
Abstract
A facile method has been developed for the synthesis of core–shell nanostructure poly(N-isopropylacrylamide) grafted on silica nanoparticles (SiO2 NPs) by using the combination of ARGET ATRP and thiol-ene click chemistry. The covalent attachment of the thermo-responsive polymer was achieved by taking advantage of the fast, robust, and high efficient thiol-ene click reaction as demonstrated by FT-IR and XPS. The ARGET ATRP provides the good conversion of the monomer in a well-controlled manner as indicated by the narrow value of PDI (1.21). The grafting amount of the polymer on SiO2 NPs was found to be ca. 18 % as determined by TGA technique. TEM images of the encapsulated SiO2 NPs suggested that the SiO2 NPs core was covered by the soft polymer layer. Interestingly, dynamic light scattering analysis revealed that the as-synthesized nanocomposites exhibited the thermo-responsive behavior with the transition temperature around 31–33 °C.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Over the past decade, new routes for the incorporation of inorganic, organic, and polymers in nanoscale size have offered promising opportunities in the preparation of high performance advanced nanomaterials [1–4]. The emergence of “click” chemistry, typically Huisgen 1,3-dipolar azide–alkyne cycloaddition and thiol-ene reaction, has attracted immense scientific interests due to its efficiency and the versatilities [5, 6]. The ease of synthesis of the alkene/alkyne and thiol/azide functionalities, coupled with tolerance to a wide variety of functional groups, stability and reaction conditions, make these coupling process highly attractive for the synthesis of complex macromolecular structures [7–9]. In addition, “click” chemistry has also greatly facilitated the post-modification of functional polymers [10, 11] and synthesis of nanocomposites [12, 13]. Recently, the combination of controlled radical polymerization (CRP) and click reaction has been regarded as a powerful strategy for the preparation of functional hybrid materials [10, 14, 15].
Two of the most successful CRP techniques are reversible addition fragmentation chain transfer (RAFT) and atom transfer radical polymerization (ATRP). Both techniques have been successfully applied for the synthesis of various homo-polymers as well as block copolymers [16–20]. Despite of such achievements, these methods suffer drawbacks that limit their use for the preparation of polymers to use in practical applications. RAFT employs a RAFT agent as a chain transfer agent, often a thiocarbonyl-based compound, to control the polymerization. If the RAFT agent is not cleaved from the polymer in post-polymerization, polymers produced via RAFT can be highly colored or toxic [21–23]. In ATRP, a transition metal catalyst (often a Cu(I)/(II) system) used with relatively high concentration to control the polymerization leads to difficulties for the purification process in order to yield products with high purity [24]. Additionally, conventional ATRP must be carried out in a completely deoxygenated atmosphere to avoid the catalyst poisoning [24]. Although there have been several advances in order to overcome these issues, still some problems remain in this technique [25]. A new technology based on “activators regenerated by electron transfer atom transfer radical polymerization” (ARGET ATRP) which uses an excess amount of a reducing agent to regenerate Cu(I) from Cu(II) to maintain an appropriate Cu(I)/(II) balance was developed by Matyjazewski and his group [26]. The inclusion of this reducing agent allows to reduce the concentration of copper to part per million (ppm) levels and in most cases removal of the low levels of catalyst is achieved simply by precipitation of the polymer. Moreover, the usage of an excess reducing agent provides tolerance of oxygen, as a result, the polymerization can be conducted in the presence of limited amounts of air [26–29]. Therefore, the combination of “click” chemistry with robust CRP techniques like ARGET ATRP may offer a superior route for the fabrication of well-defined inorganic/polymer nanocomposites.
Stimuli-responsive polymers are preferable building blocks for the fabrication and engineering of nanomaterials because they exhibit abrupt changes in their physicochemical properties corresponding to the shift in external environment. By tailoring with these kinds of polymers, the surface behavior can be tuned under external stimuli, such as ionic strength, pH, as well as temperature and thereby the release of guest molecules from the surface can be controlled [30–32]. Among these types of polymers, poly(N-isopropylacrylamide) (PNIPAm) is one of the most extensively studied polymers due to its thermo-responsive properties [33]. PNIPAm is biocompatible and it exhibits a lower critical solution temperature (LCST) at ∼32 °C that is nearby human body temperature [33]. Therefore, PNIPAm has been widely applied for biomedical applications including biosensor, tissue engineering, and controlled drug delivery [33–36]. Silicon dioxide nanoparticles, also known as silica nanoparticles (SiO2 NPs), are the core materials for a large number of materials researches due to their chemical and mechanical stability [37, 38]. Several advanced functional materials based on SiO2 NPs for opto-electronics and biomedical applications have been achieved by coating of SiO2 NPs with a layer of polymers [14, 30, 39–43]. Consequently, SiO2 NPs coatings have attracted significant scientific interest and such a huge effort has been paid in order to discover new robust surface modification protocol [44, 45]. Taking all of the above considerations into account, this paper demonstrates a facile method for the versatile and efficient functionalization of SiO2 NPs with thermo-responsive polymer PNIPAm. This approach is based on tailor-made thiol-functionalized SiO2 NPs and alkene-functionalized PNIPAm brush precursors. The novelty of the method is the combination of robust living radical polymerization and click chemistry for a facile chemical anchoring of thermo-responsive polymer onto SiO2 NPs. The ARGET ATRP offers the ability to prepare the polymers with controlled structure and compositions with click chemistry as easy handling and highly efficient strategy. This newly devised protocol requires only a ppm level of catalyst and it does not require extremely deoxygenated condition. Overall, it is a one-step process. To the best of our knowledge, this is the first report for the synthesis PNIPAm-g-SiO2 nanocomposites by “thiol-ene” click chemistry and ARGET ATRP technique.
Experimental
Materials
NIPAm (Sigma-Aldrich, 97 %) was purified by recrystallization with hexane. 2-Bromoisobutyrylbromide (2-BiBB, Sigma-Aldrich, 98 %) was distilled under reduced pressure over CaH2. 10-Undecen-1-ol (Tokyo Chemical Industry, 98 %), tetraethyl orthosilicate (TEOS, Sigma-Aldrich, 98 %), 3-mercaptopropyl trimethoxysilane (MPTS, Sigma-Aldrich, 98 %), N,N,N′,N′,N″-pentamethyldiethylenetriamine (PMDETA, Sigma-Aldrich, 99 %), ammonia solution (NH3·H2O, Junsei, 28 wt%), copper (0) (99.9 %), Irgacure 651 (Sigma-Aldrich, 99 %), and solvents (reaction grade) were used as received.
Synthesis of thiol-functionalized SiO2 NPs
SiO2 nanoparticles were prepared by sol–gel reaction according to the modified Stöber method [46, 47]. Namely, about 18 mL (0.040 mol) of TEOS was added dropwise to a mixture of absolute ethanol (300 mL), deionized water (30 mL), and 28 wt% ammonia solution (6 mL). The reaction mixture was stirred for 6 h at room temperature. Subsequently, 4 mL (8.4 mmol) of MPTS was injected into the SiO2 sol, and the reaction was allowed to proceed for another 18 h. After the reaction, the resulting SiO2 nanospheres with thiol bonds on the surface were purified by three centrifugation/redispersion cycles in acetone, ethanol, and deionized water. The SiO2 nanospheres were finally dried in a vacuum oven at room temperature overnight.
Synthesis of 10-undecenyl 2-bromoisobutyrate (UniB-Br)
The vinyl-terminated ATRP initiator, UniB-Br, was synthesized according to following procedure: 10-undecen-1-ol (1.70 g, 0.01 mol) was mixed with triethylamine (1.01 g, 0.01 mol) in 50 mL of dried toluene. To this solution, a solution of BiBB (2.30 g, 0.01 mol) in toluene (10 ml) was added dropwise for 30 min with stirring. The reaction was kept for 12 h with stirring under nitrogen atmosphere. The precipitate was filtered off using a frit funnel. The product was yellowish oil after the removal of the solvent. The oily mixture was dissolved in toluene and washed with large amount of 0.01 N HCl and cold water, respectively. After drying with anhydrous CaCl2, the solvent was removed under reduced pressure. The product, UniB-Br, was purified by SiO2 column chromatography using hexane:ethylacetate (15:1) as eluent. The final product was colorless oil with a yield of 90.5 %. 1H NMR (400 MHz, CDCl3): δ 5.80 (m, 1H, –CH=), 4.97 and 4.92 (m, 2H, H 2C=), 4.15 (t, 2H, –OCH 2–), 2.02 (m, 2H, =CH–CH 2–,), 1.92 (s, 2H, –C(CH 3)2–), 1.66 (m, 2H, –CH 2–CH2–O–), 1.26–1.37 (m, 12H, –(CH 2)6CH2–CH2–O–).
Synthesis of vinyl-terminated polymer by ARGET ATRP
The vinyl-terminated thermo-responsive PNIPAm was synthesized under mild condition by taking advantages of ARGET ATRP. The polymerization was performed in isopropyl alcohol (IPA), employing CuBr2/PMDETA as a catalyst and UniB-Br as an initiator in the presence of copper powder as a reducing agent at room temperature. In a typical process, a dry 25 mL vial equipped with a magnetic stirrer was charged with NIPAm (5.65 g, 0.01 mol), IPA (4.0 mL), copper powder (50 mg), and UniB-Br (159 mg, 1 mmol). After sealing the vial with a septum and melting the mixture, the resulting solution was degassed by nitrogen bubbling. A degassed solution of CuBr2 (1.11 mg, 5 μmol) and the PMDETA ligand (8.65 mg, 50 μmol) in IPA (1.0 mL) was introduced into the vial to initiate the polymerization. The polymerization was stopped after 24 h by removing the septum and exposing the catalyst to air. The product mixture was diluted in dichloromethane and passed through a short alumina column to remove the catalyst, followed by precipitation in excess amount of diethyl ether. Dissolution and precipitation were repeated several times until white powder was obtained. The final product was dried thoroughly under vacuum prior to characterization (conversion: 60 %). The polymer was analyzed by GPC and 1H NMR.
Surface modification of SiO2 NPs with PNIPAm using “click” chemistry
About 0.12 g of the SiO2–SH and 0.25 g (0.05 mmol) of vinyl-terminated PNIPAm were introduced into 10 mL of dichloromethane in a Pyrex tube. After the addition of sufficient amount of the photo-initiator, the mixture was homogenized by ultrasonic treatment. The tube was sealed and degassed by nitrogen bubbling for 15 min. Then, the reaction mixture was exposed to an UV (365 nm) source and stirred for 24 h with magnetic stirring to ensure the completed conversion. The resultant PNIPAm-g-SiO2 nanocomposites were precipitated in cold ethyl ether, and washed thoroughly with dichloromethane to remove the un-reacted PNIPAm chains.
Characterization
Fourier-transform infrared (FT-IR) spectroscopic analysis was carried out on an Agilent FT-IR spectrophotometer. Transmission electron microscopy (TEM) images were obtained using a JEOL BTEM with an accelerating voltage of 80 kV. Surface composition of nanocomposites was investigated by an X-ray Photoelectron Spectroscopy (XPS) (Thermo VG Multilab 2000) in ultrahigh vacuum with Al Kα radiation. The molecular weight (M n) and molecular weight distribution (PDI = M w/M n) of the vinyl-terminated polymer prepared by ARGET ATRP were characterized by gel permeation chromatography (GPC) on an HP 1100 apparatus equipped with a set of four columns (105, 104, 103, 102 Å: polymer standards service), THF was used as an eluent solvent. Poly(methylmethacrylate) samples were used as standards to construct the calibration curve. Thermogravimetric analysis (TGA) was carried out on a thermogravimetric analyzer (TA Instrument, Model 2050) at a heating rate of 10 °C/min in nitrogen. The 1H NMR spectrum of UniB-Br initiator and polymers were measured using a JNM-ECP 400 (JEOL) Spectrophotometer with deuterated chloroform as a solvent.
Results and discussion
A general strategy for the surface modification of SiO2 NPs with the well-defined thermo-responsive polymer by taking advantages of ARGET ATRP and thiol-ene “click” chemistry is presented in Scheme 1. In the process, first the thiol-functionalized SiO2 NPs were prepared. Meanwhile, the vinyl-terminated polymer was realized by the robust controlled radical polymerization. Subsequently, the covalent attachment of clickable SiO2 NPs with vinyl-terminated polymeric shells was achieved by thiol-ene chemistry.

Synthetic approach for the preparation of PNIPAm-g-SiO2 nanocomposites by the combination of click chemistry and ARGET ATRP
The vinyl-terminated ATRP initiator was prepared by the esterification of alcohol and bromide acid which is widely recognized as the most efficient method for the preparation of functionalized ATRP initiator. The 1H NMR spectrum of the vinyl-terminated ATRP initiator is depicted in Fig. 1a. 1H NMR spectrum shows the peaks at (a) 5.82 and (b) 4.99 ppm, which confirmed the attachment of vinyl-terminated groups to the initiator. By using the vinyl-functionalized initiator, further modifications to attach “ene” moiety to the polymer chains are not necessary because the vinyl group of 10-undecenyl 2-bromoisobutyrate is un-reactive to ATRP reaction [48]. The vinyl-terminated ATRP initiator was used for the synthesis of PNIPAm by a facile ARGET ATRP process. Figure 1b shows the 1H NMR spectrum of the vinyl-terminated PNIPAm. On the spectrum, characteristic signals of PNIPAm at (j) 3.98 and (k) 1.12 ppm representing methyne and methyl protons of isopropyl groups are represented. In addition, the relatively broad peaks at 1.5–2.5 ppm due to the characteristic chemical shifts of polymer backbone protons are also observed. These results contributed to verify the successful synthesis of the vinyl-terminated PNIPAm. From GPC analysis, the Mn and molecular weight distribution PDI of the vinyl-PNIPAm were determined to be 7100 kg/mol and 1.21, respectively.

1H NMR spectrum of the vinyl-terminated ATRP initiator (a), and vinyl-terminated PNIPAm (b)
Figure 2 shows the FT-IR spectra of SiO2-SH and PNIPAm-g-SiO2 nanoparticles. The FT-IR spectrum of typical thiol-modified SiO2 NPs is shown in Fig. 2a; the weak but obvious band at 2394 cm−1 corresponding to the thiol stretching vibration, whereas the bands around 2979–2926 cm−1 can be attributed to the alkyl symmetric and asymmetric stretching. The thiol-functionalized SiO2 NPs were used as the precursor materials for the “click” modification. After the “click” reaction, the disappearance of the thiol peak indicates successful consumption of thiol to the vinyl-terminated PNIPAm through radical addition. Moreover, it can also be seen that the spectrum of PNIPAm-g-SiO2 exhibited the strong absorption at 1649 and 2980 cm−1, attributable to the stretching vibration of carbonyl groups (>C=O) and polymer chain alkyl groups (–CH2, –CH3), respectively. In both case of SiO2-SH and PNIPAm-g-SiO2 spectra, the appearance of the characteristic peaks of Si–O stretching, bending, and out of plane deformation at about 1100, 940, and 450 cm−1 suggested that the covalent modification through this newly devised approach may not significantly alter the inherent chemical feature of the original SiO2 NPs. This finding is quite resembled with other reported results on encapsulation of SiO2 NPs by polymers [14, 30, 36, 39]. These FT-IR results confirm the successful covalent attachment of polymers onto the surface of SiO2 NPs via “click” process.

FT-IR spectrum of thiol-functionalized SiO2 NPs (a), and PNIPAm-g-SiO2 (b)
XPS was employed to characterize the changes in chemical composition of the nanosphere surface during the modification process. Figure 3a, b presents the XPS wide scan spectra of the SiO2-SH and PNIPAm-g-SiO2 nanocomposites, respectively. The existence of the C1s and S2p signals in the SiO2-SH spectrum was attributed to the organic composition of the silane agent attached onto the surface of SiO2 NPs. By comparison of Fig. 3b and Fig. 3a, the appearance of the signal at 400.4 eV due to N1s indicated the success of the covalent attachment of the polymer chain onto the SiO2 NP surface upon the click modification. The subsequent thiol-ene click grafting of PNIPAm brushes on the nanospheres has also caused a remarkable increasing of intensity of the C1s signal.

XPS a wide scan of the SiO2-SH, b wide scan of PNIPAm-g-SiO2, c C1s core level, and d N1s core level of PNIPAm-g-SiO2
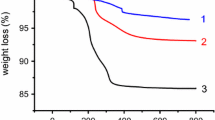
The degree of functionalization of the SiO2 NPs via the “click” protocol was investigated through TGA analysis. The TGA curves of the SiO2-SH (a), PNIPAm-g-SiO2 (b), and PNIPAm homopolymer (c) are shown in Fig. 4, as exhibiting the variation of residual masses of the samples depended on temperature. In the case of SiO2-SH NPs, while temperature was raised up to 700 °C about 10 % weight was lost may be due to the decomposition of thiol functionality. After polymer immobilization, a significant amount of weight loss was observed at the temperature range of ca. 350–450 °C. This major weight loss occurred at around 360 °C can be attributed to the decomposition of PNIPAm chain. Based on TGA, the content of PNIPAm in the composites can be estimated to be ca. 18 %.

TGA curve of SiO2–SH (a), PNIPAm-g-SiO2 (b), and PNIPAm homopolymer (c)
The TEM micrographs of bare SiO2 NPs and the modified SiO2 NPs are depicted in Fig. 5. The samples were prepared from the chloroform solution (1 wt%) by a drop-casting method. The SiO2-SH NPs exhibited an average external diameter of ca. 100 nm and showed relatively broad size distribution (Fig. 5a). After the modification, the existence of the polymer layer which was estimated to be ca. 15 nm as an outer shell (lighter color) surrounding SiO2 NPs could be clearly observed (Fig. 5b). It can be concluded that SiO2 NPs were successfully encapsulated by PNIPAm shells employing thiol-ene click chemistry.

TEM micrograph of SiO2–SH (a), and PNIPAm-g-SiO2(b) (Scale bar 200 nm)
The colloidal stability of SiO2 NPs was significantly improved by the covalent grafting of PNIPAm chains. The digital photographs of freshly prepared dispersion of SiO2-SH and PNIPAm-g-SiO2 nanocomposites in the chloroform are shown in Fig. 6a for a comparative study. The as-synthesized nanocomposites exhibited a good dispersion in chloroform, while SiO2-SH showed large aggregation. Moreover, the SiO2-SH precipitated in chloroform within a few minutes, whereas the nanocomposites remained well dispersed in chloroform for prolonged period (Fig. 6b). It could be deduced that the existence of PNIPAm chains covalently attached to SiO2 surface which had improved the colloidal stability of SiO2 NPs remarkably since they might play the role of a steric stabilizing layer to prevent agglomeration of individual particles. When the as-synthesized nanocomposites dispersed into hydrophobic solution, the severe aggregation of the NPs was minimized due to the repellant force between polymer chains existed on nanoparticles surface.

Digital photographs of SiO2–SH and PNIPAm-g-SiO2 dispersed in chloroform at preparation (a) and after 12 h (b)
The thermo-responsive property of PNIPAm-g-SiO2 nanocomposites was studied by dynamic light scattering (DLS) at a range of temperatures increasing from 25 to 39 °C. The relationship between the hydrodynamic diameter of the nanocomposites and temperature is depicted in Fig. 7. At the temperature from 25 to 31 °C, PNIPAm is hydrophilic and soluble in water; the PNIPAm chains exist in a coil state, forming a solvated, incompact nanoshell on the exterior surface of SiO2. As a result, the nanospheres diameter negligibly decreased in that range of temperature. When the temperature crossed the LCST, in the range of 31–33 °C, the hydrodynamic diameter of the core–shell nanostructure decreased dramatically because the hydrophilic chains in water underwent the inverse phase transition state to be hydrophobic. Therefore, long-PNIPAm chains collapsed toward the surface of SiO2, forming a compact closed nanoshell around the exterior surface of SiO2. These results suggest that the surface of SiO2 NPs has been tailored with the thermo-responsive behavior of PNIPAm by click reaction.

Diameter of PNIPAm-g-SiO2 nanocomposites in response to shift of temperature
Conclusion
In this study, we have demonstrated a robust protocol for the covalent immobilization of the thermo-responsive polymer PNIPAm on SiO2 NPs employing the combination of ARGET ATRP and click chemistry. The thiol-ene approach for grafting vinyl-terminated polymers is straightforward and effective to directly graft polymers to the accessible thiol on the surface of SiO2 NPs in the one-step process. The success of covalent functionalization was confirmed by surface analysis methods (FT-IR and XPS). 1H NMR spectrum of PNIPAm further confirmed the synthesis of the polymer via ARGET ATRP technique. The molecular weight and molecular weight distributions of the grafted polymer were calculated to be 7100 kg/mol and 1.21, respectively. TGA analysis revealed that the content of PNIPAm in the composites was ca. 18 %. TEM images of the bare and modified SiO2 NPs suggested that the SiO2 NP core was covered by the soft polymer layer. The colloidal stability of SiO2 NPs was significantly improved by the covalent grafting of the PNIPAm brush. The as-synthesized nanocomposites exhibited the thermo-responsive behavior as indicated by DLS which could be valuable for potential applications toward biomedical areas. Moreover, the combination of click chemistry and ARGET ATRP to construct well-defined hybrid nanostructures with a multifunctional surface is expected to allow the mimicking of more complex molecules and macromolecular structures in life science as well as for the applications in multifunctional and stimuli-responsive delivery systems.
References
Ober CK, Cheng SZD, Hammond PT et al (2008) Research in macromolecular science: challenges and opportunities for the next decade. Macromolecules 42:465–471
Hawker CJ, Wooley KL (2005) The convergence of synthetic organic and polymer chemistries. Science 309:1200–1205
Descalzo AB, Martínez-Máñez R, Sancenón F, Hoffmann K, Rurack K (2006) The supramolecular chemistry of organic–inorganic hybrid materials. Angew Chem Int Ed 45:5924–5948
Lim CW, Song K, Kim SH (2012) Synthesis of PPy/silica nanocomposites with cratered surfaces and their application in heavy metal extraction. J Ind Eng Chem 18:24–28
Tucker-Schwartz AK, Farrell RA, Garrell RL (2011) Thiol-ene click reaction as a general route to functional trialkoxysilanes for surface coating applications. J Am Chem Soc 133:11026–11029
Becer CR, Hoogenboom R, Schubert US (2009) Click chemistry beyond metal-catalyzed cycloaddition. Angew Chem Int Ed 48:4900–4908
Laurent BA, Grayson SM (2006) An efficient route to well-defined macrocyclic polymers via “click” cyclization. J Am Chem Soc 128:4238–4239
Hoskins JN, Grayson SM (2009) Synthesis and degradation behavior of cyclic poly(ε-caprolactone). Macromolecules 42:6406–6413
Eugene DM, Grayson SM (2008) Efficient preparation of cyclic poly(methyl acrylate)-block-poly(styrene) by combination of atom transfer radical polymerization and click cyclization. Macromolecules 41:5082–5084
Lowe AB (2010) Thiol-ene “click” reactions and recent applications in polymer and materials synthesis. Polym Chem 1:17–36
Campos LM, Killops KL, Sakai R et al (2008) Development of thermal and photochemical strategies for thiol-ene click polymer functionalization. Macromolecules 41:7063–7070
Amici J, Kahveci MU, Allia P et al (2012) Polymer grafting onto magnetite nanoparticles by “click” reaction. J Mater Sci 47:412–419. doi:10.1007/s10853-011-5814-z
Amici J, Allia P, Tiberto P, Sangermano M (2011) Poly(ethylene glycol)-coated Fe3O4 nanoparticles by UV-thiol-ene addition of PEG dithiol on vinyl-functionalized magnetite surface. Macromol Chem Phys 212:1629–1635
Chen J, Liu M, Chen C, Gong H, Gao C (2011) Synthesis and characterization of silica nanoparticles with well-defined thermoresponsive PNIPAM via a combination of RAFT and click chemistry. ACS Appl Mater Interfaces 3:3215–3223
Kuo S-W, Hong J-L, Huang Y-C et al (2012) Star poly(N-isopropylacrylamide) tethered to polyhedral oligomeric silsesquioxane (POSS) nanoparticles by a combination of ATRP and click chemistry. J Nanomater 2012:10
Matyjaszewski K (2012) Atom transfer radical polymerization (ATRP): current status and future perspectives. Macromolecules 45:4015–4039
Bulmus V (2011) RAFT polymerization mediated bioconjugation strategies. Polym Chem 2:1463–1472
Semsarilar M, Perrier S (2010) ‘Green’ reversible addition-fragmentation chain-transfer (RAFT) polymerization. Nat Chem 2:811–820
Boyer C, Stenzel MH, Davis TP (2011) Building nanostructures using RAFT polymerization. J Polym Sci A 49:551–595
Choochottiros C, Park E, Chin I-J (2012) Synthesis and characterization of polylactide–poly(methyl methacrylate) copolymer by combining of ROP and AGET ATRP. J Ind Eng Chem 18:993–1000
Chang C-W, Bays E, Tao L, Alconcel SNS, Maynard HD (2009) Differences in cytotoxicity of poly(PEGA)s synthesized by reversible addition–fragmentation chain transfer polymerization. Chem Commun 0:3580–3582
Nguyen TLU, Farrugia B, Davis TP, Barner-Kowollik C, Stenzel MH (2007) Core–shell microspheres with surface grafted poly(vinyl alcohol) as drug carriers for the treatment of hepatocellular carcinoma. J Polym Sci A 45:3256–3272
Stenzel MH, Barner-Kowollik C, Davis TP, Dalton HM (2004) Amphiphilic block copolymers based on poly(2-acryloyloxyethyl phosphorylcholine) prepared via RAFT polymerisation as biocompatible nanocontainers. Macromol Biosci 4:445–453
Xia J, Matyjaszewski K (1999) controlled/“living” radical polymerization. atom transfer radical polymerization catalyzed by copper(I) and picolylamine complexes. Macromolecules 32:2434–2437
Tsarevsky NV, Matyjaszewski K (2007) “Green” atom transfer radical polymerization: from process design to preparation of well-defined environmentally friendly polymeric materials. Chem Rev 107:2270–2299
Matyjaszewski K, Jakubowski W, Min K et al (2006) Diminishing catalyst concentration in atom transfer radical polymerization with reducing agents. Proc Natl Acad Sci 103:15309–15314
Matyjaszewski K, Dong H, Jakubowski W, Pietrasik J, Kusumo A (2007) Grafting from surfaces for “everyone”: ARGET ATRP in the presence of air. Langmuir 23:4528–4531
Chan N, Cunningham MF, Hutchinson RA (2008) ARGET ATRP of methacrylates and acrylates with stoichiometric ratios of ligand to copper. Macromol Chem Phys 209:1797–1805
Paterson SM, Brown DH, Chirila TV et al (2010) The synthesis of water-soluble PHEMA via ARGET ATRP in protic media. J Polym Sci A 48:4084–4092
Lien Y-H, Wu T-M, Wu J-H, Liao J-W (2011) Cytotoxicity and drug release behavior of PNIPAM grafted on silica-coated iron oxide nanoparticles. J Nanopart Res 13:5065–5075
Varga I, Szalai I, Mészaros R, Gilányi T (2006) Pulsating pH-responsive nanogels. J Phys Chem B 110:20297–20301
Zelikin AN (2010) Drug releasing polymer thin films: new era of surface-mediated drug delivery. ACS Nano 4:2494–2509
Pelton R (2000) Temperature-sensitive aqueous microgels. Adv Colloid Interface Sci 85:1–33
Heinz P, Brétagnol F, Mannelli I et al (2008) Phase transition of pNIPAM grafted on plasma-activated PEO monitored in-situ by quartz crystal microbalance. J Phys Conf Ser 100:012033
Ohya S, Nakayama Y, Matsuda T (2001) Thermoresponsive artificial extracellular matrix for tissue engineering: hyaluronic acid bioconjugated with poly(N-isopropylacrylamide) grafts. Biomacromolecules 2:856–863
Lee Y-G, Park C-Y, Song K-H, Kim S-S, Oh S-G (2012) Preparation of monodispersed PNIPAm/silica composites and characterization of their thermal behaviors. Prog Org Coat 18:744–751
Zhang S, RenLiu, Jiang J, Yang C, Chen M, Liu X (2011) Facile synthesis of waterborne UV-curable polyurethane/silica nanocomposites and morphology, physical properties of its nanostructured films. Prog Org Coat 70:1–8
Işın D, Kayaman-Apohan N, Güngör A (2009) Preparation and characterization of UV-curable epoxy/silica nanocomposite coatings. Prog Org Coat 65:477–483
Park J-H, Lee Y-H, Oh S-G (2007) Preparation of thermosensitive PNIPAm-grafted mesoporous silica particles. Macromol Chem Phys 208:2419–2427
Zhang F, Hou G, Dai S, Lu R, Wang C (2012) Preparation of thermosensitive PNIPAM microcontainers and a versatile method to fabricate PNIPAM shell on particles with silica surface. Colloid Polym Sci 290:1341–1346
Nagase K, Kobayashi J, Kikuchi A et al (2012) High stability of thermoresponsive polymer-brush-grafted silica beads as chromatography matrices. ACS Appl Mater Interfaces 4:1998–2008
Ramanan RMK, Chellamuthu P, Tang L, Nguyen KT (2006) Development of a temperature-sensitive composite hydrogel for drug delivery applications. Biotechnol Prog 22:118–125
Bouclé J, Kassiba A, Emery J et al (2002) Local electrooptic effect of the SiC large-sized nanocrystallites incorporated in polymer matrices. Phys Lett A 302:196–202
Sow C, Riedl B, Blanchet P (2010) Kinetic studies of UV-waterborne nanocomposite formulations with nanoalumina and nanosilica. Prog Org Coat 67:188–194
Ramezanzadeh B, Moradian S, Tahmasebi N, Khosravi A (2011) Studying the role of polysiloxane additives and nano-SiO2 on the mechanical properties of a typical acrylic/melamine clearcoat. Prog Org Coat 72:621–631
Stöber W, Fink A, Bohn E (1968) Controlled growth of monodisperse silica spheres in the micron size range. J Colloid Interface Sci 26:62–69
Hwang HS, Bae JH, Kim HG, Lim KT (2010) Synthesis of silica–polystyrene core–shell nanoparticles via surface thiol-lactam initiated radical polymerization. Eur Polym J 46:1654–1659
Braunecker WA, Matyjaszewski K (2007) Controlled/living radical polymerization: features, developments, and perspectives. Prog Polym Sci 32:93−146
Acknowledgements
This work was financially supported by the Grant from the Industrial Source Technology Development Program (Project No. 10035163) of the Ministry of Knowledge Economy (MKE) of Korea and by the Joint Program of Cooperation in Science and Technology through NRF Grant funded by the MEST (No. 2011-0025680).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mai, T.B., Tran, T.N., Rafiqul Islam, M. et al. Covalent functionalization of silica nanoparticles with poly(N-isopropylacrylamide) employing thiol-ene chemistry and activator regenerated by electron transfer ATRP protocol. J Mater Sci 49, 1519–1526 (2014). https://doi.org/10.1007/s10853-013-7833-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-013-7833-4