-layered double hydroxides and their Sn-containing analogues. Catal Lett 69:43–50
Intissar M, Jumas JC, Besse J-P, Leroux F (2003) Reinvestigation of the layered double hydroxide containing tetravalent cations: unambiguous response provided by XAS and Mössbauer spectroscopies. Chem Mater 15:4625–4632
Intissar M, Briois V, Besse J-P, Leroux F (2005) Evidence by XAS of tin oxide coating layered double hydroxide. Phys Scripta T115:288–290
Yang ZQ, Park SE (2007) Baeyer–Villiger reaction of adamantanone over Sn-containing hydrotalcite-like catalysts. Solid State Phenom 119:163–166
Tong DS, Zhou CH, Li MY, Yu WH, Beltramini J, Lin CX, Xu ZP (2010) Structure and catalytic properties of Sn-containing layered double hydroxides synthesized in the presence of dodecylsulfate and dodecylamine. Appl Clay Sci 48:569–574
Saber O (2007) Preparation and characterization of a new nano-structural material: Co–Sn LDH. J Phys Conf Ser 61:825–830
Al Jaafari AI (2010) Controlling the morphology of nano-hybrid materials. Am J App Sci 7:171–177
Cavani F, Trifiró F, Vaccari A (1991) Hydrotalcite-type anionic clays: preparation, properties and applications. Catal Today 11:173–301
Tongamp W, Zhang Q, Saito F (2008) Mechanochemical route for synthesizing nitrate form of layered double hydroxide. Powder Technol 185:43–48
Woo MA, Woo KT, Paek M-J, Ha H-W, Choy J-H, Hwang S-J (2011) Phosphate-intercalated Ca–Fe-layered double hydroxides: crystal structure, bonding character, and release kinetics of phosphate. J Solid State Chem 184:171–176
Fogg AM, Dunn JS, Shyu SG, Cary DR, O’Hare D (1998) Selective ion-exchange intercalation of isomeric dicarboxylate anions into the layered double hydroxide [LiAl2(OH)6]Cl·H2O. Chem Mater 10:351–355
Nakayama H, Wada N, Tsuhako M (2004) Intercalation of amino acids and peptides into Mg–Al layered double hydroxide by reconstruction method. Int J Pharm 269:469–478
Qi F, Zhang X, Li S (2013) A novel method to get methotrexatum/layered double hydroxides intercalation compounds and their release properties. J Phys Chem Solids 74:1101–1108
Jimenez-Sanchidrian C, Hidalgo J, Llamas R, Ruiz J (2006) Baeyer–Villiger oxidation of cyclohexanone with hydrogen peroxide/benzonitrile over hydrotalcites as catalysts. Appl Catal A 312:86–94
Seftel EM, Popovici E, Mertens M, Stefaniak EA, van Grieken R, Cool P, Vansant EF (2008) Sn(IV)-containing layered double hydroxides as precursors for nano-sized ZnO/SnO2 photocatalysts. Appl Catal B 84:699–705
Dvininov E, Ignat M, Barvinschi P, Smithers MA, Popovici EN (2010) New SnO2/MgAl-layered double hydroxide composites as photocatalysts for cationic dyes bleaching. J Hazard Mater 177:150–158
Kapoor MP, Matsumura Y (2004) Liquid-phase methanol carbonylation catalyzed over tin promoted nickel–aluminium layered double hydroxide. Catal Today 93–95:287–291
Gracin D, Štrukil V, Frisčić Tomislav, Halasz I, Užarevic K (2014) Laboratory real-time and in situ monitoring of mechanochemical milling reactions by Raman spectroscopy. Angew Chem Int Ed 53:6193–6197
Acknowledgements
This research was financed by the OTKA NK106234 and the TÁMOP 4.2.2.A-11/1/KONV-2012-0047 Grants. M.(Á.)S. and M.Sz. gratefully acknowledge the support of TÁMOP 4.2.4.A/2-11-1-2012-0001 National Excellence Program. All these supports are highly appreciated.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ferencz, Z., Szabados, M., Ádok-Sipiczki, M. et al. Mechanochemically assisted synthesis of pristine Ca(II)Sn(IV)-layered double hydroxides and their amino acid intercalated nanocomposites. J Mater Sci 49, 8478–8486 (2014). https://doi.org/10.1007/s10853-014-8558-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-014-8558-8