
Abstract
Nickel-based alloys are being considered as construction materials for various components in high-efficiency steam turbines with envisaged operating temperatures around 700 °C. In the present study, the steam oxidation behaviour of the nickel-based alloy 625 in the temperature range of 700–800 °C was investigated whereby exposures up to 10000 h were carried out. Gravimetric data in combination with results from a variety of post exposure analysis techniques showed in all cases the formation of protective oxide scales mainly consisting of chromia with minor amounts of outer Cr/Mn spinel and internal silica. The phases found in the bulk alloy after long-term exposure were mainly needle-shaped δ-Ni3(Nb,Mo) phase, μ-phase and Si-rich η-M6C carbide. Microstructural features and phase formation were found to be related to minor variations in the alloy composition, especially iron and silicon content. The oxidation-induced chromium depletion caused a number of microstructural changes in the subsurface depletion layer. Most important was an enrichment of the intermetallic δ-phase at the scale–alloy interface. DICTRA modelling revealed this effect to be caused by uphill diffusion as a result of a negative niobium activity gradient in the subscale chromium depletion zone. Although the available kinetic and thermodynamic data allowed qualitative explanation of the δ-phase enrichment, the databases do not correctly describe the high molybdenum solubility in the δ-phase.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In an always more energy-demanding world power plant technology has to be improved, to convert energy in a more efficient and cleaner way. The most straightforward way of enhancing thermal efficiency of a steam power plant is to increase the temperature and pressure of the steam entering the turbine. Therefore, current developments aim at steam turbines operating at temperatures around 700 °C, in order to achieve plant efficiencies of over 50 % [1, 2]. As temperature increases, common structural materials such as low-alloyed or martensitic steels are no longer suitable, due to their limited mechanical properties and steam oxidation resistance [3–8]. Nickel-based alloys provide the required combination of high-temperature creep strength, as well as thermal fatigue resistance and possess substantially higher oxidation resistance than the mentioned steels. They are thus potentially suitable construction materials for a variety of stationary and rotating steam turbine components (e.g. casings and forgings) [2, 9–11].
Although nickel-based alloys have been used satisfactorily for many years in gas turbines as well as in the aerospace industry, different challenges lay ahead for their use in steam turbines. Steam turbines have very long service lives of often more than 200000 h, which means that steam turbine components have to withstand severe mechanical loads and oxidation attack for very long times. Therefore, long-term stability of alloy microstructure and its influence on mechanical properties of the alloy have to be taken into account.
With a chemical composition of 20–23 wt% Cr, 8–10 wt% Mo, 3.15–4.15 wt% Nb and up to 5 wt% Fe [12], the nickel-based alloy 625 is being considered for application in advanced steam turbine components. The alloy is characterized by high creep strength; however, various studies [13, 14] showed that alloy 625 exhibits substantial microstructural changes when exposed at high temperatures for prolonged times. Precipitates which may form in alloy 625 during ageing at various temperatures were described in a number of papers [15–18]. Different carbides as well as intermetallic phases may precipitate depending upon the temperature and the duration of the exposure.
-
Ni3(Nb,Al,Ti) γ″-phase and δ-Ni3Nb phase have been observed in the microstructure of the alloy depending on the temperature. The γ″ phase is a coherent-ordered A3B compound in which the equilibrium intermetallic phase is the δ-phase. The latter is an incoherent orthorhombic phase which typically precipitates in the form of needle-like precipitates. The presence of γ″ or δ-phase in the alloy microstructure depends on ageing temperature [17, 19, 20]. The δ-phase can form directly or by transformation of the metastable γ″ phase [21]. The γ″ to δ transformation is accompanied with a decrease in the strength of the alloy [18, 22].
-
μ-phase is a topologically close-packed phase with a rhombohedral Co7Mo6 structure and lattice parameters: a 0 = 0.47 nm, c 0 = 2.5 nm [23]. Precipitates of μ-phase may be present in the microstructure in different forms (acicular, globular, blocky, platelet or rods) and they can nucleate from existing M6C carbides [24, 25]. The significance of the μ-phase for the mechanical properties of alloy 625 is unclear [15]. However, it was observed that precipitation of μ-phase with blocky morphology and a defect structure of twins and stacking faults leads to a loss of ductility, impact toughness and oxidation resistance [25].
-
Different carbides with a diamond-cubic structure, M23C6, M6C or MC, can be present in alloy 625. M23C6 is a Cr-rich carbide, while M6C is rich in nickel, niobium and molybdenum, and MC is rich in Nb. Vernot-Loier and Cortial [26] reported a progression of carbide precipitate stability with increasing temperature (M23C6 to M6C to MC) in alloy 625.
Different time–temperature–transformation (TTT) diagrams for alloy 625 are available in literature [27–29]. These TTT diagrams differ mainly with respect to the precipitation of M23C6 and M6C type carbides, a fact which was attributed to minor differences in the chemical composition of the alloy [30].
Because of the activities in the development of power generation systems with increased service temperatures, detailed knowledge concerning the long-term steam oxidation behaviour of nickel-based materials such as alloy 625 at service temperatures in the range 700–750 ºC envisaged for advanced steam turbines is required. However, most of the literature studies dealing with the oxidation behaviour of alloy 625 were carried out at higher temperatures (900–1000 °C) and mostly in dry atmospheres such as Ar–O2 or air, e.g. [31, 32].
The present study investigates the long-term oxidation behaviour and microstructural stability of nickel-based alloy 625 during exposure at 700–800 °C in a steam-containing environment, Ar-50 %H2O, which simulates the service environment of a steam power plant [33]. After exposure times ranging from 1000 to 10000 h, the specimens were characterized using a variety of analysis techniques including light optical metallography, scanning and transmission electron microscopy (SEM and TEM), X-ray diffraction (XRD) and secondary neutrals mass spectrometry (SNMS). In the investigations, main emphasis was put on the oxide scale formation and the microstructural changes which take place in the oxidation-affected zone beneath the surface oxide scale as well as in the bulk of the alloy. Furthermore, the influence of the minor variations in chemical composition on the microstructural stability and the steam oxidation behaviour of alloy 625 has been addressed.
Experimental
Two different batches of alloy 625 were studied, which in the following will be designated as thick-walled material and as strip material, respectively. The detailed chemical compositions of the two batches analysed by inductively coupled plasma-optical emission spectroscopy (ICP-OES) and infrared (IR) analysis are listed in Table 1. Although the amounts of the main alloying elements, i.e. Cr, Mo and Nb, are similar in both cases, the concentrations of iron and silicon vary considerably between both batches, being nonetheless within the specification limits of alloy 625 [12]. The test samples of the thick-walled material consisted of coupons of 20 × 10 × 6 mm3, whereas the strip material samples were 20 × 10 × 0.5 mm3.
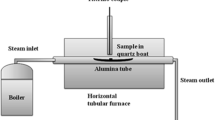
Before oxidation, the samples were ground with SiC sand paper to a 1200 grit surface finish. After cleaning and degreasing, the specimens were oxidized discontinuously at 700, 750 and 800 °C in Ar-50 %H2O. At 800 °C, the maximum exposure times were 3000 h, whereas at 700 and 750 °C, the oxidation tests were continued up to 10000 h. The model gas Ar-50 %H2O was used as test atmosphere to obtain oxidation mechanisms which are comparable to those commonly encountered in a steam turbine [33]. The reaction gas was generated by bubbling argon at atmospheric pressure through a saturator (humidifier), which was controlled at fixed temperature. The residual oxygen content in the used argon was approximately 1 ppm. The total gas pressure during the experiments was 1 bar. The linear gas flow rate was approximately 2 mm/s.
Two types of furnaces were employed: vertical and horizontal ones. For the exposures at 700 °C, the specimens were placed in alumina sample holders and heated up in a vertical furnace in dry argon with small amounts of water vapour. After reaching the desired test temperature, samples were isothermally exposed to the Ar-50 %H2O gas stream. In the horizontal furnaces, which were used for the testing at 750 and 800 °C, specimens were placed in a longitudinal alumina crucible and heated up in dry argon up to 300 °C and subsequently in Ar-50 %H2O up to the desired test temperature. In both types of experiments, the heating and cooling rate were 10 K/min. In both cases, the specimens were weighed every 250 h, i.e. after intermediate cooling to room temperature. The absolute accuracy of the used balance is ±0.01 mg.
The oxidized specimens were cross-sectioned and prepared for metallographic analyses using conventional techniques, i.e. grinding, polishing and fine polishing. Before mounting in an epoxy resin, the specimens were coated with a thin gold layer by sputtering and subsequently electrochemically coated with nickel.
The metallographic cross-sections were characterized by optical metallography and scanning electron microscopy (SEM) with energy- and wavelength-dispersive X-ray analysis (EDX/WDX). Additional analyses were carried out using transmission electron microscopy (TEM) of specimens prepared by the focused ion beam technique (FIB). Further investigations included X-ray diffraction (XRD) whereby various measurements were made on each specimen in order to establish the phase distribution as function of depth. After the first measurement, the oxide scale was ground off with SiC grinding paper (1200 grit) until eventually the non-affected bulk alloy was reached. XRD analyses were then carried out after grinding the oxide scale. The X-ray source was Cu Kα. Furthermore, selected oxidized specimens were analysed by secondary neutrals mass spectrometry (SNMS), whereby the measured depth profiles were quantified using the procedure described in Ref. [34].
Results
Oxidation behaviour
Weight change data of alloy 625 obtained during long-term oxidation in Ar-50 %H2O at temperatures between 700 and 800 °C showed that the weight change values were even up to 10000 h of exposure, for both batches very low and near the accuracy limit of measurement and the reproducibility of the discontinuous weight measurements. For this reason, an exact estimation of the oxidation rate law, i.e. the kinetics of scale thickness as function of time, was hardly possible. In spite of these difficulties, the weight change data (up to 3000 h at 800 °C as well as 10000 h at 700 and 750 °C) could with reasonable accuracy be described by a parabolic time dependence, i.e.
in which Δm is the area specific weight gain (in mg cm−2), t is the exposure time (in h) and k w is the parabolic rate constant.
Figure 1 shows an Arrhenius plot of the parabolic rate constants compared with data for some other chromia-forming materials measured at similar and/or higher temperatures in water vapour containing low pO2 environments. The data for the Cr-based materials are taken from Ref. [35], of binary Ni-25Cr from [36], alloy 625 results at 900–1000 °C from [37], and the results from the spinel forming, high-chromium steel JS3 from [38].

Parabolic rate constants for steam oxidation of alloy 625 compared with those for other chromia-forming materials in low pO2 environments (i.e. Ar-4 %H2-2 %H2O mixtures). The data for the Cr-based materials are from Ref. [35], of binary Ni-25Cr from [36]. Alloy 625 results at 900–1000 °C were taken from [37]. The results from the spinel forming, high-chromium steel JS3 are from [38]
This comparison shows that the oxidation rates were in a similar range as those found for NiCr-based alloys in low pO2 environments, whereby the oxidation rates of alloy 625 as strip material were slightly higher than those of the thick-walled material.
The alloy formed in all the tested cases a thin, protective layer (Figs. 2, 3) which, according to SNMS measurements (Fig. 4) and EDX analyses (Figs. 5, 6), mainly consists of chromia with a thin Mn-containing spinel near the scale/gas interface and some SiO2 precipitates beneath the chromia layer. The amount of SiO2 precipitates depended on the exact chemical composition of the alloy, being higher in the case of the thick-walled material due to the higher Si content of this batch. Additionally, some differences in the morphology of the Mn-containing spinel were observed. Figure 2 shows that the thick-walled alloy 625 exhibited whisker formation in the outer, spinel containing part of the oxide scale, especially at 700 °C and after long oxidation times. Cr/Mn spinel has frequently been shown to exhibit whisker morphology when formed at 800–900 °C in water vapour-rich, low pO2 gases [33, 36]. No whiskers were observed after oxidation at 750 ºC or in the case of the strip material, although SNMS analyses confirmed the presence of outer Mn in both batches at both temperatures. These differences in whisker formation might indicate that the amount and/or even the exact chemical composition of the outer Mn spinel formed on alloy 625 depend on the temperature and the exact chemical composition of the alloy.

SEM cross-sections showing surface-near microstructure of thick-walled alloy 625 after 10000 h oxidation in Ar-50 %H2O at (a, b) 700 °C and (c, d) 750 ºC

SEM cross-sections of alloy 625 (strip material) after oxidation in Ar-50 %H2O for 10000 h at (a, b) 700 °C and (c, d) 750 °C

SNMS profiles of the main constituents (Cr, Mn and Si) of the oxide scale formed on thick-walled alloy 625 during oxidation in Ar-50 %H2O at 700 °C for 10000 h

SEM cross-section showing oxide scale formed on thick-walled alloy 625 after oxidation in Ar-50 %H2O at 700 °C for 10000 h. Lower part of figure shows X-ray mappings of the different elements in the oxide scale

SEM cross-section showing oxide scale morphology of alloy 625 (strip material) after oxidation in Ar-50 %H2O at 750 °C for 10000 h. Lower part of figure shows X-ray mappings of the main oxide scale constituents
Oxidation-induced microstructural changes of alloy 625 as thick-walled material
Oxidation of alloy 625 in Ar-50 %H2O in the temperature range 700–800ºC led not only to the formation of a thin oxide layer on top of the sample but also to changes in the microstructure of the alloy.
After oxidation in Ar-50 %H2O, needle-like precipitates were found in a narrow zone beneath the oxide scale (Fig. 2) as well as more or less homogeneously distributed in the bulk material. Formation of these precipitates was observed after all oxidation tests, independent of time or temperature. The needle-like precipitates could, due to their small size, not be identified with SEM/EDX or WDX. Consequently, further investigations using transmission electron microscopy (TEM) were carried out for determining the exact chemical composition and nature of the precipitates. EDX analyses of the TEM sample indicated that the needle-like precipitates contained 71.9–75.5 at.% Ni, 13.4–15 at.% Nb, 7.7–8.6 at.% Mo and 4–4.5 at.% Cr, a chemical composition which corresponds to δ-Ni3(Nb,Mo) phase. The δ-phase in the bulk alloy does not precipitate randomly but in some preferred directions. Precipitation of δ-phase with a preferred orientation has previously been reported in other studies [15, 18, 21, 39].
TEM investigations showed that additional precipitates with a different chemical composition were also present in the bulk of alloy 625 (Fig. 7). EDX analysis of a TEM sample indicated that these precipitates contained 37.8–38.8 at.% Ni, 28.8–29.8 at.% Mo, 14.2–15.6 at.% Cr, 12.2–12.8 at.% Si and 3.7–3.8 at.% Nb. Silicon-rich precipitates, reported as a silicon-rich variant of M6C and referred as Si-rich η M6C carbide, have been previously found in alloy 625 after ageing at 750 °C [15, 40]. Si-rich η carbide has a diamond-cubic structure and a lattice parameter of approximately 1.09 nm, and it was also found in austenitic steels and other Ni-based alloys [40–45]. The presence of η carbide was confirmed by XRD profiles of alloy 625 after oxidation in Ar-50 %H2O at 700 ºC (Figs. 8, 9).

TEM (STEM/HAADF) image of thick-walled alloy 625 showing different precipitates found in the bulk alloy after 3000 h exposure in Ar-50 %H2O at 700 °C. Lower part of the figure shows X-ray mappings of the main alloying elements

XRD profiles of thick-walled alloy 625 in the as-received condition and after various times of oxidation in Ar-50 %H2O at 700 °C

XRD profiles of thick-walled alloy 625 after oxidation in Ar-50 %H2O at 700 °C for 1000 h. Black profile corresponds to the as-oxidized sample, while grey one was measured after the oxide scale was completely removed by grinding
Oxidation-induced microstructural changes of alloy 625 as strip material
After long-term oxidation in Ar-50 %H2O, δ-phase was found in the form of needle-like precipitates all over the bulk alloy, whereas some Cr-rich carbides and minor amounts of (Ti,Nb) nitrides were randomly distributed in the alloy. Additionally, a different type of precipitate was present both at the grain boundaries as well as within the grains (Fig. 10).

SEM cross-section and EDX measurements showing surface-near region of alloy 625 (strip material) after oxidation in Ar-50 %H2O at 700 °C for 10000 h. Lower part of the figure shows X-ray mappings of the main alloying elements
EDX measurements revealed that these precipitates had a chemical composition of 42.7 at.% Ni, 26.2 at.% Mo, 23.1 at.% Cr, 4.1 at.% Fe, 2.5 at.% Nb and 1.5 at.% Si. Considering isothermal sections within the Ni–Cr–Mo phase diagram [46, 47] together with μ-phase composition given in the literature for alloy 625 [15, 25], it can be concluded that these precipitates are μ-phase. Evans et al. [15] suggested that formation of μ-phase at grain boundaries in alloy 625 is favourable since it is thermodynamically more stable, and it requires less Nb than the amount needed to form M6C. Tawancy [25] suggested that the susceptibility to precipitation of μ-phase depends upon the exact iron content of the alloy. It was indicated that μ-phase forms in alloy 625 if the Fe concentration present in the alloy exceeds 3 wt% due to a decrease in the electron-to-atom ratio when Fe is substituted for Ni.
Subsurface phase transformations
Long-term oxidation of alloy 625 as thick-walled material led to the dissolution of Si-rich η-M6C carbides in the subsurface zone as can be observed in Fig. 2. The dissolution of these carbides in the subsurface zone would explain the fact that after 10000 h, no diffraction lines corresponding to this phase were observed in the XRD profiles (Fig. 8) since the penetration depth of the X-rays is limited to a few μm. Measured SNMS profiles showed a depletion/enrichment of carbon in the alloy zone immediately beneath the oxide scale. In the case of alloy 625 as strip material, μ-phase—which is present both at the grain boundaries as well as within the alloy grains—dissolved in the subsurface chromium-depleted zone (Fig. 3).
The most striking effect observed in the subscale zone after the oxidation of alloy 625 in Ar-50 %H2O was the presence of (Mo,Nb)-rich precipitates at the oxide-metal interface in both batches tested (Figs. 2, 3). EDX measurements and X-ray mappings of the main alloying elements (Fig. 10) clearly reveal this phase to be δ-phase. The EDX analyses indicate that the precipitates have a chemical composition of 68.9–70.4 at.% Ni, 9.8–10.9 at.% Nb, 12.7–13.6 at.% Mo, 5.4–5.6 at.% Cr, and 0.7–0.9 at.% Fe. The SNMS profiles in Fig. 11 show, as expected, a depletion of Cr in the subsurface zone beneath the oxide–alloy interface as a result of chromium consumption due to growth of the surface oxide scale. Apart from chromium depletion, the profiles show an enrichment of molybdenum and niobium in the immediate vicinity of oxide–scale interface.

SNMS profiles of thick-walled alloy 625 after oxidation in Ar-50 %H2O at 700 °C for 3000 h; (a) Overview of main elements Ni, Cr, O and Mo (b) elements Mo, Nb, Fe, Si and O
SNMS profiles measured after 3000 h oxidation at different temperatures (Fig. 12) showed that the Mo and Nb enrichments in the subsurface zone increases with increasing temperature, while the chromium concentration at the scale–alloy interface tends to decrease with increasing temperature. The more pronounced subsurface enrichment of Nb and Mo at 700 °C than at 750 °C found by the SNMS analysis is in good agreement with the higher extent of δ-phase formation at the scale–alloy interface observed at the latter temperature (Fig. 2). This finding illustrates that the amount, and especially the size of the subsurface δ-phase precipitates, increased with increasing temperature.

SNMS profiles of the elements Cr, Mo and Nb in thick-walled alloy 625 after oxidation in Ar-50 %H2O for 3000 h at different temperatures
Figure 13 shows SNMS concentration profiles of chromium, niobium and molybdenum after various oxidation times at 700 °C. Chromium depletion is more marked after longer oxidation times, while the enrichment of molybdenum and niobium slightly increases with time. A similar observation was made after oxidation at 750 and 800 °C. According to classical oxidation theory [48], the concentration of chromium at the oxide–metal interface should virtually remain constant and independent of time in the case of parabolic oxidation. The fact that the chromium content at the oxide–metal interface decreases with time indicates that the growth rate of the oxide scale on alloy 625 might be slightly faster than parabolic. The observed time-dependent decrease in Cr content at the metal–oxide interface might, however, also be related to the limitations of the SNMS method, since it considers all elements together and it will only reveal exact results if the various interfaces would be ideally flat, which is obviously not the case for real alloy systems. Besides, the formation of a δ-phase layer beneath the oxide scale might also influence the shape of the Cr profiles measured by SNMS. The δ-phase layer is, especially after long oxidation times, almost as thick as the oxide layer. Since the δ-phase contains only minor amounts of Cr, its presence results in an apparent Cr depletion in the SNMS profiles in the immediate vicinity of the oxide layer.

SNMS profiles of the elements Cr, Mo and Nb in thick-walled alloy 625 after oxidation in Ar-50 %H2O at 700 °C for 1000, 3000 and 10000 h
XRD analysis was used to get more detailed information on the various phases present in the near surface region and in the bulk alloy before and after long-term exposure at high temperature in Ar-50 %H2O. Figure 8 shows XRD profiles of alloy 625 (thick-walled material) in the as-received condition as well as after oxidation at 700 °C for different times. XRD diffraction lines corresponding to Cr2O3 become stronger with increasing oxidation times, as expected since the chromia layer thickened with time. Diffraction peaks corresponding to δ-phase also become stronger after longer times, indicating that a larger amount of δ-phase was present. An additional series of XRD measurements was carried out on a specimen oxidized at 700 °C for 1000 h in order to investigate the phase distribution as function of depth (Fig. 9). After the first measurement in the as-oxidized condition, the oxide scale was removed by careful grinding with SiC grinding paper (1200 grit) and then once again analysed by XRD. The analysis of the as-oxidized specimen revealed the presence of Cr2O3, η-M6C carbides, δ-phase and the γ matrix. After removing the oxide scale, δ-phase diffraction peaks were weaker but still present, indicating the presence of δ-phase in the bulk alloy, i.e. δ-phase in the form of needle-like precipitates, as shown in Fig. 2. An interesting fact from the XRD results is that the exact location of the diffraction lines for δ-phase showed a better fit to the database files of Ni3Mo than those of Ni3Nb.
Discussion
Subsurface phase transformations
The dissolution of M6C carbides observed in alloy 625 as thick-walled material (Fig. 2) together with depletion and back diffusion of carbon in the subsurface zone of the alloy was previously observed by Chyrkin et al. [37] after oxidation of the alloy at 900–1000 °C in air and in Ar-H2-H2O. A similar phenomenon, i.e. dissolution of M6C carbides in the subsurface zone, also occurred in the Co-based alloy Haynes 25 after long-term exposure in Ar-50 %H2O at 700–800 °C [38]. The M6C dissolution was in both cases thought to be the result of an indirect effect, i.e. back diffusion of carbon in the Cr gradient established across the depletion zone. Strong chromium depletion near the scale–alloy interface in chromia-forming nickel-based alloys results in an increase of the carbon activity in the γ-phase in the subscale zone [37]. The increase in C-activity results in carbon diffusing back in direction of the bulk alloy. This leads to the dissolution of the carbides in the surface-near region and to a carbon enrichment just underneath the chromium depletion zone.
In the case of alloy 625 as strip material, the dissolution of μ-phase in the subsurface zone can be explained considering that the μ-phase has an approximate chemical composition of Ni2(Cr,Mo). Since chromium is one of the main constituents of μ-phase, the continuing growth of the chromia-based surface scale and the subsequent chromium depletion has a result that μ-phase is no longer stable and consequently dissolves.
As mentioned before, the most striking effect observed in the subsurface zone after oxidation in steam in the temperature range 700–800 ºC was that δ-Ni3(Nb,Mo) phase became enriched near the alloy–scale interface, independently of the exact chemical composition of the alloy. Nb enrichment beneath the oxide scale and subsurface formation of δ-phase was previously observed after oxidation of alloy 625 at 900–1000 °C in air and in Ar-H2-H2O by Chyrkin et al. [37]. The authors found subsurface enrichment of Nb and formation of δ-phase even after oxidation at 1000 ºC in spite of the fact that at this temperature δ-phase was not present in the bulk alloy. Contrary to the results obtained after oxidation at higher temperatures (900–1000 °C), SNMS profiles as well as SEM analyses in the present study showed a subsurface enrichment of both Nb and Mo, indicating that at 700–800 °C, the δ-phase formed is Ni3(Nb,Mo) and δ-Ni3Nb is not found at higher temperatures. An enrichments of both Nb and Mo beneath the oxide layer have been previously observed in alloy 625 after oxidation of the alloy in steam-containing atmospheres at 750 °C [49], and the δ-phase as Ni3(Nb, Mo) has already been reported to form in alloy 625 when exposed in air plus 10 %H2O at 760 °C and at 871 °C for 360 days [50]. However, no explanation for this phenomenon was provided.
Formation of δ-Ni3(Nb,Mo) phase at 700–800 °C can be explained considering that, although the δ-phase is commonly defined as Ni3Nb, a δ-Ni3Mo phase can also form in the binary Ni–Mo system at temperatures below 910 °C [51]. According to the ternary Ni–Nb–Mo phase diagram [52], the δ-phase can dissolve substantial amounts of molybdenum at 900 °C, indicating that a δ-Ni3(Nb,Mo) phase is stable in a certain range of compositions (Fig. 14). Phase equilibrium diagrams of the Ni–Mo–Nb system at different temperatures were calculated with ThermoCalc using thermodynamic data from the database TTNI7 (Fig. 15). The calculations showed that at 700 °C, the δ-phase exists in the binary systems as Ni3Nb and Ni3Mo, respectively. The two phases exhibit complete miscibility which in the ternary phase diagram results in a line where δ-phase exists as Ni3(Nb, Mo).


Phase equilibrium diagrams of the Ni–Mo–Nb system at 700 and 900 °C calculated with ThermoCalc using the database TTNI7 [55]
At 900 °C, the calculated and experimentally determined ternary Ni–Mo–Nb phase diagram showed some differences. Although the three-phase fields in both diagrams are in fair agreement, the compositions at the corners of the phase fields substantially differ. For instance, the solubility of Ni in the Mo/Nb solid solution is higher in the experimental diagram than in the calculated one. Additionally, Ni can dissolve up to 10 at.% of Nb in the calculated ternary diagram, whereas in the experimental one, the limit is approximately 3 at.%. However, the most important difference between both diagrams is that, contrary to the experimentally determined ternary Ni–Mo–Nb phase diagram [52] (Fig. 14), the binary δ-Ni3Mo phase is no longer stable in the calculated Ni–Mo–Nb diagram, and the phase field in which Ni3Mo and Ni3Nb exhibit complete miscibility in the experimentally determined diagram, transforms in the calculated diagram into a line where only Ni3Nb exists, although it still has substantial solubility for Mo.
Modelling of subsurface phase transformations
Formation of the δ-Ni3(Nb,Mo) phase immediately beneath the oxide scale implies that a strong driving force exists for Nb and Mo to diffuse from the bulk alloy towards the scale–alloy interface. Chromium depletion in the subsurface zone was supposed to be the main reason for the subsurface enrichment of δ-Ni3Nb phase in alloy 625 observed by Chyrkin et al. [37], when the alloy was exposed in air and in Ar-H2-H2O at 900–1000 °C. The chromium depletion process was modelled using the finite difference programme DICTRA [53] taking thermodynamic data from the database TTNI7 adapted to ThermoCalc and kinetic data from the database MOBNI1. An outward, time-dependent flux of chromium, which simulated the chromium loss as a result of surface oxide formation and growth, was used as the input for the calculations. The calculations predicted a strong enrichment of δ-phase in the immediate vicinity of the scale–alloy interface. Further calculations made using ThermoCalc showed that the main thermodynamic driving force for the δ-phase enrichment was a decreasing niobium activity in the subscale zone as a result of chromium depletion. Since the Nb activity decreases with decreasing chromium content, niobium exhibits an uphill diffusion towards the most extensively chromium-depleted site, i.e. the scale–alloy interface, where Nb becomes enriched.
A similar approach was used in the present study to describe the findings obtained during steam oxidation at 700 °C. For the DICTRA calculations, only the phases which were experimentally identified in thick-walled material (i.e. γ, δ and M6C) were considered. As in the case of higher temperatures, the DICTRA simulations predicted a surface enrichment of Nb-rich δ-phase (Fig. 16). The input data of the time-dependent chromium flux were derived from the oxidation data of alloy 625 as thick-walled material (Fig. 1) assuming a pure chromia scale which thickens according to a parabolic time dependence. Additionally, the influence of chromium content on the niobium activity in alloy 625 at 700 ºC was calculated with ThermoCalc [54]. As found in the previous studies for the 900–1000 °C range, the niobium activity at 700 °C was also found to decrease with decreasing chromium content, thus explaining uphill diffusion of Nb towards the scale–alloy interface.

Phase distribution in near surface region of alloy 625 specimen after 1000 h oxidation at 700 °C calculated with DICTRA. A time–dependent, outward Cr-flux was assumed based on the oxidation data of alloy 625 as thick-walled material after oxidation in Ar-50 %H2O at 700 °C (see Fig. 1)
The predicted formation/enrichment of δ-phase near the alloy–scale interface modelled with DICTRA (Fig. 16) is in agreement with the experimental results. Nonetheless, the databases used by ThermoCalc and DICTRA considered δ-phase as Ni3Nb, whereas the experimental results showed that at 700–800ºC, the δ-phase has a chemical composition of Ni3(Nb,Mo), whereby the Nb/Mo atomic ratio was, according to EDX measurements, approximately 1.75. Since at 700 °C the δ-phase can dissolve substantial amounts of molybdenum, molybdenum also diffuses towards the oxide scale–alloy interface and becomes incorporated in the phase, leading to a subsurface enrichment of both Nb and Mo (Fig. 11).
The fact that DICTRA/ThermoCalc consider δ-phase as Ni3Nb and not as Ni3(Nb,Mo) is directly related to the phase distribution of alloy 625 calculated by ThermoCalc. The phase equilibria as function of temperature for both investigated alloy 625 batches was calculated with ThermoCalc using thermodynamic data from the database TTNI7 [55] (Figs. 17, 18). According to the calculations, the thermodynamically stable phases for both compositions at 700–800 °C in the bulk alloy are γ-, δ- and μ-phase as well as TiN and M23C6 carbides. The main differences in the calculations for both batches lay in the predicted amount of each phase. More δ-phase is expected to form in the strip material, and the phase is stable in a wider temperature range than in the thick-walled material. Additionally, σ-phase appears as thermodynamically stable phase in alloy 625 as strip material at 800–900ºC.

Phase distribution of thick-walled alloy 625 as function of temperature calculated with ThermoCalc using the database TTNI7 [55]. Also, given are the calculated compositions in at.% of the different phases at relevant temperatures

Phase distribution of alloy 625 (strip material) as function of temperature calculated with ThermoCalc using the database TTNI7 [55]. Also, given are the calculated compositions in at.% of the different phases at relevant temperatures
ThermoCalc predicts the formation of μ-phase for both batches tested although the experimental results showed that μ-phase does not always form in alloy 625. From the Ni–Mo–Nb diagram at 700 °C, it can be seen that the stability field of μ-phase is very small. Nonetheless, the μ-phase is stable in a wide range of compositions in the Ni–Cr-Mo and the Ni–Cr-Nb ternary diagrams at 700 °C. Apparently, the addition of chromium to the Ni–Nb–Mo system leads to an increase of the stability field of μ-phase in such a way that ThermoCalc assumes that a three-phase field consisting of γ, μ-phase and δ-phase exists. Since μ-phase contains a substantial amount of Mo (approximately 26 at.%), ThermoCalc assumes that Mo remains in the μ-phase rather than getting incorporated in the δ-phase.
Summary and conclusions
Oxidation of two alloy 625 batches in Ar-50 %H2O at 700–800 °C for oxidation times up to 10000 h resulted in the formation of a thin chromia-based surface scale with an outer Cr/Mn spinel and internal SiO2. The alloy showed very low weight change values even after oxidation up to 10000 h. No substantial differences were observed in the oxidation behaviour of the two studied batches apart from slight changes in the amount of SiO2 precipitates and in the morphology of the outer Cr/Mn spinel.
Exposure of alloy 625 at 700–800 °C resulted in changes in the bulk alloy microstructure. Independent of the studied batch and, therefore, sample thickness and individual chemical composition of alloy 625, δ-phase as Ni3(Nb,Mo) was found in the bulk alloy in the form of needle-like precipitates together with a number of other precipitates. In the case of the thick-walled material, those precipitates were mainly Si-rich η-M6C carbide. In the bulk of the strip material, Cr-rich carbides, some randomly distributed (Ti, Nb)-nitrides and minor amounts of other precipitates, identified as μ-phase, were found.
An enrichment of δ-Ni3(Nb, Mo) phase immediately beneath the oxide scale occurred in both tested batches. The reason for the δ-phase enrichment beneath the oxide scale is an uphill diffusion of Nb in the chromium-depleted zone. The activity of Nb decreases with decreasing chromium content, and therefore niobium has to diffuse towards the oxide scale. Additionally, it was observed that the chemical composition of the δ-phase at 700 °C differs from that observed at higher temperatures (900–1000 °C), since at 700–800 °C, the δ-phase can dissolve substantial amounts of molybdenum. This provides a driving force for an outward diffusion of molybdenum from the bulk alloy towards the scale/alloy interface. Apart from the subsurface enrichment of δ-Ni3(Nb,Mo) phase, it was observed that, depending on the batch considered, η-M6C carbides or μ-phase dissolved in the subsurface chromium-depleted zone.
References
Maurer GE, Patel AD (2010) Alloy design and processing challenges for advanced power systems: an alloy producer’s perspective. Adv Sci Technol 72:22–30
Maziasz PJ, Wright IG, Shingledecker J, Gibbons TB, Romanosky RR (2005) Defining the materials issues and research for ultra-supercritical steam turbines. In: Advances in materials technology for fossil power plants 2004: proceedings from the fourth international conference (ASM International), proceedings from the fourth international conference, pp 602–622
Ennis PJ, Quadakkers WJ (2007) Mechanisms of steam oxidation in high strength martensitic steels. Int J Press Vessels Pip 84:75–81
Nickel H, Ennis PJ, Quadakkers WJ (1997) Candidate materials for advanced power generating plants and high temperature heat exchangers. Trans Indian Inst Met 50:681–689
Ehlers J (2001) Oxidation von ferritischen 9–12 % Cr-Stählen in wasserdampfhaltigen Atmosphären bei 550 bis 650 °C, PhD thesis, RWTH Aachen
Ehlers J, Young DJ, Smaardijk EJ, Tyagi AK, Penkalla HJ, Singheiser L, Quadakkers WJ (2006) Enhanced oxidation of the 9%Cr steel P91 in water vapour containing environments. Corros Sci 48:3428–3454
Quadakkers WJ, Ennis PJ, Zurek J, Michalik M (2005) Steam oxidation of ferritic steels—laboratory test kinetic data. Mater High Temp 22:47–60
Zurek J, Michalik M, Schmitz F, Kern TU, Singheiser L, Quadakkers WJ (2005) The effect of water-vapor content and gas flow rate on the oxidation mechanism of a 10%Cr-ferritic steel in Ar–H2O mixtures. Oxid Met 63:401–422
Blum R, Vanstone RW (2004) Materials development for boilers and steam turbines operating at 700 °C. In: Proceedings 4th international conference advances in materials technology for fossil power plants—EPRI conference, Hilton Head Island, South Carolina, pp 116–136
Shingledecker JP, Wright IG (2006) Evaluation of the materials technology required for a 760 ºC power steam boiler. In: Proceedings to the 8th liege conference on materials for advanced power engineering liege, Belgium, pp 107–120
Essuman E, Meier GH, Zurek J, Hansel M, Norby T, Singheiser L, Quadakkers WJ (2008) Protective and non-protective scale formation of NiCr alloys in water vapour containing high- and low-pO2 gases. Corros Sci 50:1753–1760
INCONEL® alloy 625, Datasheet, Special Metals Corporation (2013)
Ferrer L, Pieraggi B, Uginet JF (1991) Microstructural evolution during thermomechanical processing of alloy 625. In: Loria EA (ed) Superalloys 718, 625 and various derivatives. The Minerals, Metals and Materials Society, New York, pp 217–228
Brooks JW, Bridges PJ (1988) Metallurgical stability of Inconel alloy 718. In: Reichman S, Duhl DN, Maurer G, Antolovich S, Lund C (eds) Superalloys 1988. The Metallurgical Society, Warrendale, PA, pp 33–42
Evans ND, Maziasz PJ, Shingledecker JP, Yamamoto Y (2008) Microstructure evolution of alloy 625 foil and sheet during creep at 750 °C. Mater Sci Eng A 498:412–420
Mathew MD, Parameswaran P, Rao KBS (2008) Microstructural changes in alloy 625 during high temperature creep. Mater Charact 59:508–513
Palanichamy P, Mathew MD, Latha S, Jayakumar T, Bhanu K, Rao SK, Mannan SL, Raj B (2001) Assessing microstructural changes in alloy 625 using ultrasonic waves and correlation with tensile properties. Scripta Mater 45:1025–1030
Cortial F, Corrieu J, Vernot-Loier C (1995) Influence of heat treatments on microstructure, mechanical properties, and corrosion resistance of weld alloy 625. Metall Mater Trans A 26:1273–1286
Mathew MD, Murty KL, Rao KBS, Mannan SL (1999) Non-destructive evaluation of the effect of aging on tensile properties of alloy 625. In: Non-destructive evaluation. Minerals, Metals & Materials Soc, Warrendale, pp 35–42
Mathew MD, Rao KBS, Mannan SL, Shing R (1999) Recovery of creep properties of alloy 625 after long term service. In: Sakuma T, Yagi K (eds) Creep and fracture engineering materials and structures. Trans Tech Publications, pp 537–547
Sundararaman M, Mukhopadhyay P, Banerjee S (1988) Precipitation of the δ-Ni3Nb phase in two nickel base superalloys. Metall Mater Trans A 19:453–465
Eiselstein HL, Tillack DJ (1991) The invention and definition of alloy 625. In: Loria EA (ed) Superalloys 718, 625 and various derivatives. The Minerals, Metals and Materials Society, Warrendale, pp 1–14
Forsyth JB, D′Alte Da Veiga LM (1962) Structure of µ-phase Co7Mo6. Acta Crystallogr A 15:543–546
Zhao K, Lou LH, Wen Y, Li H, Hu ZQ (2004) Nucleation and growth of μ phase. J Mater Sci 39:369–371
Tawancy HM (1996) Precipitation characteristics of μ-phase in wrought nickel-base alloys and its effect on their properties. J Mater Sci 31:3929–3936
Vernot-Loier C, Cortial F (1991) Influence of heat treatments on microstructure, mechanical properties of and corrosion behaviour of alloy 625 forged rod. In: Loria EA (ed) Superalloys 718, 625 and various derivatives. The Minerals, Metals and Materials Society, Warrendale, pp 409–422
Schnabel E, Schuller HJ, Schwaab P (1971) The precipitation and recrystallization behavior of the nickel base alloy INCONEL alloy 625. Prakt Metallogr 8:521–527
Crum JR, Adkins ME, Lipscomb WG (1986) Performance of high nickel-alloys in refinery and petrochemical environments. Mater Perform 25:27–33
Floreen S, Fuchs GE, Yang WJ (1994) The metallurgy of alloy-625. In: Superalloys 718, 625, 706 and various derivatives. Minerals, Metals & Materials Society, Warrendale, PA, pp 13–37
Wang YS, Guan XM, Ye HQ, Bi J, Xu AS (1980) Effect of silicon on grain boundary carbide precipitation and properties of a cobalt-free wrought nickel-base superalloy. In: Tien JK (ed) Superalloys. ASM, Metals Park OH, pp 63–70
Buscail H, Rolland R, Issartel C, Rabaste F, Riffard F, Aranda L, Vilasi M (2011) Effects of water vapour on the oxidation of a nickel-base 625 alloy between 900 and 1100 °C. J Mater Sci 46:5903–5915
N’Dah E, Hierro MP, Borrero K, Perez FJ (2007) Study of the cyclic oxidation resistance of superalloy IN-625: lifetime predicted by COSP-modelling program. Oxid Met 68:9–21
Quadakkers WJ, Zurek J (2010) Oxidation in steam and steam/hydrogen environments. In: Richardson JA (ed) Sheir’s corrosion. Elsevier, Amsterdam, pp 407–456
Quadakkers WJ, Elschner A, Speier W, Nickel H (1991) Composition and growth mechanisms of alumina scales on FeCrAl-based alloys determined by SNMS. Appl Surf Sci 52:271–287
Quadakkers WJ, Piron-Abellan J, Shemet V, Singheiser L (2003) Metallic interconnectors for solid oxide fuel cells—a review. Mater High Temp 20:115–127
Zurek J, Young DJ, Essuman E, Hänsel M, Penkalla HJ, Niewolak L, Quadakkers WJ (2008) Growth and adherence of chromia based surface scales on Ni-base alloys in high- and low-pO2 gases. Mater Sci Eng A 477:259–270
Chyrkin A, Huczkowski P, Shemet V, Singheiser L, Quadakkers J (2011) Sub-scale depletion and enrichment processes during high temperature oxidation of the nickel base alloy 625 in the temperature range 900–1000 °C. Oxid Met 75:143–166
Garcia LF, Chyrkin A, Huttel T, Bohme C, Barnikel J, Gruner D, Schmitz F, Quadakkers WJ (2012) Oxide scale formation and subsurface phase transformations during long-term steam exposure of the cobalt base alloy 25. Mater Corros 63:878–888
Kirman I (1969) Precipitation in Fe–Ni–Cr–Nb system. J Iron Steel Inst 207:1612–1618
Zimmermann U, Grimmer H (1987) On the formation of η-’ carbides during high-temperature corrosion of 3 nickel-based alloys in nuclear process heat helium. Mater Sci Eng 87:131–136
Kenik EA, Maziasz PJ, Swindeman RW, Cervenka J, May D (2003) Structure and phase stability in a cast modified-HP austenite after long-term ageing. Scripta Mater 49:117–122
Maziasz PJ (1979) Formation of diamond-cubic Eta phase in type-316 stainless-steel exposed to thermal aging or irradiation environments. Scr Metall 13:621–626
Leitnaker JM, Klueh RL, Laing WR (1975) Composition of Eta carbide phase in 2 1/4 Cr-1 Mo steel. Metall Trans A 6:1949–1955
Lee EH, Rowcliffe AF, Kenik EA (1979) Effects of Si and Ti on the phase-stability and swelling behavior of AISI-316 stainless-steel. J Nucl Mater 83:79–89
Williams TM, Titchmarsh JM (1981) Silicon-rich phases in austenitic alloys. J Nucl Mater 98:223–226
Raghavan M, Mueller RR, Vaughn GA, Floreen S (1984) Determination of isothermal sections of nickel rich portion of Ni–Cr–Mo system by analytical electron-microscopy. Metall Trans A 15:783–792
Gozlan E, Bamberger M, Dirnfeld SF, Prinz B, Klodt J (1991) Topologically close-packed precipitations and phase-diagrams of Ni–Mo–Cr and Ni–Mo–Fe and of Ni–Mo–Fe with constant additions of chromium. Mater Sci Eng A Microstruct Process 141:85–95
Wagner C (1956) Oxidation of alloys involving noble metals. J Electrochem Soc 103:571–580
Gorman DM, Higginson RL, Du H, McColvin G, Fry AT, Thomson RC (2013) Microstructural analysis of IN617 and IN625 oxidised in the presence of steam for use in ultra-supercritical power plant. Oxid Metals 79:553–566
Deodeshmukh VP (2012) Long-term oxidation and chromia vaporization behavior of high-temperature foil alloys in water vapor containing environment. In: 8th international symposium on high-temperature corrosion and protection of materials, Les Embiez, paper 158
Okamoto H (1991) Mo–Ni phase diagram. J Phase Equilibria 12:703
Villars P, Prince A, Okamoto H (1995) Handbook of ternary alloy phase diagrams. ASM International, Metals Park OH
Borgenstam A, Engstrom A, Hoglund L, Agren J (2000) DICTRA, a tool for simulation of diffusional transformations in alloys. J Phase Equilibria 21:269–280
Jansson B, Schalin M, Selleby M, Sundman B (1993) The thermo-calc database system. In: Bale CW, Irons GA (eds) Computer software in chemical and extractive metallurgy. Canadian Institute of Mining, Metallurgy and Petroleum, Montreal, pp 57–71
TTNI7 Database, Thermo Tech Ni-Based Superalloys Database v7.0. (2006)
Okamoto H (1992) Nb–Ni phase diagram. J Phase Equilibria 13:444–445
Acknowledgements
The authors are grateful to Mr. H. Cosler, Ms. A. Kick, and Mr R. Mahnke for carrying out the oxidation tests, Mr. V. Gutzeit and Mr. J. Bartsch for optical microscopy, Dr. D. Grüner, Dr. E. Wessel and Ms. D. Esser for SEM and TEM investigations, Mr. M. Ziegner for XRD analyses and Mr. M. Borzikov for SNMS measurements. The Central Chemistry Department (ZEA) is acknowledged for carrying out the ICP-OES analyses.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Garcia-Fresnillo, L., Chyrkin, A., Böhme, C. et al. Oxidation behaviour and microstructural stability of alloy 625 during long-term exposure in steam. J Mater Sci 49, 6127–6142 (2014). https://doi.org/10.1007/s10853-014-8344-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-014-8344-7