
Abstract
The use of papers in packaging and development of novel technological applications for paper substrates largely depends on the control of the hydrophilic properties of the cellulose fibres and improvement of the water-repellent properties. This review provides an actual summary of available literature on theoretical concepts and practical methods to improve the hydrophobicity of cellulose fibres and paper webs. In the first part, the interaction of water with cellulose fibres and paper webs is described at different levels ranging from the molecular scale over the micro- to macroscale fibre properties towards the interactions with porous substrates. The concepts for hydrophobicity and superhydrophobicity applied to cellulose fibres are reviewed, considering the surface chemistry and topographical features. In the second part, current techniques for hydrophobization based on sizing or direct fibre surface functionalization are described. Besides traditional sizing procedures, novelties in nanoparticle applications as hydrophobic sizing agent are reviewed. Novel trends in physical, chemical and nanotechnological fibre surface modifications or surface coatings are reviewed to turn the wetting properties into the superhydrophobic regime. The main future concern in controlling cellulose wettability lies in the development of sustainable modification techniques based on renewable resources such as biopolymers and green chemistry.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Paper, paperboard and linerboard are preferred packaging materials with good mechanical strength, flexibility and low cost. For various end-user applications, papers require a broad range of properties with respect to liquid interactions: while tissues and towels are desired to take up water quickly, cupstock and packaging papers should resist water. The specific control of interactions between liquids versus fibrous surfaces is relevant for designing windshields, waterproof clothing and self-cleaning textiles. Recently, specific interest has grown in tuning the resistance of paper substrates towards liquids for packaging, food storage, medical industry, printing industries, microfluidics or bioassay devices. The interaction of liquids to paper surfaces is critical for several processes in paper processing and utilisation such as, e.g. gluing, printing or coating.
Cellulose is an attractive renewable and biodegradable resource, but the hydrophilic nature makes it sensitive to water and moisture adsorption. This is a main hurdle in application and durability of cellulose fibres. As the basic paper structure consists of a heterogeneous fibre web with given porosity and surface roughness, there has been a lot of efforts to fully control and understand the water adsorption and transport by modeling or experimental research. The high degree of chemical and physical surface heterogeneities often complicate the analysis, but the hierarchical fibre structure simultaneously provides opportunities to enhance the paper surface functionality. Several processes and additives are applicable to achieve desired paper properties and protect against water, e.g. by bulk modification, surface treatment or structuring of papers and cellulose fibres. As the main commercial benefits of paper substrates include light weight and recyclability, the modifications should likely not conflict with those advantages while providing a platform for surface functionalization.
The control over interactions between water and paper fibres is a fundamental issue during the entire paper formation, but this review paper will mainly focus on end-user properties of paper surfaces. The hydrophobization of cellulose by internal and surface sizing might be insufficient to meet the requirements for water resistance of packaging materials. Then, conventional barrier coatings should become rather thick, with accumulation of chemicals and/or poor recyclability that change the environmental impact. The chemical modification of cellulose fibres received much attention in composite formulations, but it remains limited in connection with paper applications. In the past, the chemical modification of paper fibres targeted improvements of strength. Nowadays, the surface functionality of physically and chemically modified fibres should also create water protection. In this review, some basic principles on water interactions and water uptake of cellulose fibres and paper webs are first considered together with basic ideas on wettability and hydrophobicity. Next, common and innovative methods improving the water resistance of papers are described, with emphasis on recent nanotechnological surface modifications. These should be critically evaluated for a future generation paper products in terms of sustainability, compatibility and applicability in paper industry.
Basic concepts
Hydrophilicity and solvent interactions of cellulose
Due to the intrinsic hydrophilicity of cellulose, paper fibres are sensitive to water interactions but they are simultaneously insoluble in water. Only few organic and inorganic impurities (usually less than 1 % for bleached cellulose) form a water-soluble extract [1]. The surface of cellulose fibres consequently builds an interface in contact with water. The fundamental interactions between cellulose and solvents, or specifically water, and the role of the intrinsic molecular structure are therefore first reviewed in this section.
The insolubility of cellulose in aqueous media is mostly related to strong intra- and intermolecular hydrogen bonding between cellulose molecules, but the role of crystallinity should also be considered [2]. For better understanding the solubility pattern of cellulose, some fundamental physicochemical aspects such as intermolecular interactions were reviewed from kinetic and thermodynamic viewpoint [3]: it seems that cellulose is significantly amphiphilic and hydrophobic interactions should simultaneously be implemented to explain solubility mechanisms. Once a cellulose fibre is in contact with water or solvent, the molecules attach relatively fast to the outer polymer layer before they further penetrate into the surface, which leads to the instantaneous formation of a gel-like layer affecting further solvent interactions. Therefore, the role of time effects in polymer diffusion should be considered simultaneously with thermodynamic interactions.
The reactivity of the different hydroxyl groups towards solvents and adsorption of water at the surface correlates with the degree of organisation and conformation of the macromolecular cellulose chains [4]. As cellulose is a semi-crystalline material, the reactivity towards crystalline and amorphous sites should be considered separately. The structure and accessibility of different cellulose allomorphs, and particularly the hydrogen bonding system, were investigated extensively [5]. In case of perfect ordering, some hydroxyl groups in the molecular side chains are engaged in a hydrogen bond with the oxygen atom in the six-ring membered structure belonging to the adjacent glucose ring. Therefore, these hydroxyl groups are inaccessible for water penetration and almost nonreactive, as could be determined by 31P NMR spectroscopy [6]. For example, the surface hydroxyl groups in highly crystalline cellulose (e.g. bacterial cellulose) are almost inaccessible in contrast to cotton fibres with a less perfect structure [7]. However, the role of specific crystalline structures and surface morphologies in cellulose should be considered more in detail including its hierarchical organisation (Fig. 1a). Although there is some confusion in the field of cellulose research about precise definitions, the cellulosic components of a wood fibre wall structure are the cellulose molecule, the elementary fibril (3.5 nm), the microfibril (about 10–35 nm) and the cellulose fibres (about 20 μm) [8]. When assuming a model structure of organised molecular cellulose chains into native Iβ allomorph crystalline microfibrils (Fig. 1b), three families of crystalline faces with about the same area are identified, e.g. by X-ray diffraction [9, 10] or models [11]. For each crystal face, different roughness, accessibility of the hydrophilic and hydrophobic groups, as well as surface and attachment energies were predicted [12]. As such, many properties including adsorption and adhesion correlate to molecular interactions at the level of microfibrillar surfaces. The microfibril faces were used as model surfaces for absorption studies by molecular modeling [13]: the (200) hydrophobic face has many interacting sites without specific geometry for absorption (Fig. 1c), while the hydrophilic (110) and \( (1\overline{1} 0) \) faces have interaction sites with a more constrained orientation for the absorbed molecules (Fig. 1d). The absorption sites on the latter faces are thus more specific due to topological characteristics and induce specific orientations for the solvent molecules to the surface. As a result, wetting of the crystalline faces was different for cotton cellulose [14] or cellulose Iβ [15–17]. The structure of the cellulose surface may be different to the bulk due to a local reorientation of the crystal faces. In general, the (110) and \( (1\overline{1} 0) \) faces are likely oriented parallel near the surface as seen by NMR analysis [18, 19], dynamic NMR studies [20], AFM analysis [21] or gas chromatography [22]. On the other hand, the hydroxyl groups on amorphous cellulose sites have equivalent accessibility and consequently react equally. Therefore, the amorphous phase has privileged sites and topography for solvent penetration, as locally characterised by maximum van der Waals and electrostatic interactions with favourable absorption energies [13].

Structure and organisation of cellulose: a representation of a possible molecular architecture of the cellulose molecule, showing its relationship to the microfibrils and to the total cell wall, b schematic representation of a cross-section of the Iβ crystal structure with three crystal structures (model) (from [12]), c morphology of \( (1\overline{1} 0) \) crystal surface (from [13]), d morphology of (200) crystal surface (from [13])
The situation under atmospheric or humid conditions causes permanent water uptake of cellulose fibres under different forms, depending on the moisture content (Fig. 2a). The constitutive water is the water that remains present under zero relative moisture content, and is strongly bonded to the cellulose fibre surface through electrostatic or hydrogen bonds. The monolayer of constitutive water affects the subsequent absorption phenomena that might become more homogeneous over the entire surface. On top of the primary water monolayer, imbibed or sorbed water is held by the fibre under an environment of 100 % relative humidity. Additionally, the imbibed water is either retained in the fibre wall itself (into so-called microreticular pores) or in the pores of the cell wall (into so-called macroreticular pores). Finally, the free water is kept by fully saturated fibres and includes the inter-fibre water in the pores and intra-fibre water in the lumen. This water is not chemically bonded but kept by capillary forces. Depending on the interaction between water and cellulose fibres, it is generally categorised as unbound (or bulk) and bound water. The bound water is associated with the cellulose surface and divided into freezing water (in the pores of the fibre wall) and nonfreezing water (chemically bonded to the hydroxylic and carboxylic acid groups) as in Fig. 2b [23, 24]. The nonfreezing water includes the first one to three layers of water adjacent to the surface and it does not freeze as the motion of water molecules is restricted: the molecules have a configuration resembling ice in association with the surface, as detected by DSC [25]. The freezing water can freeze at temperatures below the usual freezing point due to so-called freezing point depression, and its amount can be determined by thermoporometry [26, 27] or heat capacity measurements [28]. The most important feature in this respect is a critical pore size of 40 Ǻ on the fibre surface that can carry 100 % nonfreezing water, while larger pores contain both freezing and nonfreezing water [29]. In general, the microfibrils and nanopores on the fibre surface have important effects in mediating interactions [30]. Another model for the interaction between cellulose fibres and water was developed [28, 31], where a swollen fibre is considered as a mixture of (i) partially water-soluble polymer with water at the surface, and (ii) an insoluble polymer phase.

Interaction and organisation of water at the surface of cellulose fibres: a various forms of water as cellulose fibres are progressively wetted (adapted from [42]), b schematic layer of water building on top of a cellulose fibre: capillary water (W c), bulk water (W b), freezing bound water (W f), nonfreezing water (W nf) (adapted from [24])
The different types of water can be determined by differential thermal analysis and NMR analysis as a function of relative humidity [32, 33]. From thermogravimetric analysis, the highest content of bound water was determined for cellulose with least crystallinity [34]. The final water content of cellulose films can be determined by solvent exchange and quartz balance [35]. The adsorbed water molecules also influence the hydroxyl arrangement and can be distinguished from the initial monolayer using ultrahigh frequency or near-infrared spectroscopy [36, 37]. The mechanisms and side effects of water absorption and desorption on pristine cellulosic fibres are well documented [38]. However, the absorption of idealised cellulose surfaces differs from papers due to water penetration, surface flatness and stability of the substrate.
Water interactions of fibrous paper webs
The interaction of water with a cellulose fibre web is controlling almost all technological processes and applications in paper science. At first, water interactions happen during the formation of paper sheets in the papermaking process. Under water-swollen conditions of the paper fibres, water is the major component of the cell wall and present in a microporous gel of hemicelluloses and lignin in between the cellulose constituents. During removal of hemicelluloses and lignin by pulping, the water content increases and completely occupies the space of the fibre cell wall. We do not go into further detail on the water interaction and retention capacity of paper webs in the wet-end technology [39]. Second, water interactions after drying the paper web happen in contact with the environment or play a role in converting processes and post-processing such as coating application from water-based media. These become also important when considering paper substrates for microfluidic devices [40]. Then, the complexity and heterogeneity of the paper structure at different levels should be considered, including the surface as well as bulk morphologies [41].
The water transport mechanisms into paper and the influences of fundamental variables have been discussed in many publications [42, 43]. The water transport into paper is generally based on a diffusion model [44], with following mechanisms: (i) penetration in the capillaries of the sheet, (ii) surface diffusion along the capillary walls, (iii) diffusion through the fibres and (iv) vapour phase transport through the fibres [45, 46]. The dominating mechanism depends on the network structure, external pressure, time and hydrophobic fibre properties. Quantitative information on the diffusive motion of water and solvents was obtained from NMR studies [47], studying influences of local charges and hydrophobicity at the fibre surface. The charge effects did not play a primary role, but nanopores and microsized solvent pools within the cellulose fibres critically influenced the diffusion rates. In this respect, the interactions with water were inferior to these with other solvents due to strong hydrogen bonding that hinder the exchange with the fibre surface. Several properties of the paper web such as the ability to hold water [water retention value (WRV)], dimensional stability, surface roughness and strength change during contact and water transport. Therefore, a full model describing the net transport of water into a real paper substrate becomes more complicated and should include time-dependent changes of the porous structure and water–fibre interactions [48].
The water penetration into a paper web is one specific case of fluid flow in porous media. The bulk permeability of porous media can generally be related to the porosity under certain conditions. However, the paper porosity is difficult to define and can be examined by experimental methods such as microscopy, solvent exchange, solute exclusion, gas absorption, mercury porosimetry, air permeability, freeze-drying, tomography, etc. Considering the dried paper web as a 3D porous structure, it consists of a network with randomly distributed capillaries. An overview of flow and diffusion mechanisms into paper and their influencing parameters was given before [49]. Some theories describing the dynamics of droplets and films onto porous substrates were developed [50–52]. Several analytical models predict the permeability of general porous media [53], fibre porous media [54, 55] and particularly wet paper sheets [56] or unbeaten sulphite pulp handsheets [57]. The available permeability models are mainly developed for homogeneous fibre networks with fibres oriented in one specific direction, randomly oriented in the plane of the material, or isotropically oriented in a space [58]. In a realistic model for effective water permeability through paper webs, however, inhomogeneous solid volume fractions, fibre orientations and diameters should be considered: these nonuniformities can be included using reconstructed microstructures obtained from imaging-based techniques over the entire thickness (Fig. 3a) [59]. In a particular case, it was shown that the transverse permeability of a fibrous medium is independent of in-plane fibre orientation, while it increases with deviation of the fibres’ through-plane angle from zero [60]. After calendaring, the transformation of the solid volume fraction into a specific web profile along the thickness is important [61]: the permeability decreases with higher directionality and compression of the paper web. Fibre webs of identical solid volume fraction, however, exhibited almost identical permeability regardless of the fibre orientations. Specifically for hydrophobic papers, the penetration mechanisms are mainly affected by diffusion, capillary effect and external pressure [62]. The water diffusion through papers was measured in plane and thickness directions by NMR pulsed field gradients, and fitted by a simple two-component diffusion model (Fig. 3b) [63]: the diffusion coefficients were not affected by fibre dimensions, internal structure or chemical composition, but were consistently higher along in-plane direction, with a difference between a slow and a fast diffusion coefficient. The main phenomena of water penetration, i.e. absorption and fibre swelling, are described below.

Water penetration into paper, considered as a 3D porous fibre network, a model for reconstructed microstructure and simulation of flow through the fibre network (isotropic view) [59], b diffusion coefficients for water through a paper network comparing in-plane and through-plane diffusion with two components including a fast diffusion and slow diffusion component, (filled circle) fast in-plane, (filled triangle) slow in-plane, (open circle) fast through-plane, (open triangle) slow through-plane (data summarised from [63])
The water absorption in paper is dominated by penetration under capillary pressure, interfibre penetration and molecular diffusion. Therefore, the absorption mainly depends on the surface tension and capillary size of water in contact with the paper fibre. The gradual uptake of water by a porous substrate is referred to as a surface tension driven flow or wicking [64]. The capillary pressure under wicking is generally created on the capillary walls of a porous medium at the interface of the wet and dry matrix. Water absorption into the paper originates from the balance between mutual attraction of the molecules in the liquid medium (cohesion) and attraction of the liquid molecules to the solid medium (adhesion): under a positive capillary pressure (i.e. when the contact angle between the liquid phase and the solid phase is less than 90°), the water moves into the porous substrate. A theoretical equation for the penetration depth during wicking of nonpolar liquids into vertical parallel capillaries was put forward by the Lucas–Washburn model [65, 66]. This model is based on the dynamic motion of an infinite amount of fluid into an empty capillary, while the porous medium is represented as a bundle of aligned capillaries with the same radii. The equation combines the Navier–Stokes and Laplace equations, assuming a laminar flow of a Newtonian fluid with constant contact angle: as a result, the penetration depth changes with the square root of time, in agreement with diffusion controlled processes. The Washburn model has been traditionally used to describe liquid penetration into paper: it provides a first-order approximation with a constant instead of dynamic contact angle and it considers an average pore size diameter. Therefore, it does not account for swelling and pore size distribution effects in paper, or it cannot be used for chemically reactive fibres. The rewetting of paper is a nonequilibrium process that prevents the validity of the Washburn model. The capillary rise effects were later corrected with dynamic contact angles, by modifying a constant term in the Washburn model as a function of the liquid type and velocity of the moving liquid front [67]. The dynamic contact angles and inertia were also explicitly used to describe the penetration kinetics in porous media by the two-liquid method [68]. In modified Lucas–Washburn models, the effects of inertia and viscous forces were also introduced as an impulse drag [69], or as a kinetic term [70]. Similar equations were improved multiple times by incorporating a term for initial contact effects, continuous flow, kinetic energy [71] or using the full expression of the Navier–Stokes equation [72]. In another approach, new theoretical models for capillary water sorption in combination with the swelling of paper in thickness direction were proposed [73], assuming that the pore radii in a swelling porous medium decrease linearly with time [74]. However, this assumption cannot always be validated with experimental results and depends on the absorptivity of the medium or eventual increase in thickness of the paper sheet [75]. In a similar way, the Davies–Hocking model [76] states that the available surface for sorption diminishes during absorption. Based on the single capillary model, dynamic wicking into porous substrates was also developed where the interface between the drop and the paper is considered as a bundle of single capillaries with the same radii [77]. This equation has been further improved by incorporating an additional gravity term and inertial forces that counteracts water uptake [78]. Others changed the aspect ratio of the capillaries to calculate deviations from simple capillary geometries [79]. Overall, the penetration path of a droplet depends on the droplet size, i.e. the ratio of droplet to void volume: for small volumes, the vertical wicking into the pore volume directly below is predominant, while for larger volumes, the radial spreading onto the paper becomes important. The radial spreading thus explains a regime of slow drop penetration rates by redistribution of liquids from the large into small pores [80]. The liquid penetration into real fibre networks such as paper strips was explicitly investigated [81]: in this model, the random penetration into capillaries was introduced by a pore tortuosity factor. It was concluded that the liquid penetration distance was proportional to the square root of time as suggested by the Lucas–Washburn equation, but the final penetration depth was lower for high tortuosity. The relationships between the wicking flow rates into porous fibre networks were experimentally studied for various orientations along and across the fibres [82]. In multiple paper structures, the wicking shows an enhancement in the beginning of the experiment, while gradually diminishing with time [83]. The penetration of water into paper in terms of a theoretical diffusion model was evaluated together with surface roughness effects, which requires experimental data measured by, e.g. dynamic sorption [84]. In recent years, a new approach has been used to model wicking in porous fibre media based on Darcy’s law by relating the average penetration velocity of a liquid to the pressure gradient [85]. Wicking experiments under external pressure indicated, however, that the predictions of the Darcy law were inconsistent over a broad range of externally applied liquid pressures [86]: this may be caused by the fact that the wicking parameters are affected by the applied pressure as well as change during the wetting of compressive cellulose wipes. The Darcy model was later modified for spreading of low vapour pressure liquids on paper, and showed good agreement with experiments on water absorption [87] and radial penetration of liquids in thin porous substrates [88]. While the Washburn model only considers a 1D absorption with assumption of a laminar flow, the Darcy law can be extended to model 2- and 3D flows in more complex geometries [89]. Therefore, it can be used for numerical simulation of flows in porous media and more complex situations, where, e.g. a single-phase flow behind the liquid front is introduced. A new model based on the Darcy law for wicking in paper-like swelling media also accounts for the rate of change in porosity by matrix swelling and liquid absorption [90]. For practical applications, the short-term penetration and absorption into porous substrates is important in coating and printing, considering fluid properties, surface forces and pore geometries with complex connections and arrangements [91].
The swelling of cellulose fibres induces fibre rising and sheet roughening at meso- and microscale [92, 93]. The swelling is generally ascribed to penetration of water molecules between hydrogen-bonded fibrils in the fibre wall. The water molecules diffuse through amorphous parts of the cellulose matrix and break-up intermolecular hydrogen bonds. As the amount of bonded water increases, the degree of internal bonding of the fibre wall decreases in parallel. The increase in intermolecular distance of the cellulose chains finally causes swelling, which occurs as a result of water entering in parallel with a reduction in osmotic pressure. Consequently, there is a swelling equilibrium where the difference in chemical potential due to concentration gradients is balanced by the osmotic pressure. Another theory assumes that swelling is induced by the displacement of morphological features (lamellae) in the delignified fibre cell wall and submorphological swelling is caused by bonded water [94]. The hydration and swelling of pulp fibres could be followed by DSC analysis [25]. As the structure and molecular arrangement of paper fibres are influenced by swelling, the physical and transport properties (e.g. wicking and wettability) change. The degree of swelling depends on the amount of acidic groups in the carbohydrate fraction that enhance the interaction with water. As a result, the substitution of hydroxyl groups by hydrophobic groups, e.g. by acetylation decreases swelling. After subsequent swelling and drying cycles, the cellulose or paper base structure has permanently changed and the physical and chemical properties are reduced upon rewetting [95]: consequently, the absorption and strength of rewetted fibres are lower. During drying, the cellulose loses both free and bound water and start to shrink. It has modified to a hard and stiff structure that does not re-swell to recreate the never-dried state.
Wettability and liquid interactions of paper
The wetting phenomenon represents the ability of a liquid to make contact with a solid surface, resulting in spreading or confinement of the liquid on the solid. The equilibrium state depends on the balance of adhesive and cohesive forces between the liquid and solid at a molecular level. The direct surface forces at a water–cellulose interface could be measured for thin spin-coated films, taking into account the swelling [96]: the interactions for a water-swollen cellulose layer are dominated by steric rather than electrostatic forces, due to the dangling tails of cellulose chains and the presence of microfibrils extending about 100 nm above the surface.
The acid–base chemistry controls wetting phenomena and interactions between liquids and solid surfaces [97]. In general, the degree of wetting depends on the formation of acid–base adducts between the liquid and functional groups on the solid surface. The work of adhesion has thermodynamic contributions of acid–base forces (i.e. all the electron donor–acceptor interactions such as hydrogen bonds) and Lifshitz-van der Waals (LW) forces (i.e. dispersion, dipolar and induction forces). The surface tension of cellulose films, the interfacial tension with liquid droplets and the work of adhesion can be calculated from the van Oss-Good approach [98]: the main interactions were detected between the electron donor (or Lewis base) γ − components of the cellulose and the electron acceptor (acid) γ + components of the contacting liquid. A negative value for the acid–base interfacial tension was found for contacts between water and native cellulose, indicating that the cellulose surface absorbs the water molecules by hydration that leads to a low solid surface tension γ LW. The spreading film pressure is consequently negative, which is the equilibrium film pressure of the adsorbed vapour of the liquid onto the solid substrate. In general, the dispersive component of the surface-free energy is not strongly influenced by different surface sizing. The basicity component is indicative for the alkaline nature of paper, while the acidity component varies for different sizing agents [99].
The static wetting and surface energy of paper is macroscopically determined by the contact angle of a test liquid. The effects of wetting on porous substrates are schematically represented in Fig. 4. On physically smooth/rigid and chemically homogeneous/insoluble substrates, the contact angle is determined by the thermodynamic equilibrium between the interfacial forces according to the Young-Dupré equation [100], and measured at the three-phase contact line among air, liquid and solid. The liquid–vapour interfacial tension γ LV is related to the interfacial tension γ SV at the solid–vapour interface and γ SL at the solid–liquid interface through the equilibrium contact angle, assuming that the system reaches a global minimum in energy surrounded by infinitesimally close nonequilibrium states in the energetic field. However, the wetting of fibrous and paper surfaces is complicated by physical heterogeneities (e.g. surface roughness due to uneven fibres and porosity) and chemical heterogeneities (e.g. hydrophobic domains with variable size and concentration after internal sizing or coating). Thus, the situation of equilibrium contact angles [101] should be compared to the situation of constrained wetting [102] to understand the parameters influencing the wetting degree. These surface heterogeneities were included in models by Wenzel [103] and Cassie and Baxter [104], as further explained. The Wenzel model is a thermodynamic model for the droplet state on a rough surface, assuming that the water droplet makes contact over the entire surface area and only applies for systems with no hysteresis. This means that it applies, e.g. for porous media with radial grooves where the droplet contact line moves reversibly, but the applicability for paper substrates is limited. Surfaces with random roughness were considered separately in a modified form of the Wenzel equation that includes a factor for surface texture in addition to the conventional roughness factor [105]. The Cassie–Baxter model also accounts for chemical surface heterogeneities such as entrapment of air pockets in between the roughness asperities. The wetting of paper is further complicated by liquid spreading and absorption into the fibrous network. Therefore, time- and pressure-dependent wetting experiments are required. The dynamic wetting experiments with advancing and receding liquid fronts are more representative for heterogeneous surfaces [106], and the effects of fluid front roughening in liquid flow through paper were recognised [107]. General experiments on wetting of porous structures were further optimised using other techniques such as the capillary rise method and film flotation [108]. The wetting of papers was also studied during condensation under ESEM conditions to monitor effects in the water-swollen state [109]. Other wetting behaviour of papers was recorded by high-power ultrasound [110]: a higher wetting speed was then related to high liquid absorption rates within the pores and fibre swelling. The escape of gasses from the surface roughness volume and from pores was the main physical process for light- to medium-sized papers (contact angles 40°–70°).

Schematic representation of interaction between a water droplet and a porous paper substrate, including wetting mechanisms (taking into account surface roughness and chemical surface heterogeneity) and absorption mechanisms (taking into account radial spreading and vertical penetration by analytical models or experimental models (inset from [142])
The dynamic droplet interactions and flow have been studied besides static conditions on paper. The macroscopic hydrodynamic behaviour of liquids was originally treated apart from the microscopic scale, but it was later corrected for surface roughness, adsorption and contamination [111]. The fundamental wetting dynamics and droplet spreading have been reviewed [112], including different regimes of partial and complete wetting [113]. The process of wetting takes some time from the moment that a liquid droplet is put into contact with a paper substrate until the substrate has completely wetted. The wetting time depends on the flow properties and viscosity of the liquid on the one hand, while it depends also on the structure of the solid substrate. The wetting kinetics on smooth surfaces follow a theoretical power-law model [114, 115]. The liquid spreading on rough surfaces also follows a simple power-law model, but mostly applied on an empirical basis [116]. The dynamics of droplet spreading toward an equilibrium state can be described by hydrodynamics [117, 118] or molecular kinetics [119], differing in the mode of energy dissipation at the droplet contact line. For interpretation of the contact line dynamics, the droplet size has to be considered (Fig. 5). The hydrodynamic theory is based on physical chemistry and statistical physics, considering long-range force interactions over the contact line [120–122]. This theory leads to a solution of the Navier–Stokes equation for a fluid as described in the Cox equation [123]. The molecular-kinetics theory better describes the droplet relaxation on homogeneous and smooth surfaces [124]. The contact line motion is then defined by statistic kinetics of multiple events at molecular level within the three-phase contact zone. This theory leads to an equation that relates the radial velocity of the wetting line to the dynamic contact angle. The mechanisms that control the overall wetting on homogeneous surfaces are governed by microscopic processes near the contact line, including a local energy dissipation factor attributed to molecular friction [125]. The wetting onto heterogeneous and rough surfaces might furthermore be characterised by nonequilibrium stick and jump movements of the contact line, changing with the relative orientation of the contact line towards the grooves [126]. Under extreme conditions of high-speed jet coating on paper, the motion and location of the contact line depends on the fluid dynamics of the jet impingement [127]. Pinning of the contact line hinders the liquid spreading under certain conditions of surface heterogeneity, and might disappear on rough surfaces when the grooves are deep enough to entrap air beneath the droplet [128].

Contact lines formed by water droplets of different volumes: a 0.1 μl, b 0.2 μl, c 4 μl, d 8 μm on a handsheet paper substrate (from [203])
The dynamics of wetting in combination with absorption measurements of water droplets on porous surfaces and paper are scarcely studied. In one study, the molecular-kinetic theory was combined with relaxation of the drop volume according to the Darcy law to describe absorption [51]: this model implies that the porous structure is only filled vertically, i.e. the radial flow within the pores is neglected and filling at a particular vertical capillary starts if the wetting front reaches that radius. In another study, the dynamic absorption of a droplet on paper was studied by introducing a model for the area covered by the spreading liquid as a function of time [129]: this model only applies for impermeable fibres. When also hysteresis effects are considered, the penetration happens with a fixed contact line and the radial penetration always precedes spontaneous penetration [130]. For dynamic spreading and absorption of droplets on sized papers with physical and chemical heterogeneity [131], two sequential phenomena result in a time-delay before absorption actually starts: (i) first, the water partially wets the surface up to a pseudo-equilibrium situation; and (ii) second, the absorption into the bulk starts as the drop has wetted to a certain extent. The pseudo-equilibrium or metastable contact angle is a function of the chemical surface heterogeneity and was also affected by the surface roughness due to the entrapment of air rendering the surface more hydrophobic. The wetting rates were lower than for pure hydrodynamic wetting: the wetting dynamics were retarded by surface roughness but did not explicitly depends on chemical surface heterogeneities. Also other empirical studies confirmed that the onset of liquid penetration after contact between a liquid and paper is postponed by a wetting delay [132].
The wetting and dynamic spreading of droplets onto coated papers received more attention due to importance in printing. Spreading is influenced by surfactants and/or solvents, as the latter molecules migrate to the liquid–air interface and affect the solid–liquid interface energy [133]. However, the time-dependent behaviour of a water droplet onto coated papers remains influenced by the underlaying paper substrate and cellulose microstructure [134]. The ink-jet printing coatings include single layer coatings, complex multi-layered structures, or have a top-coating of an emulsified polymer that forms a specific surface structure with microcracks to improve ink acceptance. A high-rate wetting regime was then attributed to capillary flow into those microcracks. Consequently, the coating porosity and eventually the bulk density should both be included in models [135], to estimate the dynamic wetting and absorption [136]. During wetting experiments for water and ethylene glycol droplets onto ink-jet papers, only a regime of low wetting rates was correlated with pseudo-equilibrium contact angles [137] and deviations between hydrodynamic and molecular-kinetic models were attributed to surface energy effects (acid/base) rather than roughness effects. For coated offset papers, the hydrodynamic model provides best fit at low spreading rates, while the molecular-kinetic theory can be applied over a broader velocity range [138]. Such pigmented paper coatings behave in first approach as a porous layer: the wetting and penetration on porous layers was studied for different layer thickness [139], and droplet sizes [140]. In parallel, the droplet interactions on porous substrates were studied by considering absorption and dewetting separately as a function of the pore saturation [141]. In practice, the wetting of real coating structures is often more complex and requires network modeling [142]: this approach is used as a design tool for paper coatings by implementing a network of voids with rectangular cross-sections of different aspect ratio and elliptical connections. As such, the anisotropy of a pigmented paper coating can be adequately simulated. Especially, the influences of inertia on wetting and absorption of coated papers may cause deviations from the Lucas–Washburn equation in porous media, although it was found that the effect of inertia is very small for small pores (<1 μm) [143]. When the inertia of the fluid is taken into account (e.g. hexane as a model for ink solvents), the permeation and wetting of a paper coating network changes after few milliseconds as the liquid enters the voids. The anisotropy only influences the permeation when inertia of the wetting fluid is taken into account. On the other hand, the effect of different void structures was investigated by including more anisotropic features that result in a fast advancing wetting front through the smallest voids. This wetting behaviour is mainly caused by the fluid inertia and appears as a preferential flow into specific voids. The preferential flow assumed in simulations is much larger compared to the theoretical Lucas-Washburn model. However, the liquid absorption and wetting on coated papers is inhomogeneous as studied in detail by applying small droplet volumes and short-time absorption measurements [144]. Then, influences of coating layers with different composition and thickness are revealed: while the pore structures of thin and thick coatings may be different, it could not fully explain wetting variations.
The previous studies consider the interactions between liquid and paper after contact between both has been established, while the initial contact is actually made during impact of a liquid drop onto the paper surface. Those effects were studied separately [145], including the subsequent stages of droplet spreading and recoil before reaching an equilibrium contact state. In a first approach for smooth and homogeneous model paper surfaces, the drop impact velocity largely influences the droplet spreading [146]. By comparing different hydrophobic and hydrophilic model surfaces, sized papers show a unique switch during droplet impact: the substrates behave in a hydrophobic manner during droplet impact or hydrophilically during recoil. On the other hand, unsized papers show significantly different dynamic contact angles and impact behaviour than the sized samples: the water absorption and swelling of the cellulose fibre then contributes to higher adhesion of the water, almost immediately after drop impact. In parallel, smooth cellulose films and papers show similar impact dynamics, suggesting that the surface energy plays a more dominant role than roughness [147].
Hydrophobicity and superhydrophobicity
In parallel with the above considerations, water diffusion and absorption into cellulose lead to distortions of the paper structure. The water diffusion into cellulosic materials can be slowed down by the creation of external or internal barrier layers that reduce the interactions with water and protect the hydrophilic cellulose surface. This is generally done by creating hydrophobic domains on the paper surface (coating or surface sizing) and within the bulk (internal sizing or fibre treatment). As a result, the chemical and morphological heterogeneity of paper substrates further increases. The effects of micro- and macroscale roughness are first discussed, together with the chemical surface composition.
Based on the general contact angle theory of Wenzel [103], the surface hydrophobicity can be improved by: (i) reducing the surface energy, and (ii) increasing the surface roughness of an initially hydrophobic surface, i.e. increasing the surface area on a microscopic scale. So far, the lowest surface-free energy is reported for trifluoromethylene group (CF3)-terminated surfaces with an equilibrium contact angle of 100° for water on flat surfaces [148, 149]. A surface with aligned fluorinated groves presents the lowest surface energy of 6.7 mJ/m2 and has a maximum dynamic water contact angle of about 120° [150]. The apparent water contact angle θ on a rough surface can be described by Wenzel model in Eq. 1, indicating that the equilibrium contact angle θ eq on a corresponding flat surface (equilibrium contact angle according to the Young equation) is multiplied by a roughness factor r:
The roughness factor r is defined as the ratio of the actual surface area to the geometrically projected surface area. The roughness parameter S rd gives the effective surface area with respect to the projected area as a percentage increment [151], and relates to the roughness factor r as given in Eq. 2:
The roughness enlarges the wetting or nonwetting behaviour: the apparent contact angle decreases at higher roughness on hydrophilic surfaces (θ eq < 90°), while the apparent contact angle increases at higher roughness on hydrophobic surfaces (θ eq > 90°). The hydrophobicity of a paper surface can thus be improved by increasing surface roughness: during wetting of paper sheets, surface distortions and eventual rising of the paper fibres due to swelling may already introduce additional roughening [152]. The Schuttleworth–Bailey model [153] often describes better the hydrophobicity on paper as it takes into account the macroscale roughness of the surface profile, according to Eq. 3:
where θ is the apparent contact angle and θ eq is the equilibrium contact angle. The maximum roughness slope on the surface can be calculated directly from the surface profiles with peak height h and radius R. The latter model was explicitly used to describe the higher hydrophobicity on sized papers that were patterned by drying in contact with specific wire geometries [154]. As a result, the hydrophobicity improved for papers with the most uneven pattern, while the contact angle was lower for papers with the finest pattern. The contact angles on patterned papers agree with general observations that the contact angle is higher for large-scale (micrometre scale) roughness as introduced by Schuttleworth and Bailey. On the other hand, for small-scale roughness (nanometer scale) the theories of Wenzel still apply [155] by considering the increase in surface area as a network of small capillaries. In addition to the microscale roughness, the hydrophobicity can be further increased by adding a level of roughness at the nanometer scale [156], or so-called fractal surface profiles [157]. Otherwise, the surface roughness can be specifically tuned by paper coating, where a multi-scale roughness profile can be created combining micro- and nanoscale features [158]: it is known that the surface roughness parameters depend on the sampling area, but they can be adequately scaled by considering the correlation length as an extrapolation parameter that was determined for coated papers analysed by optical profilometry (microscale roughness) or AFM (nanoscale roughness) (Fig. 6). The multi-scale roughness profile increases the total surface area and allows for the entrapment of air in between the water droplet and the surface. Depending on the wetting regime, the water droplet either penetrates in between the roughness asperities (Wenzel regime, see Fig. 4) or it remains staying on top of the roughness peaks (Cassie–Baxter regime, see Fig. 4). The effects of chemical surface heterogeneity are considered in the Cassie–Baxter model in Eq. 4:
where θ is the apparent contact angle, θ s is the equilibrium Young’s contact angle and f is the solid–liquid contact area fraction. The occupation of a surface fraction (1 − f) by air pockets augments the apparent contact angle. The validity of the Wenzel and/or Cassie–Baxter wetting model is under debate by several authors [159–161]. From physical standpoint, the Cassie–Baxter model does sometimes not predict correct contact angles on rough surfaces as confirmed by microscopy [162], while the model could be adapted for sawtooth surfaces [163]. From chemical viewpoint, the Cassie–Baxter model may only apply to macroscopic chemical heterogeneities [164]. For physical heterogeneities such as surface roughness, the contact angle mainly depends on the roughness along the triple line, and not on the surface ratios of the heterogeneities beneath the drop [165–167]. The macroscopically measured water contact angle onto paper is probably a mean value of local contact angles resulting from surface roughness and hydrophobicity.

Extrapolation of roughness data between optical profilometry and AFM for coated papers as a function of the sampling area and correlation length, a calculation of the correlation length β for the different sampling areas, b roughness parameters Sa (open triangle) and Sz (open square) and software extrapolation values (filled square) as a function of the correlation length β (from [158])
Also the droplet motion should be considered to determine the wetting state. If a drop spreads onto a surface, it has a larger contact angle (i.e. advancing contact angle θ a) than when it recoils from the surface with a lower contact angle (i.e. receding contact angle θ r). The contact angle hysteresis (i.e. difference between the advancing and receding contact angle during dynamic contact angle measurements) is one important criterion for (super-)hydrophobic surfaces, as it is a measure for the adhesion strength of a droplet to the surface. A relationship between hysteresis and surface hydrophobicity is given by Eq. 5, with k is the constant, g the gravity, m and w are mass and contact diameter of the droplet and γ LV is the surface tension [168]:
The hysteresis depends on two properties, including (i) a metastable state energy, and (ii) a barrier energy for the drop moving from the one into another metastable state [169]. From physical viewpoint, hysteresis exists when the separation energy for a liquid and a solid interface is larger than the energy required to form a new interface [170]. The physical or mechanical hysteresis becomes stronger when the contact area between the solid and liquid increases. As the overall contact area increases with roughness, the wetting of the additional surface area is expected to enlarge the hysteresis effect [171–173]. However, the superposition of a micro- and nanoscale roughness profile may also decrease the wetting and hysteresis. From chemical viewpoint, hysteresis relates to molecular interactions at the interface and variations in molecular arrangements on the substrate [174]. The chemical hysteresis is therefore strongly influenced by heterogeneities due to polar and nonpolar moieties at the surface [167, 175, 176]. The hysteresis can be influenced by physical or chemical surface modification to tune the interfacial interactions. The chemical hysteresis onto hydrophobic papers can specifically be modified by altering the active surface groups, while the physical hysteresis can be simultaneously changed by surface roughening [177]. The Wenzel and Cassie–Baxter equations were combined to predict the transition from a Wenzel-type to a Cassie-type of wetting [178]: for a hydrophobic surface, the contact angle hysteresis increases with roughness in the Wenzel state, and it decreases with roughness in the Cassie state. Two analytical formulas for hysteresis on rough surfaces were developed to describe the sticky and slippy behaviour [179].
Superhydrophobic surfaces have usually contact angles above 150°, but the wetting dynamics and contact angle hysteresis should also be considered as important parameters when using papers for self-cleaning and anti-adhesive properties. A systematic review on self-cleaning surfaces with hydrophobic or hydrophilic coatings was published [180]. The superhydrophobicity relates to a transition between different wetting states [181], and can theoretically be achieved through a smart combination of surface chemistry (reduction in surface energy) and structure (increase in surface roughness). This idea is practically derived from examples of plants such as Lotus leaves [182–184], or insects such as the wings of butterflies and legs of the water strider [185, 186]. The surface microstructure and composition of Lotus leaves was characterised many times [187–189]. The higher contact angle on heterogeneous surfaces explicitly relates to the presence of micro- to nanoscale hierarchical roughness structures [190] or fractal surfaces [191], and provides self-cleaning properties [192]. The concept has successfully been transformed into bionic nano-engineering [193] for the formation of hierarchical synthetic surfaces with similar properties [194–196]. The contribution of surface roughness on steady and sliding contact angles for superhydrophobic surfaces is well documented [197]. Several routes are followed to create superhydrophobicity, such as (i) increasing the roughness of an initially hydrophobic substrate, (ii) increasing the hydrophobicity of an initially rough substrate or (iii) modifying a surface with a low surface energy chemical that simultaneously provides an inherent roughness. As such, superhydrophobic surfaces with tunable hysteresis were made on metals [198], glass [199] and polymers [200]. The techniques were initially limited to rigid substrates, as the structuring of soft substrates was supposed to be destroyed by deformation.
The first idea for a superhydrophobic polyester fabric appeared in a patent of 1945, based on the use of a silicone coating [201], with a restriction that the fibre has to be tightly woven and presents adequate roughness [202]. Cellulosics with tunable sticky and roll-off properties were fabricated more recently [203]. Superhydrophobic paper surfaces are generally categorised depending on the degree of droplet adhesion and hysteresis (Fig. 7) [204, 205]: (i) the ‘roll-off’ superhydrophobic papers have a hysteresis below 10° and droplets consequently spontaneously roll off the surface, while (ii) the ‘sticky’ superhydrophobic papers have a hysteresis above 10° with consequently adhering droplets. The differences in superhydrophobicity can also be demonstrated from models predicting the hysteresis [206] and work of adhesion [207]. General surface roughening techniques include mechanical, physical or lithographical approaches that can be applied on a (rather small) range of substrates. Other techniques such as electrospinning, chemical vapour deposition, nanoparticle coating, sol–gel processing, solution casting, layer-by-layer (LbL) deposition, colloidal assembly become more versatile and can be applied to different substrates [195]. For paper substrates, the inherent fibre morphology offers an approach to create roughness at multiple levels (micro- to nanosize). Paper webs already have an inherent microscale roughness due to exposure of cellulose fibres and cavities at the surface, in combination with a nanoscale roughness due to the coverage of cellulose fibres by microfibrils with diameters of 3–30 nm. The latter fibre morphology at the surface depends on the processing and especially calendaring of the paper surface: as such, the morphology of tissue or filter papers will have other effects than office or packaging paper. When using the cellulose fibre morphology as a multi-scale roughness component, good knowledge of the cellulose structure and behaviour is required as summarised in previous paragraphs. In addition, inherent chemical heterogeneity is provided by the alternation of crystalline (e.g. microfibrils) and amorphous (e.g. matrix surrounding the microfibrils) domains, and can be boosted by preferential etching. The complex structure of cellulose fibres and papers often inhibit the use of common techniques for surface modification due to absorbance, swelling, inhomogeneity, porosity or thermal stability. Practical examples for paper surface modifications are presented later.

Hydrophobicity by paper sizing
Papers are commonly protected against water by treating the pulp with hydrophobic agents. The effects of internal sizing prior to sheet formation were reviewed [208]. It is not the goal of this review paper to go into great detail on the internal sizing processes and effects, but some findings are mentioned for completeness and as a reference hydrophobicity for standard paper grades. Although the hydrophobic character of internal sizing improves the bulk resistance against water penetration, it often does not provide a sufficient water barrier, e.g. for packaging papers. Other influences of internal sizing on dewatering, retention of fillers/fibres and possible improvement of the paper strength exist, but are not further considered here. A comprehensive overview of sizing methods and agents is well covered [209]. The internal sizing is done by wet-end fibre modifications, using nonreactive agents or synthetic agents that chemically react with the cellulose hydroxyl groups to form stable ester linkages. For internal sizing, the dynamics of wetting are crucial as colloidal substances are first adsorbed onto the fibres and consequently wet the fibre surface under drying.
Traditional internal sizing for paper substrates
The nonactive sizing ingredients include rosin (resin from pine trees, Fig. 8a) that is added during acidic sizing. The rosin is usually added together with aluminium salts (Al2(SO4)3·2H2O) to facilitate the distribution and fixation onto cellulose fibres [210]. In order to optimise the interaction with the cellulose fibre, the hydrophobic rosin–aluminium precipitates are positively charged and should be formed in situ [211]. The mechanisms of premixing the rosin sizing and processes for optimization as a function of the solution charge and particle size were developed [212].

Traditional internal sizing agents for paper. a abietic acid as a component of rosin, b alkene ketene dimer (AKD), c alkenyl succinic anhydride (ASA)
The chemically reactive agents include alkene ketene dimer wax (AKD, Fig. 8b) [213] and alkenyl succinic anhydride (ASA, Fig. 8c) [214], which are used for neutral or alkaline sizing and have better compatibility with the papermaking process. Sizing at neutral pH allows using calcium carbonate as additional filler and provides better storage durability than acid sizing. Both AKD and ASA products are available in good dispersion qualities [215, 216], and react with cellulose through a covalent ester bonding that results in either chemical or physical absorption on the surface [217, 218]. In the presence of water, the hydrolysis of sizing agent and the formation of corresponding ketones reduces the sizing efficiency as there remains a fraction of unbound sizing agent [219, 220]. The AKD is usually added as a polyelectrolyte emulsion and interactions with the paper web include the following mechanisms: (i) retention of AKD particles on the pulp fibres, (ii) spreading over the fibre surface in a monolayer film and (iii) re-conformation and covalent bonding. The spontaneous spreading of an AKD melt over the cellulose surface was investigated to establish the mechanisms and driving forces responsible for the dynamic wetting of AKD [221]. Two wetting stages generally occur, including (i) the balancing between interfacial forces and viscous dissipation towards an equilibrium state (rapid decrease of contact angle over time), and (ii) an equilibrium shift due to hydrolysis of AKD vapour molecules physically absorbed at the cellulose surface (slow decrease of contact angle over period of hours). The amount of physically absorbed AKD depends on the chemical substrate composition. While the retention and sorption mechanisms of the wax particles to cellulose and re-conformation with covalent bonding to the fibres are well understood [222, 223], a complete spreading of AKD onto smooth cellulose model surfaces was not observed. Therefore, questions arose on the general mechanisms and efficiency of internal sizing. In a later study, only partial wetting of AKD to cellulose was confirmed by fitting of the wetting behaviour to the Hoffmann–Tanner equation. The degree of wetting also depends on the temperature [224], as a lower viscosity at high temperature improves the wetting velocity. Later, the slow monolayer spreading and diffusion was better understood in parallel with a redistribution of AKD at the fibre surface [225]. The amount of bound AKD in wet paper significantly increases after drying. Nevertheless, the reacted AKD only constitutes a very small portion of the totally retained AKD. Therefore, migration of AKD through the paper bulk towards the surface might cause a variation of contact angles over time, which can be stabilised in the presence of precipitated calcium carbonate (PCC) [226]. For fully sized AKD samples, a maximum advancing water contact angle of 110° was achieved [227], while the AKD wax also enhances the contact angle when added as a modification aid in regenerated paper fibres [228]. The hydrophobization of filter papers by AKD also provides water contact angles of 110°–125°, while the contact area fraction between the water drop and the paper surface was 51 % [229]. However, dynamic contact angle measurements revealed strong heterogeneity for AKD sized fibres, while higher and more stable hydrophobicity was observed after removal of unbound sizing by extraction [230]. In comparison, ASA has lower retention than AKD but it covers better the cellulose surface through formation of hydrolyzed products that may consequently provide higher contact angles [231, 232]. As such, ASA sized samples had maximum contact angles of 90°, which could be improved to 100° or 104° in combination with a styrene-based sizing [233]. The hydrophobic efficiency could be further improved by vapour deposition of AKD and ASA, resulting in typical esterification and a much faster reaction time [234]. The sizing efficiency of ASA can be optimised by regulating the amount of dissolved Ca2+ and HCO3 − ions, PCC, pH and temperature [235].
Recent developments in internal sizing
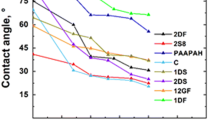
The efficiency of common sizing products may be improved by simultaneous hydrophobization and roughening at micro- to nanoscale. This can be done by introducing novel processing methods for AKD and/or novel sizing agents (Fig. 9). By controlling the fractal growth of AKD crystals, a rough surface with contact angles of 174° was obtained in contrast with smooth AKD surfaces having contact angles of only 109° [157]. Otherwise, AKD with an average particle size of 1–2 μm was deposited from a rapidly expanding supercritical carbon dioxide solution [236], resulting in contact angles up to 173°. This technique was also applied in combination of AKD with a crystallising wax from organic solvents, leading to lower stick–slip of the advancing contact angle [237].

Novel methods for internal sizing of papers, including new processing methods: a fractal growth of AKD crystals [157], b rapidly expanding supercritical CO2 [236], c rapidly expanding supercritical CO2 in combination with crystalline wax, or new sizing products [237], d fatty acid anhydrides [241], e styrene maleic anhydride [247], f styrene maleimide [248], g polystyrene-based micelles (from [246]), h modified talc (from [244])
In recent decades, new hydrophobic sizing products have been introduced such as synthetic cationic or anionic polymers, fluorinated polyurethanes [238], fatty acids with different chain lengths [239], quaternary ammonium salts [240], fatty acid anhydrides [241], amines [242], ethylene copolymers [243] or surface-modified amphiphilic talc [244]. The sizing with fluorochemicals having a linear or branched monofunctionalized fluoroalkyl chain or based on perfluoropolyether needs to be limited because of environmental concerns, and alternatives are currently developed. Synthetic copolymers, such as styrene copolymers, have the advantage over rosin that they bear cationic groups directly on the polymer chain and can be added without need of alum as fixation agent. Hence, about 80 % of the styrene copolymer is retained in the pulp, resulting in good hydrophobicity with contact angles of around 105° [245]. Micelles of polystyrene-based cationic copolymers were synthesised from styrene and the cationic comonomer vinylbenzyl trimethylammonium chloride. These copolymers are completely self-retaining on the fibre surface and can be used as internal sizing agents in a broad pH range without hydrolysis or sizing reversion [246]. Especially, the copolymers of anionic or cationic styrene maleic anhydride (SMA) with low molecular weights [247], or styrene maleimide [248], received attention as hydrophobic additives in the wet-end papermaking. The SMA could be used in bleached pulps with ferrous chloride or alum as mordants, but its efficiency mainly depends on the interaction of the polymer with the lignin component of the paper fibres. A higher ratio of styrene to maleimide improves the hydrophobicity, and has also a positive effect on sizing at both low and high concentrations (0.5–3 %). However, the internal sizing efficiency of maleimides decreases at high concentrations because of retention problems attributed to excessive cationic charges. A styrene/butadiene cationic latex was successfully used as hydrophobic sizing and retention agent for clay-loaded papers [249]. However, the possibilities of internal sizing remain limited and eventual reinforcement of the paper bulk with additional fibres did not significantly alter the sizing effect [250].
Surface sizing for paper substrates
Barrier coatings are traditionally developed from latex binders mixed with dedicated fillers or (functional) additives such as kaolin, calcium carbonate or alumina trihydrate (Fig. 10). Plate-like inorganic fillers are typically added together with a neutralising agent to improve the barrier properties by enlarging the diffusion path through the coating [251]. The effect of different binder types for coatings with latex and kaolin additives has been evaluated [252], by differentiating the surface fractions covered by each component. The apparent contact angles according to the Wenzel equation indicated a higher hydrophobicity for higher contents of styrene–butadiene latex, with some differences depending on whether a latex with high or low glass transition temperature was used, as the latter clearly provides a different topography. Both contributions of local wettability on the latex fraction (contact angle 109°) or kaolin fraction (contact angle 33°) were combined by means of the Cassie–Baxter equation in order to calculate a contact angle for the composite coating. Furthermore, the surface tensions were determined by using the Owens–Wendt, Wu and van Oss approaches, where both surface chemistry and surface roughness influence the dispersive and polar components. In parallel, the liquid absorption rate decreases with a higher amount of latex due to the lower porosity. Also during a continuous extrusion coating process of polyethylene, fillers such as montmorillonite have been added for better barrier performance [253].

Recent developments in surface sizing products for improving water repellency and creating superhydrophobic paper surfaces: a kaolin, b aluminium trihydrate, c precipitated calcium carbonate (PCC), d ground calcium carbonate (GCC), e surface coverage of kaolin and styrene-butadiene latex (from [252]), f microfibrillated cellulose (MFC), g PCC with fatty acid coating (from [274]), h PCC with amino function (from [277]), i PCC with microfibril binder (from [278])
Polysaccharide additives such as microfibrillated cellulose (MFC) are expected to further improve the barrier properties [254, 255]. The terminology in literature, often referred to as nanocellulose is often misleading, but we focus here on the use of cellulose microfibrils produced by a microfluidizer and resulting in diameters of 10–50 nm and several micrometres length. The MFC has been recently considered as a valuable additive in papermaking for improving the physical and mechanical properties of pulp [256]: the strength of handmade paper could be increased by 35 % due to reinforcement and application of MCC suspension as external additive. On the other hand, MFC is a film-forming material showing also promising properties as a barrier film [257], and is a good candidate to improve the barrier properties in cellulosic materials [258]. As already demonstrated, the MFC additives create good resistance against oxygen and oil when deposited on paper [259] or incorporated within paper coatings form an aqueous dispersion [260]. By investigating the mass transfer properties, the barrier properties of MFC films were attributed to their structure with a porous and closely packed fibre network, while the film cross-sections form dense layers with almost no porosity. However, the water contact angle values of MFC films remain in the hydrophilic region with a maximum of 68° for hybrid MFC films with silesquioxane [261]. As a main concern, the final barrier performance of MFC films and additives strongly depends on the relative humidity: dry MFC films have excellent oxygen barrier properties, but a dramatic decrease in these properties was observed at higher water contents [262]. There has been limited number of studies published so far presenting water uptake of neat MFC films [263]. The diffusion of water is rather controlled by the surface than by the core, probably because the barrier effect mainly relates to the presence of water at the surface during the sorption kinetics [264]: as such, the MFC has much better barrier properties than cellulose nanowhiskers through a combination of favourable entanglements and different surface chemistry. After homogenization, the MFC with high lignin contents have a higher water vapour transmission rate (WVTR), even with a higher initial contact angle, most likely due to large hydrophobic pores in the film [265]. Therefore, the physical properties and barrier properties of the MFC should be further improved by hydrophobic surface treatments, while preserving the dense network morphology of the cellulose fibrils. Indeed, the combination of a dense network structure and hydrophobic surface can combine hydrophobicity with good oxygen barrier properties. By heterogeneous acetylation, the contact angle of MFC increases upon reaching a certain reaction time and some decrease following the further acetylation was confirmed depending on the degree of substitution (DS) [266]. Other techniques for surface hydrophobization of MFC were reported, such as silylation [267], silazanation [268], surface grafting with different chemicals [269], absorption of cationic surfactants [270] to name only few. The pre-treatment with 2,2,6,6-tetramethylpiperidinyl-1-oxyl (TEMPO)-mediated oxidation resulted in a dense network with even better oxygen barrier levels than recommended for packaging, in parallel with better hydrophobicity [271]. The surface modification of MFC is a relatively new field of research with limited number of published research that was summarised in a recent review paper [272].
The fabrication of superhydrophobic paper coatings may include mineral additives [273], such as silica, calcium carbonate and clay. The minerals can be applied either as filling agents in the bulk (wet-end) or as a coating (dry-end): in both cases, good dispersibility in an aqueous medium is generally required. Moreover, the minerals often have to be hydrophobized and/or their surfaces need to be patterned in order to induce a certain roughness that improves the superhydrophobicity. The dispersibility of the latter hydrophobized minerals even becomes more challenging and needs to be developed in parallel with suitable binder systems. The surface sizing of paper with microsized PCC may deliver a certain roughness required for improved water resistance in combination with a fatty acid (stearic acid) coating [274]: in that case, the PCC surfaces were pre-treated with a thin layer of a calcium salt to provide good compatibility after mixing with a polymer latex binder. The hydrophobic additive is applied in combination with a traditional styrene–acrylate copolymer latex, while the hydrophobicity of the coated paper can be further improved by immersing it consequently in a potassium stearate solution. In parallel, a two-step dip-coating process for paper modification was performed: (i) first, within an aqueous suspension of PCC; and (ii) second, in a solution of AKD. However, the dipping of calcium carbonate-coated papers in a stearate solution was most critical in permanently increasing the contact angle. Multilayer-coated papers with ground calcium carbonate (GCC), kaolin, PCC and mineral blends were evaluated [275], where spreading of macroscale droplets over short timing was mostly hindered by the higher surface roughness and porosity of the coating. Indeed, the number of coating layers containing kaolin does not directly relate to the contact angle [276], but the actual ratio of the surface coverage between polar mineral additives and nonpolar binder latex influences the surface energy. Otherwise, nonconventional hydrophobic clays with fine particle sizes were modified by dehydroxylation and anchoring an amino functional additive to the mineral surface in order to control the dispersibility under certain conditions [277]. The hydrophobic clays were used as filler in water-based coatings that include a binder of styrene–butadiene latex and a co-binder of waxy corn starch. The mobility and repellency of water droplets could thereafter be modified by different printing inks. The effect of cellulose microfibrils as a binder for the PCC was studied [278], and it was concluded that the retention and consequently the formation of a rough surface coating improves in the presence of nanofibre additives leading to superhydrophobic papers (Fig. 11). The MFC was successfully used for creating hierarchical superhydrophobic surfaces, by airbrushing the solvent-based fibres followed by quick drying and subsequent fluorochemical surface modification [279].

SEM images of a AKD sized filter paper, b filter paper dip-coated with PCC without using cellulose microfibrils as binder and then sized with AKD, c filter paper dip-coated with PCC with added cellulose microfibrils as binder and then sized with AKD subsequently, d the interaction between PCC particles and cellulose microfibrils when the slurry was coated on a glass slide, e cellulose microfibrils and nano- and microfilm patches of the recombined microfibrils connecting and holding the PCC particles together (from [278])
Hydrophobicity by fibre surface engineering
While most industrial hydrophobization techniques rely on internal or surface sizing of paper products, we further focus in this review on innovative surface modifications for individual cellulose fibres or papers. The compatibilization and hydrophobization of natural fibres by surface engineering has tremendously increased over the last years, mainly to incorporate them as fibrous reinforcement in bio-based composites [280, 281]. Similar surface treatments and/or hydrophobic coatings may also apply to improve the performance and water resistance of paper fibres. Various techniques for creating (super)hydrophobic surfaces are well known for general substrates, but they often cannot be simply transferred to paper substrates as cellulose is easily damaged under severe chemical, physical and thermal treatments. An overview of chemical, physical and nanotechnological methods applied to cellulose fibre surfaces will be presented, focusing on the different techniques and resulting surface morphologies (Fig. 12).

Overview of surface engineering for hydrophobic cellulose fibres or papers: methods and common products
Chemical surface modifications
Graft polymerization is a successful tool for the chemical modification of cellulose surfaces. Some features of the cellulose structure and grafting reactivity were reviewed, together with different techniques [282]. The methods for modification of cellulose surfaces by grafting include: (i) ‘grafting from’ by coupling polymerizable monomers to the surface after the creation of reactive sites at the fibre surface, (ii) ‘grafting onto’ by coupling a mono-functional polymer to the surface or (iii) coupling of functional bridging molecules between the cellulose and polymer [283]. As such, cellulose has been modified with various hydrophobic polymers for desirable surface properties.
The “grafting from” reaction is mainly performed by free-radical polymerization or reversible addition–fragmentation chain transfer (RAFT) [284]. Vinyl monomers with long paraffin chains were grafted from cellulose by direct irradiation, resulting in reduced swelling and improved water droplet imbibition of the fibres as a function of the grafting percentage [285]: the homopolymerization of the grafting agent could be suppressed and the grafting reaction was favoured with an almost linear increase in yield, depending on the amount of styrene. Acrylic acid and acrylonitrile were grafted from cellulose pulp with ceric ammonium nitrate as an initiator, also reducing absorption and swelling [286]. Specifically, biodegradable polymers such as polycaprolactone were grafted from different types of cellulose and pulp (Fig. 13a), reducing the polar surface energy to almost zero [287]: as such, the water penetration lowered and contact angles progressively increased from 90° to 95° as the grafts had higher molecular weight. Also starch-grafted cellulose fibres were formed through hydrogen bonding among cellulose, starch and ammonium zirconium (IV) carbonate, followed by crosslinking [288]: the increase in surface coverage by a starch hydrogel significantly improves the WRV and can be tuned as a function of reaction conditions (pH, temperature), or composition (starch and crosslinker amount).

Example of chemical modification of cellulose fibres by ‘grafting from’, resulting in hydrophobic cellulose surfaces with a polycaprolactam (from [287]), b poly(ethyl acrylate) (from [290]), and superhydrophobic cellulose surfaces with c hierarchical graft-on-graft architectures with fluorine and long alkyl chains (from [292])
Surface-initiated atom transfer radical polymerization (ATRP) was used for modifying pulp and cellulose fibres and/or cellulose crystals in combination with, e.g. polystyrene [289]. For this reaction, the wood pulp and Kraft cellulose fibres may serve as initiator for grafting of, e.g. poly(ethyl acrylate) (Fig. 13b) [290], while the cellulose fibres and microfibrils were also modified with butyl acrylate [291]. In one study with ATRP, a polymer layer with micro- to nanoscale binary roughness was grown on the cellulose surface by grafting glycidyl methacrylate with a 2-bromoisobutyryl bromide initiator (Fig. 13c) [292]: as such, a complex branched ‘graft-on-graft’ architecture was formed and followed by a post-functionalisation with fluorine [293], or alternatively with siloxane and sufficiently long alkyl chains (C15H31) [294] to obtain superhydrophobic cellulose with contact angles of 170°. As an advantage, the grafted species have a well-defined molecular weight, molecular weight distribution and chain ends that allow for precise control of the surface properties, while the cellulose fibrillar structure remains existing. As a disadvantage, the nanoscale roughness induced through deposition of a soft polymer film easily undergoes deformation that limits robustness and durability.
The ‘grafting onto’ reaction includes modifications of the hydroxyl groups that consequently improve the water resistance of cellulose [295]: e.g. etherification was done with hydroxypropyl and esterification was done with hexanoate, while the esterified fibres generally show best hydrophobicity [296]. Many esterification reactions use perfluorined agents to maximise the hydrophobicity by, e.g. pentafluorobenzoylation [297], trifluoropropanoylation [298] or trifluoroacetylation [299, 300]. However, the hydrolytic stability of the perfluorinated cellulose strongly depends on pH or vapour humidity due to the general fragility of CF3 moieties attached to the ester group. The most popular grafting method in surface hydrophobization introduces perfluorinated methyl and/or methylene groups. Often, the perfluorination is done in combination with silane coupling agents: the bonding of cellulose with silicon provides organic–inorganic hybrid materials as a broad platform for surface modification. Especially, several fluorine-bearing alkoxysilanes were synthesised and grafted onto cellulose with contact angles of 115° to 130° [301]: the alkoxysilanes are favourable coupling agents and the grafting reaction proceeds homogeneously over the entire thickness of a cellulose fibre mat. After modification of the cellulose surface with a silane coupling agent, the introduced ethoxy groups can be further hydrolysed under acidic conditions to attach specific (e.g. perfluorinated) side chains, which provide a hydro- and lipophobic character (Fig. 14a) [302]. The silanization of cellulose could also be done in a gas–solid reaction using more environmentally friendly conditions with very short reaction times (30 s) and concentrations, while preserving the original fibre ultra-structure in combination with a nanoroughness at the surface (Fig. 14b) [303]. Similar silanization reactions were done by a solution-immersion or chemical vapour deposition process to form self-assembled silane monolayers with maximum contact angles of 130° depending on the introduction of either fluorine or long alkyl chains [304]. After immersion of a model cellulose surface with controllable porosity into a fluorinated trichlorosilane solution, a thin adsorbed monolayer film (<10 nm) with local atomic fluorine content up to 53 % and surface energy of 15 mN/m was formed [305]: then, the cellulose surface porosity could be additionally influenced by structured silicon templates. Nowadays, more tendencies exist for grafting polymers onto cellulose without fluorinated moieties. The chemical vapour deposition at low temperature was used to deposit polymethylsiloxane from a saturated trichloromethane atmosphere, resulting in a covalently attached layer with uniform microscale roughness and nanoscale protuberances [306]. Before the silanization reaction, cellulose may also first be immersed in a silica sol for binding the silica as a coating onto the cellulose surface over a water glass cross-linker that is deposited by sol–gel method (Fig. 14c) [307]: as such, the superhydrophobicity receives better durability.

Example of chemical surface modification of cellulose by silanation under different conditions and creation of a structured cellulose surface with a (3-isocyanatopropy)triethoxysilane followed by acid hydrolysis (from [302]), b trichloromethylsilane in presence of controlled humidity (from [303]), c mixed organosilanes and cross-linker (from [307])
Traditionally, papers are modified by graft (co-)polymerization with styrene and/or acrylonitrile (co-)monomers [308]: the grafted polymer chains can penetrate over the entire sheet thickness, changing the pore structure and water penetration. Recently, more sustainable grafting methods were developed by introducing renewable materials. A heterogeneous esterification with fatty acids was used to homogeneously cover paper fibres with undecylenate, undecanoate, oleate and stearate with a given DS [309]: the nonpolar surface energy of cellulose significantly decreases (lowest for oleate), while the donor–acceptor characteristics had hardly changed and the acid–base interactions are still present within the esterified layer. Especially, the partial esterification of cellulose fibres with fatty acids is applied to introduce long aliphatic chains with hydrophobic properties (Fig. 15) [310]: it was found that the surface coverage by fatty acids increases for higher chain lengths and is little affected by the DS. The penetration depth of the reaction with fatty acids (up to C22 long) could be controlled in favourable swelling media such as DMF, in contrast with toluene as nonswelling medium [311]. Consequently, the polar surface component of the fibres decreased for an almost constant dispersive surface energy, as a function of treatment time. Other sustainable methods consider the recycling of the used solvent: therefore, the esterification of cellulose with succinic anhydride was performed in ionic liquids in combination with an appropriate catalyst, resulting in a specific DS of the cellulose surface with improved hydrophobicity [312].

Example of chemical surface modification of cellulose using renewable resources such as fatty acids for esterification (left) and homogeneous surface coverage of a paper surface (right) (from [310])
Through LbL absorption, a rough coating may form on cellulose surfaces. By consecutively treating the cellulose fibres with oppositely charged polyelectrolytes (PEL), multiple layers are deposited by electrostatic interaction. As such, desired surface architectures and chemical properties can be obtained that influence dry and wet adhesion, friction and wettability. This technique was also applied for wood fibres [313], enhancing hydrophilicity, adhesion and network strength at the level of local fibre–fibre joints [314]. For cellulose fibres, the alternating layers of poly(allylamine) or poly(ethylene oxide) with poly(acrylic acid) reduced the wettability, depending on the outermost layer and pH of adsorption (Fig. 16a) [315]: in parallel, the fibres with low wettability provide higher tensile strength according to AFM pull-off tests, as the degree of molecular mobility in the outermost layer defines the degree of rigidity and dry fibre adhesion. As such, cellulose surfaces were created with high contact angle hysteresis (100° on smooth surfaces, up to 150° on rough surfaces), depending on the number of PEL layers and ionic strength of the deposition solution [316]. In general, only relatively hydrophilic PEL can be directly dissolved in water and hence, the hydrophobicity by absorption of unimeric polymer solutions is limited. More hydrophobic PEL solutions can be created from kinetically trapped aqueous nanoparticles: as such, the adsorption of amphiphilic polyelectrolytes results in the formation of polymeric nanoparticles with variable coverage of the cellulose surface, while additional thermal annealing provides hydrophobicity with contact angles of θ a = 160° and θ r = 120° [317]. The superhydrophobicity was also created by LbL deposition of a poly(diallyldimethyl-ammonium chloride) or poly(DADMAC) including TiO2 particles, followed by the modification with a thin fluorosiloxane film that results in contact angles of 162° [318]. Similarly, the multilayer deposition of cationic poly(DADMAC) and anionic silica nanoparticles followed by a fluorination creates superhydrophobic wood fibres with water contact angles of above 150° and sliding angles below 5° (Fig. 16b) [319]. Alternatively, five layers of poly(allylamine hydrochloride) and poly(acrylic acid) were adsorbed, followed by the adsorption of paraffin wax : the contact angle measured 60 s after a drop of water was applied to the sheet was about 138°. If the paper sheets were cured at 160 °C, the contact angle was 150° (Fig. 16c) [320]. Especially for LbL-coated paper sheets with alternating layers of poly(allylamine hydrochloride) and kaolin, the surface changes from hydrophilic to hydrophobic with increased number of multilayers and if the terminating layer was kaolin clay [321]. The LbL deposition is also used to form a bilayer structure of alternating PEL and montmorillonite from an aqueous solution (Fig. 16d): the water vapour barrier properties of these coatings improved by layer thickness and high ionic strength [322].

Examples of layer-by-layer (LbL) absorption techniques a general representation for LbL deposition on single cellulose fibre (from [315]), b deposition of cationic poly(DADMAC) in combination with anionic silica nanoparticles followed by a fluorination treatment on paper sheet (from [319]), c deposition of poly(allylamine hydrochloride) and poly(acrylic acid) followed by the adsorption of paraffin wax (from [320]), d alternating layers with montmorillonite (from [322])
A simple modification for fibres or large paper areas includes solution casting, immersion or dip-coating. As a disadvantage of wet-chemical processes, the solution treatment may cause shrinkage of the paper and dissolution of the printing ink. On the other hand, the interaction between cellulose and polymers may induce strengthening with better protection and durability. However, the strength of papers treated in solvent mixtures sometimes reduces due to a reduction in hydrogen bonding within the dried paper network [323]. Several monomer solutions were used for solvent treatments, such as, e.g. ethyl-cyanoacrylate in acetone or toluene: immediately after impregnation and solvent evaporation, the acrylate crosslinks with the cellulosic hydroxyl groups in the presence of moisture and it forms a hydrophobic polymer shell, while preserving the structure of the entangled fibre network [324]. In addition, carnauba wax flakes and PTFE nanoparticles were dispersed as additives in the solution: as such, tunable contact angles with a maximum of 160° and hysteresis of 10° were obtained for about 45 wt% PTFE, while the carnauba wax induced hydrophobicity with no self-cleaning properties (Fig. 17a). Similarly, papers were hydrophobized by immersion into an acetic acid solution of methacrylate with hydrolyzed reactive silanol coupling agent [325]: after the evaporation of acetic acid, thermal treatment, washing and drying, contact angles were about 110° through the reaction of hydrophobic propyl and methacryloxy groups with the silane coupling agent. The silane coupling of cellulose involves a condensation reaction between the silanol and the hydroxyl groups of cellulose [326, 327]: the coupling agent acts as crosslinker and simultaneously undergoes self-crosslinking that improves the physical paper strength. Alternative impregnation techniques include dip-coating of paper sheets in waterborne epoxy emulsions with fluorinated methacrylate (Fig. 17b) [328]: the adhesion of polymer chains was subsequently optimised after thermal curing through covalent binding with the cellulose fibres, and should provide better robustness and stability than traditional polymer grafting or nanoparticle deposition on cellulose. The resulting low surface energy provides superhydrophobicity in combination with superoleophobicity: the surfaces have sticky contact angles of 152° and good adhesion of the water even after holding the surface upside down. A traditional aqueous-based process was compared with a dry technique in an inert atmosphere for fluorination reactions (Fig. 17c, d) [329]: during admicellar polymerization with a surfactant and fluoro-monomer in solution, a thin patchy-like fluoropolymer film was formed onto cellulose with contact angles of 125°, in contrast with lower contact angles of 117° after direct fluorination with elemental fluorine gas due to different fluorine concentrations and surface structures.

Physico-chemical surface modifications and surface patterning
Plasma processes are preferred to tune the wetting properties of cellulose surfaces, as it provides good versatility and does not involve solvents. During the RF-plasma treatment of paper surfaces, a SiCl4 plasma induces high wettability after short treatment times and relatively low power, irrespective of the original hydrophobic sizing of the paper [330]: the conversion of Si–Cl into Si–OH bonds upon exposure of the surface to atmospheric moisture then enhances the hydrophilicity. Similarly, a hydrogen or oxygen plasma reduces the cellulosic hydroxyl groups and also improves the water wettability of resin-rich hydrophobic papers [331]. During DC-plasma in air and enzymatic cellulose treatment, the cellulose fabrics become hydrophilic due to breakage of the glycosidic bond (enzymatic treatment), in combination with specific oxidation and surface etching (plasma treatment) removing the noncellulosic compounds [332]. The atmospheric cold plasma modification by dielectric-barrier discharge changes the polar and dispersive surface energies of bleached Kraft pulp, leading to higher contact angles (33°–43°) and a lower polar surface energy at higher plasma intensity [333]: this relates to oxidation and surface cleaning effects removing contaminants. These observations agree with studies under low-pressure plasma, but some effects of fibre cross-linking under high-pressure plasma may simultaneously improve the wet-strength/stiffness of pulp.
Other plasma treatments of cellulose fibres include etching processes, where the amorphous parts are eroded and the crystalline parts are conserved. As such, the selective etching under appropriate plasma conditions may generate a specific surface roughness, in parallel with the intrinsic hierarchical structure of cellulose as detailed in a previous paragraph. Basic studies on plasma-assisted etching of paper surfaces provide insights in interfibre bonding, internal fibre structure and coating distribution [334]. The selective etching of amorphous domains under mild oxygen conditions only affects the morphology of the top surface layer, and develops the features of crystalline cellulose microfibrils. As such, a two-step plasma process may consider (i) oxidative etching to generate given roughness, followed by (ii) plasma-enhanced deposition of a pentafluoroethane film (±100 nm), to create superhydrophobic surfaces with controllable sticky and roll-off properties following the Cassie–Baxter wetting state [335]: contact angles of 160° with hysteresis of 3° (roll-off) or contact angles of 144° with hysteresis 79° (sticky) were created, depending on the etching and plasma deposition conditions (Fig. 18) [203]. This process was used for patterning paper into microarrays to control the mobility of microliter droplets in combination with desk-top printing. The adhesion and directional mobility of water droplets could be further tuned by adding chemical heterogeneity [336].

Illustration of cellulose fibres after plasma processing at two magnifications of ×5000 (top) and ×20000 (bottom): a, b untreated cellulose fibre; c, d oxygen-etched cellulose fibre; e, f oxygen-etched and fluorine-coated cellulose fibre (from [203])
Plasma polymerization or plasma-induced grafting of hydrophobic polymers on cellulose fibres is used to improve the hydrophobicity and barrier properties. Some basic plasma processes for compatibilizing cellulose surfaces in fibre-reinforced composites and theoretical background were reviewed [337]. However, most plasma treatments include fluorine for reducing the surface energy, as illustrated by some of the following examples. The plasma treatment of sisal chemi-thermomechanical pulp (CTMP) with fluorotriethylsilane improves hydrophobicity of the pulp in wet conditions [338]. The plasma polymerization of pentafluoroethane films on paper and regenerated cellulose renders long-term stability (Fig. 19a) [339]: constant contact angles of 104° for over 1 h require a minimum film thickness of 70 nm that completely covers the surface and the near-surface fibres. In general, the fluorocarbon films have good wetting resistance and low water vapour uptake, but they show significant moisture diffusivity: the moisture is not chemically bonded and consequently penetrates easily through the fluorine coating, while the final moisture absorption in the paper bulk further affects hydrophobic resistance. Other films from perfluoromethylcyclohexane monomers were plasma-polymerized onto filter papers: the degree of film permeation and water absorption depends on deposition time and pressure, and the degree of hydrophobicity is controlled by the fluorine content (Fig. 19b) [340]. By comparing different fluorine containing monomers, the efficiency of CF2 and CF3 groups is different and the hydrophobicity abruptly increases at fluorine concentrations above 15 %. The surface treatments with CF4 plasma generate coatings with fluorine content up to 51 % and contact angles of 147°, after relative short treatment times of 30 s [341]. In parallel, some molecular fragmentation of the fluorine monomers occurred in the plasma [342]. The plasma treatment also affects molecular fragmentation of the cellulose and consequently reduces the surface stability with various effects depending on the felt- or wire-side of the paper [343]. The change in surface conformation of cellulose surfaces with a CF4 plasma film upon contact with water was studied in great detail [344]: from ESCA measurements, a decrease in the fluorine to carbon ratio was noticed at the surface upon contact with water (to 10 % of original ratio), or moisture (to 70 % of original ratio). The fluorine could be partly recovered after heat treatment, showing that the fluorine has not leaked into the medium but that changes are due to a rearrangement of the polymer segments at the cellulose surface under hydration. The interactions with water for untreated hydrophilic cellulose strongly relate to the effects of swelling and chemical interaction (solvation) of water and polymer molecules as defined by the Flory–Huggins interaction parameter. The interactions with water for modified hydrophobic cellulose are mainly due to the interfacial energy between both phases. As a more environmentally friendly method, butyl acrylate and ethyl-hexyl acrylate were grafted under atmospheric cold plasma onto cellulose fibres with contact angles up to 130° (Fig. 19c) [345].

As a recent application, the plasma treatment of papers with a patterned mask allows for the selective control of local wettability and liquid penetration by introducing a hydrophilic pattern onto a hydrophobic surface [346]. By optimising plasma times and intensities, channels with well-defined borders are developed and over-etching can be avoided. The plasma-treated areas are consequently wetted by aqueous solutions and used for transport along and within channels by capillary penetration, according to the Washburn model neglecting the gravity [347, 348]. Papers with a hydrophobic wax coating could also be structured by CO2 laser treatments to form hydrophilic patterns with minimum feature sizes of 60 μm [349]: the hydrophilicity developed after contact with atmospheric oxygen at the surface, in combination with a physical surface modification due to melting and re-solidification (Fig. 20a). Alternatively, patterns with thinner lines and better resolution can be made by photolithography using UV-curing and photoresist materials based on epoxy [350] and polyisoprene derivates [351]. The top–down methods compete with a bottom–up process of ink-jet printing hydrophobic patterns with, e.g. AKD-heptane solution: the printed patterns have contact angles of above 110° after curing. The deposition of small picoliter hydrophobic ink droplets in combination with a fast evaporating solvent, results in well-defined patterns that are obtained by a single printing step (e.g. for microfluidic sensors). In general, the chemistries for cellulose hydrophobization can be used for printing a hydrophilic–hydrophobic contrast forming liquid penetration channels. As such, micropatterned papers are fabricated by printing hydrophobic walls that extend through the thickness of the paper (Fig. 20b) [352]: e.g. wax printing with a solid ink results in typical line widths of 2 mm [353–355], and the subsequent curing at 130° allows for melting and penetration of the wax melt to form a hydrophobic pattern [356]. Other printing techniques with PDMS hydrophobic inks including vinyl groups need lower curing times in oven or by infrared [357]: the latter hydrophobic barrier layer provides contact angles of 142° for a single print layer, or about 134° on multiple print layers, depending on the smoothness and coverage by PDMS predicted by the Cassie–Baxter law. The PDMS inks are preferred because they contain no solvent, have short curing times and their oil consistency promotes good flow and penetration into paper.

Methods for patterning paper into hydrophobic/hydrophilic areas by a laser treatment, showing the hydrophilic paper areas that are nontreated (top) and hydrophobic paper areas that are treated (bottom) (from [349]), b wax printing with a top-view (top) and cross-section (bottom) of the hydrophilic wax domains (from [352])
The electrostatic spraying method was used to create differential superhydrophobicity and hydrophilicity on each side of a cellulosic sample [358]. Therefore, solutions of polyvinylfluoride [(–CHF–CHF–) n ] and fluorinated silane molecules [CF3(CF2)7CH2CH2Si(OCH3)3] were sprayed from a dimethylformamide solution onto cotton fabric or paper, resulting in a coating with fine particles or circular beads depending on the PVDF concentration (Fig. 21). A one-step electrospraying process requires large amount of silica located at the surface to provide high hydrophobicity. A two-step coating process with first deposition of PVDF and subsequent deposition of silica was more promising due to more effective location of silica at the surface, resulting in contact angles of maximum 162°. Besides the chemical composition, the surface roughness and superhydrophobicity was mainly affected by the diameter of the sprayed droplets, which can be tuned by the spraying parameters: the smaller particles yield higher hydrophobicity for similar surface roughness and chemical composition. The general surface roughness is defined by the vertical deviations of the surface profile, but the local particle diameter should be considered as an important horizontal dimension of the surface profile. A sprayed surface structure with bumps and/or honeycomb-like pores has appropriate roughness for hydrophobicity [359], but the spike-like surface morphologies additionally allow air entrapment.

Electrospraying of a coating containing polyvinylidene fluoride (PVDF) and fluorinated silane molecules, a coating morphology at low PDVF content, b coating morphology at high PDVF content (from [358])
Flexible wetting of papers can be controlled by stimulated electrowetting with a voltage-induced change of contact angles between 40° and 160° [360, 361]. Therefore, a layered structure is deposited on glassine paper with a metal layer as ground electrode, a dielectric intermediate layer and a fluoropolymer top layer (Fig. 22a): as a result, papers can be used as e-papers, displays, lenses, transistors or (bio)medical assays. However, the smoothness and pore size of the paper substrate is a primary selection criterion for good performance with fast switching times between the different wetting states. Another process for reversible wetting was created by self-assembly of photo-sensitive monolayers onto titania pre-coated cellulose nanofibre surfaces [362]: the as-prepared monolayer with trifluoromethoxy-phenlyazophenoxy pentanoic acid has almost superhydrophobic properties, while it transfers into an extremely hydrophilic state after exposure to UV light irradiation and the original hydrophobicity is recovered after dark storage (Fig. 22b). The reversible wetting mechanisms are attributed to conformational transformation of the photo-sensitive molecules under UV light.

Illustration of methods for flexible wetting by a electro-stimulated wetting (from [363)], b UV-induced wetting (from [362])
Surface coating
Sol–gel methods were favourably used for the deposition of inorganic coatings on cellulose substrates [363], where a silica coating serves as primary carrier layer to covalently bind hydrophobic groups of cotton specifically at low temperatures (Fig. 23a) [364]. In general, it is known that sol–gel coatings have a favourable nanosized roughness for the formation of superhydrophobic surfaces and can be applied by spincoating or air-brushing of metal alkoxides and eventually a fluoropolymer toplayer (Fig. 23b) [365]. As a disadvantage, the silane coupling agents are sensitive to pH and can be destroyed by swelling of cellulose during the reaction. Moreover, the cellulose surface is less reactive than inorganic oxides onto which silanes with coupling agent are usually applied.

Illustration of hydrophobic cellulose surfaces modified by sol–gel process, a silica coating (from [364]), b metal-alkoxy coating with fluoropolymer top layer (from [365]), c sol–gel in the presence of silica nanoparticles (from [377]), d cross-linked sol gel in the presence of polycarboxylic acid (from [379]), e sol–gel with long alkyl chains (from [380]), f sol–gel with self-assembled alkyl-silane on primary titania film attached to cellulose (from [383])
The sol–gel technique was specifically used in paper science for conservation [366], better ink-jet printing quality [367], control of dye leaching [368], or improved tensile strength and good water repellence (contact angle 120°) in combination with titanium butoxide catalyst [369]. The chemical interactions between the silica and cellulose fibres involve hydrogen bonding between silanol and hydroxyl functional groups [370]: however, the bonding and adhesion of the coating is often weak and is destroyed in liquids [371]. Therefore, a more stable polymeric network should be formed on the cellulose fibres: the hydrolysis of the alkoxysilanes during the sol–gel reaction can be followed by a polycondensation among these groups with residual alkoxy to form a network structure. This method can eventually be used in combination with silane coupling agents, or with synthesised silane polymers to promote the cross-linking and the stability of the coating on cellulosic textiles [372]. It is also used in combination with nonfluorinated alkylsilane on cotton substrates [373], which influences the reactivity of the sol–gel especially for modification of hydrophilic cellulose [374]. The sol–gel method can easily be applied to form hybrid deposits onto cellulose together with fluorocarbons [375], urea [376] or silica nanoparticles (Fig. 23c) [377]. The hybrid organic–inorganic coatings deposited in combination with polycarboxylic acids may improve the durability of the hydrophobic cellulose [378], as the acid works as a catalyst in preparing the sol–gel and serves as a cross-linker in promoting esterification between the silica layer and cellulose: e.g. polycarboxylic acids such as butanetetracarboxylic acid (BTCA) promote the anchoring of a silica nanoparticle coating onto cellulose with contact angles of 137° (Fig. 23d) [379]. By changing the organic components in the sol–gel in terms of the alkoxysilane precursor chain length, the surface energy and wetting on paper could be adjusted: the longer alkyl chains generally reduce the wettability (contact angle 80° for short to 100° for long chains) and make the absorptivity of papers more effectively. The hydrophobic effect of silica sols with long alkyl chains is well established and relates to a specific surface conformation (Fig. 23e) [380, 381]. The properties and processing of the sol–gel can be changed to tune the coating thickness and homogeneity. After deposition of sol–gel coatings by spraying and thermal curing, the paper surface could be mostly covered by silica and the cellulose was almost inaccessible for water, except near the fibre–fibre intersections that were only partially covered. The deposition of an ultrathin coating by the sol–gel method has indeed the advantage that the original structure of the cellulose fibres is narrowly followed, as demonstrated for a 2.5-nm titania film [382]. Subsequently, the sol–gel nanocoating can be functionalized by a solution-immersion process in order to form an outermost monolayer of self-assembled alkyl-silane that further improves the hydrophobicity (Fig. 23f) [383]: with this combination, the contact angle increases from 57° for an ultrathin titania sol–gel coating to 155° for the silane modified sol–gel coating with good long-term stability over a broad pH range. The high contact angles and self-cleaning properties are attributed to self-assembly of the monolayer coating without altering the initial fibre morphology.
The atomic layer deposition was used, e.g. for coating a thin aluminium oxide or zinc oxide layer on cellulose and consequently controlling the hydrophilic/hydrophobic wetting transitions [384]: the hydrophobicity was intrinsically improved by the formation of trimethylaluminium bonds and the wetting properties were finally affected by the exposure to air and carbon absorption. The RF-magnetron sputtering under argon gas was used for the deposition of a fluorocarbon coating onto regenerated cellulose films [385]: as such, a porous coating surface with undulated islands of about 100 nm grows vertically from the cellulose surface and includes nanometer particles. By changing the sputtering conditions, the static water contact angle was maximised at 95° under low power and high pressures, in agreement with the high concentration ratios [F]/[C] and THE presence of double bonds.
New developments in nanoparticle depositions
The functionalization of paper and cellulose with nanoparticles has become an emerging area that hugely increases the number of surface functionalities, as shown by a recent overview of various properties [386]. Especially for tuning hydrophobicity, a nano-engineering approach allows to mimic the Lotus-leaf effect, by inducing chemical modification and dual-scale surface roughness on cellulose for water repellency in paper and fabric applications [387].
Metallic nanoparticles are frequently deposited onto cellulose or paper surfaces. For example, a reduction reaction was used to cover cotton textiles with gold nanoparticles [388], but a direct chemical bond between gold and cellulose was not demonstrated. Some degree of hydrophobicity was obtained by dip-coating cellulose sheets in a superparamagnetic nanoparticle solution and cyanoacrylate, where manganese ferrite colloidal nanoparticles were deposited to provide contact angles of 120° to 140° [389]: after ageing of the solution, particle agglomeration consequently provides coatings with higher roughness and contact angles than fresh solutions (Fig. 24). Similarly, a thin hydrophobic film with titania nanoparticles was formed by dip-coating and drying [390]. The photocatalytic activity of titanium dioxide also breaks down pollutants and microorganisms, while providing superhydrophobicity and high opacity [391]: after hydrophobic modification of the titanium dioxide particles with a coupling agent, they effectively provide superhydrophobic properties when added to the pulp slurry under beating. In general, the small reactivity and low interaction between inorganic nanoparticles and cellulose is one concern when embedding nanoparticles into papers. To improve the reactivity and adherence, nanoparticles can be further coupled with an epoxy group (Fig. 25a) [392], or carboxylic acid can be used as a crosslinking agent that forms an ester linkage with the cellulose for embedding silica nanoparticles in a nanocomposite coating (Fig. 25b) [393, 394]. The nanoparticle deposition is often used in combination with pre- or post-functionalisation of their surface by grafting. The most preferred method for superhydrophobicity is the fluorination of nanoparticle surfaces, as studied for silica nanospheres [395]: the effects of nanoparticle size and surface coverage in an array of silica nanospheres were investigated and optimised in terms of a Cassie–Baxter wetting model. Superhydrophobic papers were also created by modifying silica nanoparticle coatings with perfluorooctyltriethylsilane [319]. Similarly, fluorinated alkylsilane-coated silica nanoparticles were prepared by a co-hydrolysis reaction [396]. A multi-step nano-engineering process was applied by adsorbing amorphous silica particles onto cellulose, followed by grafting of the nanoparticles with a fluorine-bearing silane coupling agent (Fig. 25c) [280]: this processing route is an alternative for the previously mentioned direct grafting of cellulose surfaces, where the increase in fibre surface roughness by nanoparticle deposits obviously favours superhydrophobicity. The latter particle adsorption and surface roughness on cellulose could be specifically controlled by the deposition of multiple LbL coatings, providing good stability and finally a positive surface charge. Similar properties could also be obtained after functionalization of nanoparticles in the presence of perfluorooctylated quaternary ammonium silane coupling agent [397], or with nonfluorinated hydrophobic polymers such as epoxy and subsequent grafting with SMA (Fig. 25d) [398]. After the in situ introduction of silica nanoparticles, subsequent hydrophobization with polydimethylsiloxane (PDMS) renders the cellulose superhydrophobic (contact angles 155°), while hydrophobization with perfluoroalkyl also provides additional oleophobicity (contact angle 140° for sunflower oil) [399]. Otherwise, mesoporous silica nanoparticles were formed in a one-pot co-condensation reaction and functionalized with tridecafluoro-octyltriethoxysilane [400]: the nanoparticle surface properties can be well controlled as a higher amount of organosilane during the reaction leads to a more radially branched wormhole-like mesoporosity, a decrease in the surface area, pore volume and amount of surface silanol groups, and an enrichment of the surface with more fluorocarbon moieties.

Optical microscopy of cellulose sheets treated with a solution of MnFe2O4 nanoparticles in toluene, a dip-coating in fresh solution, b dip-coating in aged solution with agglomerated nanoparticles (from [389])

Nanoparticle deposition onto cellulose fibres or paper substrates and further functionalization, a in the presence of an epoxy group (from [392]), b in the presence of carboxylic acid used for additional esterification of embedded nanoparticles (from [393]), c by adsorption of SiO2 nanoparticles and perfluorination (from [280]), d by modification of SiO2 nanoparticles with epoxy groups and grafting with styrene maleic anhydride (from [398])
In parallel with the techniques presented before, the stepwise decoration of cellulose fibres with metallic nanoparticles can be done from a sol–gel, e.g. using silica sol from tetraethoxysilane under alkaline conditions followed by hydrophobization with hydrolyzed hexadecyltrimethoxysilane (Fig. 26) [401]: both the higher surface roughness and the presence of long alkyl chains then provide contact angles as high as 155°, and acceptable properties after several washing cycles. Another two-step reaction for the in situ growth of silica nanoparticles from a sol–gel results in monodisperse particles that create a dual-scale roughness profile on cotton fabrics with contact angles up to 150° [402]. A vapour deposition reaction was used for adding Al2O3 nanoparticles onto a cotton fabric, followed by a molecular vapour deposition of tri(decafluoro-1,1,2,2,-tetrahydrooctyl)-trichlorosilane [403]: as such, the dynamic contact angles were above 150° and hysteresis was low. A direct deposition under audio frequency plasma created nanoparticulate coatings onto a cotton fabric, in combination with a heat treatment to increase superhydrophobicity [404].

Formation of a superhydrophobic surface on cellulose fibre based on coating with a silica sol and long-chain alkyl groups (from [401])
A solution-immersion method was used for the formation of polymethylsilsesquioxane coatings with a micro- to nanoscale structure onto cellulose, by the deposition of a nanoparticle layer under reaction of potassium methyl siliconate and CO2 (Fig. 27) [405]. The morphology of these nanostructures and also their covering density was strongly affected by the reaction duration. The reaction conditions consequently influence the surface hydrophobicity of cotton fabrics, which can change between sticky and slippery states [406]. The polymethylsilsesquioxane coatings deposited from water glass form a dense heterogeneous and cross-linked network of nanofilaments, and provide a certain roughness profile to the surface with good robustness of the superhydrophobic properties [407].

The spray deposition of hydrophobic silica nanoparticles from an alcohol suspension created a coating with high water repellency and transparency onto papers [408], depending on the aggregation state of the particles and alcohol solvent. After the immobilisation of silane groups onto the silica nanoparticle surfaces (e.g. with dodecyltrichloro- or octadecyltrichloro-silane), a suitable nanoparticle suspension for spray deposition could be produced. It could introduce suitable low surface energy and surface roughening to paper surfaces in a single step process, with future potentials for industrial upscaling (Fig. 28a) [409]: water droplets with contact angles of 163° and almost no sticking effects were demonstrated for highly transparent papers. After spraying a nanocomposite film or organoclays, the wetting characteristics can be further manipulated through ink-jet or laser printing of solid grey patterns [410]: for different ink intensities (0–85 %), the mobility of water droplets can be tuned by changing the amount of ink/toner. A thermal liquid flame spraying processes under atmospheric conditions is used to deposit SiO2, TiO2, Al2O3 or ZrO2 nanoparticles from a liquid precursor in isopropyl alcohol, together with hydrogen or oxygen as combustion gas (Fig. 28b) [411]: as a result, packaging surface materials rendered appropriate superhydrophobic as the wettability increased due to surface roughness on the micro- and nanoscale level [412, 413]. The composition and thickness of such sprayed nanocoatings can be tailored from the feed rate of the precursor solutions, burner distance and line speed. Moreover, this coating is active after UV illumination and becomes completely wettable by photocatalytic decomposition [414].

Carbon nanotubes (CNT) have been used in a pristine and/or surface-modified form to mimic the Lotus-leaf effect on cotton textiles, with water contact angles above 150° (Fig. 29) [415]: the affinity of CNT with cellulose substrates was improved by grafting with a 8–10 nm shell of poly(butylacrylate) [416], and afterwards CNT were deposited by dip-coating under ultrasonic irradiation. A uniform, free-standing nanohybrid buckypaper was made with a high range of CNT content (13–70 %), using polymer single crystal-decorated CNT as precursor from a uniform nanohybrid suspension [417]. The superhydrophobicity and high surface water adhesion mimics the rose petal effect.

Illustration of surface modification of cellulose fibre by deposition of carbon nanotubes (CNT) using ultrasonic irradiation procedure (from [415])
Recently, aqueous dispersions of organic nanoparticles with good hydrophobicity were developed as environmentally friendly and recyclable modifiers for paper substrates. The aqueous system has been adapted with favourable rheological properties and is consequently compatible with common paper coating technologies. The SMA copolymers are known for good ink receptivity and are commonly used for internal sizing, without presenting specific hydrophobicity at the paper surface. After transformation into nanoparticles by imidization, the location of hydrophobic sites within the nanoparticles can be well controlled. First, the synthesis was optimised to obtain stable particle dispersions with maximum solid content and appropriate viscosity [418]. These materials were successfully applied by a bar-coating process onto paper and paperboard surfaces to study the chemical and morphological properties and printability (Fig. 30a) [419]. An important factor in this analysis is the multi-scale modeling of roughness parameters [158]: by combining the roughness measurements with a compositional analysis, a calibration model for tuning wettability of nanoscale-coated paper surfaces can be predicted as a function of the chemical composition and nanoscale surface roughness, resulting in maximum contact angles of 150°–160° according to the Wenzel model (Fig. 30b) [420]. A multi-scale roughness profile can be created by means of polymer nanotechnology in combination with fibrous surfaces to form superhydrophobic paper surfaces similar to the Lotus-leaf effect [421]. The application of nanoscale structures allows precise control of the location of the desired functionalities: e.g. the hydrophobic imide groups are precisely located at the outer surface of the nanoparticles [422]. This is an advantage in respect to the incorporation of bulk polymers, which likely diffuse into the bulk of the paper and are consequently not present at the surface.

Deposition of organic styrene(maleimide) nanoparticles onto a paper surface and hydrophobic properties, a micrograph of a 2-μm-thin porous coating and AFM topography, b relation of the contact angle with chemical surface composition (degree of imidization of the nanoparticles) and nanoscale surface roughness influencing the roughness parameter according to the Wenzel model (from [420])
New developments in sustainable methods
The growing demand for ‘green’ products and ‘sustainable’ paper processing requires environmentally friendly and economic modification processes that are compatible with the natural image of cellulose. The barrier properties and hydrophobicity of papers is nowadays mainly controlled by adding wax or fluorine derivates, as illustrated in previous paragraphs. However, waxed surfaces are difficult to glue and require additional corona or plasma treatment for preserving good printing properties [423], while waxes also tend to migrate during the drying process and form a continuous film holding the fibres together and reducing repulpability. The poor recyclability is a major drawback: the presence of wax affects bonding of recycled pulps, as about 1–2 % wax decreases the pulp fibre strength by around 10 %, or they can only be removed after sophisticated treatments [424]. On the other hand, the Food and Drug Administration (FDA) regulations limit the fluorine content in paper and paperboards in contact with aqueous and fatty foods to be within 0.09–0.26 % depending on the paper weight [425]. Otherwise, the use of solvents should be replaced by water-based suspensions and renewable resources such as fatty acids and biopolymers should be incorporated.
Fatty acids and derivatives from vegetable oils may favourably serve as hydrophobic moieties for modification of pulp and cellulose surfaces. The fatty acid chemistry was used for internal sizing of never-dried cellulose pulp [426], including aluminium sulphate and sodium soaps of various fatty acids with long alkyl chains up to C18. While the length of the aliphatic fatty acid chain positively affected hydrophobicity, the papers treated with fatty acid soaps show an opposite trend with contact angles depending on the degree of saturation and number of hydroxyl groups. In general, the addition of fatty acid soaps into the pulp had a limited effect, as they are restricted to the fibre surface, while the internal part retained its original hydrophilicity in combination with a relatively high WRV. Therefore, the WRVs could be improved in combination with good hydrophobicity (contact angles up to 117°) by formation of a cellulosic fibre network, e.g. by cross-linking the cellulose fibres with citric acid in a specific concentration range and subsequent thermal curing at 150 °C. The triglycerides from plants were also successfully applied to produce hydrophobic cellulose fibres by transesterification (Fig. 31a) [427]. In parallel, cellulose nanocrytals (i.e. nanowhiskers) were hydrophobized via covalent grafting of castor oil, with the polar component of the surface energy reduced from 21.5 mJ/m2 to almost zero with water contact angles of 96° [428]. Recently, a new category of hybrid nanoparticles with different encapsulated vegetable oils were synthesised: they can be adsorbed onto single cellulose fibres to improve the hydrophobicity (Fig. 31b), or used as additives in paper coatings with contact angles up to 160° (Fig. 31c) [429]. The nanoparticles favourably adsorbed onto cellulose with good adhesion attributed to hydrogen bonding, confirmed by spectroscopy.

Hydrophobicity of cellulose fibres and papers provided by vegetable oil derivatives, a direct transesterification between cellulose and triglycerides (from [427]), b adsorption of nanoparticles with incorporated vegetable oils onto a single cellulose fibre [421], c adsorption of nanoparticles with incorporated vegetable oils onto a paper surface
Biopolymers are gaining importance for paper modification and recently, several researchers started to tune the water resistance properties using, e.g. starch, cellulose derivates, chitosan, alginate, wheat gluten, whey proteins, polycaprolactone, poly(lactic) acid (PLA) and polyhydroxy-alkanoates, as reviewed before [430, 431]. After coating a paper with a chitosan solution, the water barrier properties were improved as observed by NMR-relaxometry analysis (Fig. 32a) [432]. A blend of chitosan–palmitic acid (Fig. 32b) or chitosan–dipalmitoylchitosan (Fig. 32c) resulted in even better hydrophobicity and stability for packaging paper [433]. The chitosan was combined in bilayer coatings with palmitic acid, resulting in reduced water vapour permeability rate (WVPR) and water absorption capacity [434]. Several layers of chitosan and water-soluble chitosan coatings had also a remarkable impact on the water vapour permeability of the coated papers [435], depending on the number of deposited chitosan layers.

Two other biopolymers received attention for improving the water resistance and water barrier properties of papers and paperboards. A PLA coating may be used by providing smooth surfaces for one-way paper cups [436, 437]. Also polyhydroxyalkanoate (PHA) is favourably used for sizing and coating of papers, and several copolymers of polyhydroxybutyrate (PHB) were developed as sizing agents for paper [438]. The adhesion of PHB onto cellulosic surfaces should be controlled and can be enhanced after acetylation of the cardboard with acetic acid [439], while the water barrier properties finally depend on the concentration of PHB in the coating. In addition, a polyhydroxybutyrate–valerate (PHBV) copolymer showed better oxygen and water vapour resistance than pure PHB [440]. The PHB is also used in a phase-separation technology to generate an adequate micro/nanosized structure after precipitation on cellulose, which creates superhydrophobic properties [441]: the wettability of the substrates (contact angle 153°) can subsequently be controlled by adequate argon plasma treatment in certain regions under application of a mask.
The blending or assembly of biopolymer coatings remarkably improved the moisture barrier properties and WVTR up to 95 % [442]: e.g. whey protein isolate/cellulose and poly(vinyl butyral)/zein coatings were used in a bilayer configuration with beeswax. The blending of a whey protein isolate or chitosan together with poly(ε-caprolactone) further improved the water barrier properties of coated paper by lowering the WVTR by 70–90 % [443]. A blend of an enzyme (laccase) and hydrophobic phenolic compounds resulted in best water resistance when lauryl gallate was used [444], as many other additives additionally needed thermal curing to reduce the water absorption. In a more bio-inspired assembly of lignin in conjunction with cationic polyelectrolyte layers adsorbed to cellulose surfaces, the adhesion of water towards hydrophilic substrates decreased and thus the hydrophobicity improved [445]. Bioinspired superhydrophobicity could be created onto rough polylactide surfaces [446, 447], or chitosan films [448] using a phase inversion-based deposition method.
The field of biopolymer coatings recently expanded and a variation of renewable resources is currently investigated. However, suitable processing of the biopolymer blends remains a most critical issue for further process industrialization.
Conclusions and outlook
Due to the hydrophilic nature of cellulose fibres, a hydrophobic barrier is needed to extend the range of paper products and include articles that will be in contact with liquids. The interaction of cellulose and papers with water is extensively studied at different levels ranging from the molecular cellulose structure, the organisation and properties from a microfibrillar to macroscale fibre level, towards the behaviour of cellulose films and porous structure of the paper web itself. Therefore, several phenomenological and analytical models have been developed and continuously improved during last decades by introducing supplementary relevant parameters, resulting in some modeling studies of liquid interactions with porous structures. Besides the phenomena of penetration under capillary pressure, interfibre penetration and molecular diffusion also effects of gravity and fibre swelling are considered in different environments. Besides static interactions of liquids in relation with surface energy, mainly the dynamics of wetting in combination with flow and absorption of liquid droplets has been extensively investigated for paper. Due to the inhomogeneity of paper, however, the wettability studies are more difficult and mostly based on experimental observations with consequent analysis of the surface conditions and capillary action in porous media. As such, the surface features can be tuned from an experimental approach in order to create desired static and dynamic water contact angles. The specific surface chemistry, especially in combination with the inherent hierarchical fibre morphology and surface roughness, contribute to multi-scale roughness features enhancing (super)hydrophobic properties.
The traditional methods of internal sizing and surface sizing provide a first barrier against water, but often cannot meet the requirements for modern packaging applications. Therefore, the progress in internal or surface sizing and effects of some functionalized additives were reviewed. Recent advances include micro- to nanosized structuring of internal sizing agents and surface sizing additives in parallel with the development of new copolymer formulations: new nanocellulose and clay additives are promising additives for further improving the water resistance after dedicated surface modification. However, the direct chemical and physical modifications of the cellulose fibres and/or use of surface coatings are needed for creating higher hydrophobicity or superhydrophobicity. Therefore, a broad overview is given for possible surface hydrophobization of cellulose, including graft polymerization, LbL absorption, solution casting or dip-coating, plasma modification, electrostatic spraying, sol–gel coating, atomic layer deposition. New developments in nanoparticle surface depositions include metallic and organic nanoparticle modifications. However, more sustainable alternatives for the traditional treatments with fluorine derivates are needed. Renewable feedstocks for surface hydrophobization include, e.g. vegetable oils, fatty acids and a range of biopolymers.
Apart from the overview above, the development of 100 % bio-based nanocomposite coatings for papers that combine superhydrophobicity with good processability and functionality is an emerging and recent domain of scientific research. This can likely be achieved by combining selected bio-based polymers and/or polymer blends as a matrix with dedicated functional bio-based filler materials at the micro- to nanometer scale levels. Therefore, a main future challenge lies in the development of suitable interface modification techniques that create compatible mixtures, where the barrier properties are provided by exploiting the specific morphology at different scale lengths and distribution in the coating. Therefore, both the surface morphology and the chemistry need to be controlled independently. The creation of new coating systems is a challenging task that requires a multidisciplinary approach with backgrounds in materials science, polymer chemistry, nanotechnology and mechanical sciences. Moreover, novel applications are not limited to innovative paper products but there exists lots of overlap with the need for water-resistant textile materials, composites, electrospun membranes, etc.
References
Strachan J (1938) Nature 141:332
Suzuki T, Nakagami H (1999) Eur J Pharm Biopharm 47:225
Medronho B, Romano A, Miguel MG, Stigsson L, Lindman B (2012) Cellulose 19:581
Mazeau K, Heux L (2003) J Phys Chem B 107:2394
Ciolacu D, Popa VI (2010) Cellulose allomorphs: structure, accessibility and reactivity. Nova Science Publishers, New York. ISBN 978-1-61668-323-8
Filpponen I, Argyropoulos DS (2008) Ind Eng Chem Res 47:8906
Verlhac C, Dedier J, Chanzy H (1990) J Polym Sci A 28:1171
Chinga-Carrasco G (2011) Nano Res Lett 6:417
Terinte N, Ibbett R, Schuster KC (2011) Lenzing Ber 89:118
Wada M, Okano T, Sugiyama J (1997) Cellulose 4:221
Viëtor RJ, Mazeau K, Lakin M, Perez S (2000) Biopolymers 54:342
Mazeau K (2011) Carbohydr Polym 84:524
Mazeau K, Vergelati C (2000) Langmuir 18:1919
Venkatarama P, Ashbaugh H, Johnson GP, French AD (2010) In: Proceedings of the national cotton council beltwide cotton conference, January 5–7, 2010, New Orleans, Louisiana, p 1577
Biermann O, Hadicke E, Koltzenburg S, Muller-Plathe F (2001) Angew Chem Int Ed 40:3822
Mazeau K, Rivet A (2008) Biomacromolecules 9:1352
Li Y, Lin M, Davenport JW (2011) J Phys Chem C 115:11533
Viëtor RJ, Newman RH, Ha MA, Apperley DC, Jarvis MC (2002) Plant J 30:721
Larsson PT, Wickholm K, Iversen T (1997) Carbohydr Res 302:19
Bergenstrahle M, Wohlert J, Larsson PT, Mazeau K, Berglund LA (2008) J Phys Chem B 112:2590
Kuutti L, Peltonen J, Pere J, Teleman O (1995) J Microsc 178:1
Da Silva Perez D, Ruggiero R, Morais LC, Machado AEH, Mazeau K (2004) Langmuir 20:3151
Nakamura K, Hatakeyama T, Hatakeyama H (1981) Text Res J 51:607
Bechtold T, Manian AP, Öztürk HB, Paul U, Široká B, Široký J, Soliman H, Vo LTT, Manh HV (2013) Carbohydr Polym 93:316
Maloney TM, Paulapuro H (1998) Nord Pulp Paper Res J 13:31
Kaewnopparat S, Sansernluk K, Faroongsarng D (2008) AAPS PharmSciTech 9:701
Hatakeyama T, Tanaka M, Hatakeyama H (2010) J Biomater Sci Polym Ed 21:1865
Pouchly J, Biros J, Benes S (1979) Makromol Chem 180:745
Deodhar S, Luner P (1980) ACS Symp Ser 127:273
Hubbe MA, Rojas OJ, Lucia LA, Jung TM (2007) Cellulose 14:655
Pelton R (1993) Nord Pulp Paper Res J 8:113
Heikkinen S, Alvila L, Pakkanen T, Saari T, Pakarinen P (2006) J Appl Polym Sci 100:937
Ogiwara Y, Kubota H, Hayashi S (1970) J Appl Polym Sci 14:303
Vyas S, Pradhan SD, Pavaskar NR, Lachke A (2004) Appl Biochem Biotechnol 118:177
Kittle JD, Du X, Jiand F, Qian C, Heinze T, Roman M, Esker AR (2011) Biomacromolecules 12:2881
Svedas V (1998) J Phys D Appl Phys 31:1752
Svedas V (2000) Appl Spectrosc 54:420
Banik G, Bruckle I (2011) Restaurator 31:164
Hubbe M, Heitmann JA (2007) BioResources 2:500
Li X, Ballerini DR, Shen W (2012) Biomicrofluidics 6:011301 (13 pp)
Chinga-Carrasco G (2009) J Microsc 234:211
Hoyland RW, Field R (1976) Paper Technol Ind October:213
Scallan AM (1977) Transactions of BPBIF Symposium 1. Oxford
Oliver JF, Agbezuge L, Woodcock K (1994) Colloid Surf A 89:213
Eklund D, Lindström T (1991) Paper chemistry: an introduction. DT Paper Science Publications, Grankulla
Ramarao SV, Massoquete A, Lavrykov S, Ramaswamy S (2003) Dry Technol 21:2007
Ouriadov A, Newling B, Batchelor SN (2008) J Phys Chem C 112:15860
Salminen P (2008) Ph.D. thesis, Abo Akademi University
Wu N, Hubbe MA, Rojas OJ, Park S (2009) BioResources 4:1222
Alleborn N, Raszillier H (2007) Tappi J 6:16
Clarke A, Blake TD, Carruthers K, Woodward A (2002) Langmuir 18:2980
Davies SH, Hocking LM (1999) Phys Fluids 11:48
Yazdchi K, Srivastava S, Luding S (2011) Int J Multiphase Flow 37:956
Tamayol A, Bahrami M (2009) Int J Heat Mass Transf 52:2407
Nabovati A, Llewellin EW, Sousa ACM (2009) Composites A 40:860
Reverdy-Bruas NR, Serra-Tosio JM, Chave Y, Bloch JF (2001) Dry Technol 19:2421
Nilsson L, Stenström S (1997) Int J Multiphase Flow 23:131
Stylianopoulos T, Yeckel A, Derby JJ, Luo XJ, Shephard MS, Sander EA, Barocas VH (2008) Phys Fluids 20:123601
Jaganathan S, Tafreshi HV, Pourdeyhimi B (2008) Sep Sci Technol 43:1901
Tahir MA, Tafreshi HV (2009) Phys Fluids 21:083604 (5 pp)
Zobel S, Maze B, Tafreshi HV, Wang Q, Pourdeyhimi B (2007) Chem Eng Sci 62:6285
Salminen PJ (1988) Tappi J 9:195
Perkins EL, Batchelor WJ (2012) Carbohydr Polym 87:361
Kissa E (1996) Text Res J 66:660
Lucas R (1918) Kolloid Z 23:15
Washburn EV (1921) Phys Rev 17:273
Siebold A, Nardin M, Schultz J, Walliser A, Oppliger M (2000) Colloid Surf A 161:81
Lavi B, Marmur A, Bachmann J (2008) Langmuir 24:1918
Bosanquet CH (1923) Philos Mag Ser 6:525
Joos P, Van Remoortere P, Bracke M (1990) J Colloid Interface Sci 136:189
Ichikawa N, Satoda Y (1994) J Colloid Interface Sci 162:350
Moshinskii AI (1997) Colloid J 59:62
Hoyland RW (1977) Transactions of the 6th fundamental research symposium. Pulp and Paper Fundamental Research Society, Oxford
Schuchardt DR, Berg JC (1990) Wood Fiber Sci 23:342
Wiryana S, Berg JC (1991) Wood Fiber Sci 23:456
Davies SH, Hocking LM (2000) Phys Fluids 12:1646
Middleman S (1995) Modeling axisymmetric flows: dynamics of films, jets, and drops. Academic Press, San Diego
Szekely J, Neumann AW, Chuang YK (1971) J Colloid Interface Sci 35:273
Ridgway CJ, Grane PAC, Schoelkopf J (2002) J Colloid Interface Sci 252:373
Danino D, Marmur A (1994) J Colloid Interface Sci 166:245
Hodgson KT, Berg JC (1988) J Colloid Interface Sci 121:22
Williams JG, Morris CM, Ennis BC (1979) Polym Eng Sci 14:413
McDonald P (2006) Dissertation, Georgia Institute of Technology, Atlanta
Oliver JF, Mason SG (1977) J Colloid Interface Sci 60:480
Masoodi R, Pillai KM, Varanasi PP (2007) AIChE J 53:2769
Masoodi R, Pillai KM, Varanasi PP (2010) J Eng Fibers Fabr 5:49
Gillespie T (1958) J Colloid Sci 13:32
Borhan A, Rungta KK (1993) J Colloid Interface Sci 158:403
Masoodi R, Tan H, Pillai KM (2011) AIChE J 57:1132
Masoodi R, Tan H, Pillai KM (2012) AIChE 58:2536
Bousfield DW, Karles G (2004) J Colloid Interfac Sci 270:396
Enomae T, Lepoutre P (1997) J Pulp Paper Sci 23:1
Azimi Y, Kortschot MT, Farnood R (2011) Appita J 64:428
Stone JE, Scallan AM (1968) Cellul Chem Technol 2:343
Wistara N, Young RA (1999) Cellulose 6:291
Neuman RD, Berg JM, Claesson PM (1993) Nord Pulp Paper Res J 8:96
Berg JC (1993) Nord Pulp Paper Res J 8:75
Erbil HY (1997) Turk J Chem 21:332
Lyne MB (1993) Nord Pulp Paper Res J 8:120
Young T (1805) Philos Trans R Soc Lond 95:65
Marmur A (1996) Colloids Surf 116:55
Marmur A (1996) Langmuir 12:5704
Wenzel TN (1949) J Phys Colloid Chem 53:1466
Cassie A, Baxter S (1949) Trans Faraday Soc 40:546
Huh C, Mason SG (1977) J Colloid Interface Sci 60:11
Taniguchi M, Belfort G (2002) Langmuir 18:6465
Dubé M, Chabot B, Daneault C, Alava M (2005) Pulp Paper Can 106:178
Holownia D, Kwiatkowska I, Hupka J (2008) Physicochem Probl Miner Process 42:251
Liukkonen A (1997) Scanning 19:411
Stor-Pellinen J, Haeggström E, Luukkala M (2000) Ultrasonics 38:953
Cazabat AM (1989) Nord Pulp Paper Res J 2:146
De Coninck J, De Ruijter MJ, Voué M (2001) Curr Opin Colloid Interface Sci 6:49
De Ruijter MJ, De Coninck J, Oshanin G (1999) Langmuir 15:2209
Von Bahr M, Tiberg F, Yaminsky V (2001) Colloids Surf A 193:85
Von Bahr M, Tiberg F, Zhmud BV (1999) Langmuir 15:7069
Apel-Paz M, Marmur A (1999) Colloids Surf A 146:273
Tanner H (1979) J Phys D Appl Phys 12:1473
Lopez J, Miller CA (1976) J Colloid Interface Sci 56:460
Blake TD (1993) In: Berg JC (ed) Wettability. Surfactant science series, vol 49. Marcel Dekker, New York, p 251
De Gennes PG (1985) Rev Mod Phys 57:827
Kistler S (1993) In: Berg JC (ed) Wettability. Marcel Dekker, New York
Huh C, Scriven LE (1971) J Colloid Interface Sci 35:85
Cox RG (1986) J Fluid Mech 168:169
Blake TD, Clarke A, De Coninck J, De Ruijter MJ (1997) Langmuir 13:2164
Bertrand E, Blake TD, De Coninck J (2009) J Phys Condens Matter 46:464124
Rodriguez-Valverde MA, Ruiz-Cabello FJM, Cabrezio-Vilchez MA (2008) Adv Colloid Interface Sci 138:84
Arzate A, Tanguy PA (2005) Nord Pulp Paper Res J 20:217
Herminghaus S, Brinkmann M, Seemann R (2008) Annu Rev Mater Res 38:101
Kissa E (1981) J Colloid Interface Sci 83:265
Marmur A (1997) J Colloid Interface Sci 186:462
Modaressi H, Garnier G (2002) Langmuir 18:642
Lyne MB, Aspler JS (1982) In: Hair M, Croucher MD (eds) Colloids and surfaces in reprographic technology, vol 20. American Chemical Society, Washington, DC, p 385
Tag CM, Toiviainen M, Juuti M, Rosenholm JB, Backfolk K, Gane PAC (2012) Transp Porous Media 94:225
Aydemir C (2010) Int J Polym Mater 59:387
Ridgway CJ, Gane PAC (2003) Nord Pulp Paper Res J 18:24
Ridgway CJ, Gane PAC (2002) Colloids Surf A 206:217
Järnström J, Järn M, Tag CM, Peltonen J, Rosenholm JB (2011) J Adhes Sci Technol 25:761
Järn M, Tag CM, Järnström J, Rosenholm JB (2010) J Adhes Sci Technol 24:567
Marmur A (1992) Adv Colloid Interface Sci 39:13
Marmur A (1988) J Colloid Interface Sci 122:209
Bacri L, Brochard-Wyart F (2001) Europhys Lett 56:414
Bodurtha PA, Matthews GP, Kettle JP, Roy IM (2005) J Colloid Interface Sci 283:171
Schoelkopf J, Gane PAC, Ridgway CJ, Matthews GP (2000) Nord Pulp Paper Res J 15:422
Thorman S, Ström G, Hagberg A, Johansson PA (2012) Nord Pulp Paper Res J 27:459
Kannangara D, Zhang H, Shen W (2006) Colloids Surf A 280:203
Asai A, Shioya M, Hirasawa S, Okazaki T (1993) J Imaging Technol 37:205
Kannangara D, Shen W (2008) Colloids Surf A 330:151
Hare EF, Shafrin EG, Zisman WA (1954) J Phys Chem 58:236
Pittman AG, Sharp DL, Ludwig BA (1968) J Polym Sci A 6:1729
Nishino T, Meguero M, Nakamae K, Matsushita M, Ueda Y (1999) Langmuir 15:4321
Peltonen J, Jârn M, Areva S, Linden M, Rosenholm JB (2004) Langmuir 20:9428
Stor-Pellinen J, Haeggström E, Karppinen T, Luukkala M (2001) Meas Sci Technol 12:1336
Schuttleworth R, Bailey GLJ (1948) Discuss Faraday Soc 3:16
Wagberg L (2000) Nord Pulp Paper Res J 15:598
Oliver JF, Huh C, Mason SG (1980) Colloids Surf 1:79
Mammen L, Deng X, Untch M, Vijayshankar D, Papadopoulos P, Berger R, Riccardi E, Leroy F, Vollmer D (2012) Langmuir 28:15005
Onda T, Shibuichi S, Satoh N, Tsujii K (1996) Langmuir 12:2125
Samyn P, Van Erps J, Thienpont H, Schoukens G (2011) Appl Surf Sci 257:5613
Giljean S, Bigerelle M, Anselme K, Haidara H (2011) Appl Surf Sci 257:9631
Gao L, Mc Carthy TJ (2007) Langmuir 23:3762
Mc Hale G (2007) Langmuir 23:8200
Oliver JF, Huh C, Mason SG (1977) J Adhes 8:223
Wolansky G, Marmur A (1999) Colloids Surf A 156:381
Neumann AW, Good RJ (1972) J Colloids Interface Sci 38:341
Pease DC (1945) J Phys Chem 49:107
Bartell FE, Shephard JW (1953) J Phys Chem 57:211
Bartell FE, Shepard JW (1953) J Phys Chem 57:455
Furmidge CGL (1962) J Colloid Sci 17:309
Youngblood JP, McCarthy TJ (1999) Macromolecules 32:6800
Israelachvili JN (2010) Intermolecular and surface forces, 3rd edn. Elsevier, Amsterdam
Chen W, Fadeev AY, Hsieh MC, Oner D, Youngblood J, McCarthy TJ (1999) Langmuir 15:3395
Extrand CW (2002) Langmuir 18:7991
Oner D, McCarthy TJ (2000) Langmuir 16:7777
Chen YL, Helm CA, Israelachvili JN (1991) J Phys Chem 95:10736
Priest C, Sedev R, Ralston J (2007) Phys Rev Lett 99:026103
Extrand CW (2003) Langmuir 19:3793
Li L, Breedveld V, Hess DW (2013) Colloid Polym Sci 291:417
Dettre RH, Johnson RE (1965) J Phys Chem 69:1507
McHale G, Shirtcliffe NJ, Newton MI (2004) Langmuir 20:10146
Parkin IP, Palgrave RG (2005) J Mater Chem 15:1689
Verplanck N, Coffinier Y, Thomy V, Boukherroub R (2007) Nanoscale Res Lett 2:577
Barthlott W, Neinhuis C (1997) Planta 202:1
Neinhaus C, Barthlott W (1997) Ann Bot Lond 79:667
Otten A, Herminghaus S (2004) Langmuir 20:2405
Gao X, Jiang L (2004) Nature 432:36
Wagner T, Neinhaus C, Barthlott W (1996) Acta Zool 77:213
Sun TL, Feng L, Gao XF, Jiang L (2005) Acc Chem Res 38:644
Burton Z, Bhushan B (2006) Ultramicroscopy 106:709
Bhushan B (2012) Langmuir 28:1698
Nosonovsky M, Bhushan B (2008) J Phys Condensed Matter 20:225009
Shibuichi S, Onda T, Satoh T, Tsujii N (1996) J Phys Chem 100:19512
Bhushan B, Koch K, Young YC (2008) Appl Phys Lett 93:093101
Liu Y, Chen X, Xin JH (2006) Nanotechnology 17:3259
Tang KG, Yu JH, Zhao YY, Liu Y, Wang XF, Xu RR (2006) Mater Chem 16:1741
Roach P, Shirtcliffe NJ, Newton MI (2008) Soft Matter 4:224
Feng L, Li S, Li Y, Li H, Zhang L, Zhai J, Song Y, Liu B, Jiang L, Zhu D (2002) Adv Mater 14:1587
Miwa M, Nakajiam A, Hashimoto K, Watanabe T (2000) Langmuir 16:5754
Meng H, Weng S, Xi J, Tang Z, Jiang L (2008) J Phys Chem C 112:11454
Zhao B, Collinson MM (2010) Chem Mater 22:4312
Han J, Wang X, Wang H (2008) J Colloid Interface Sci 326:360
Norton FJ (1945) US Patent Number 2386259, Serial Number 452,885
Gao L, McCarthey TJ (2006) Langmuir 22:5998
Balu B, Kim JS, Breedveld V, Hess DW (2009) Design of superhydrophobic paper/cellulose surfaces via plasma enhanced etching and deposition, contact angle: wettability and adhesion, vol 6. Elsevier, Leiden, p 235
Balu B, Breedveld V, Hess DW (2008) Langmuir 24:4785
Balu B, Kim JS, Breedveld V, Hess DW (2009) J Adhes Sci Technol 23:361
Kusumaatmaja H, Yeomans JM (2007) Langmuir 23:6019
Xiu Y, Zhu L, Hess DW, Wong CP (2008) J Phys Chem 112:11403
Hubbe MA (2006) Bioresources 2:106
Gess JM, Rodrigues JM (2005) The sizing of paper, 3d edn. Tappi Press, Atlanta. ISBN 1-59510-073-3
Wang F, Tanaka H, Kitaoka T, Hubbe MA (2000) Nord Pulp Paper Res J 15:416
Strycker LY, Thomas BD, Matjivic E (1973) J Colloid Interface Sci 43:319
Zou Y, Hsieh J, Wand TS, Mehnert E, Kokoszka J (2004) Tappi J 3:16
Lindström T, Larsson PT (2008) Nord Pulp Paper Res J 23:202
Tyagi M, Bhadra K, Goswami S, Agarwal NK (2007) Ippta J 19:143
Mattson R, Sterte J, Ödberg L (2001) In: Barker CF (ed) The science of papermaking.: transactions of the 12th Fundamental Research Symposium, Oxford, p 393
Kumar AA, Panlaj K, Sanjeev G (2012) Res J Chem Environ 16:31
Eklund D, Lindström T (1991) Paper chemistry: an introduction. DT Paper Science Publications, Grankulla
Roberts J (1991) Paper chemistry. Chapmann and Hall, New York, p 114
Ödberg LT, Lindström T, Liedberg B, Gustavsson J (1987) Tappi J 70:135
Zule J, Dolenc J (2005) Mater Technol 39:1
Garnier G, Wright J, Godbout L, Yu L (1998) Colloids Surf A 145:153
Lindström T, Sonderberg G (1986) Nord Pulp Paper Res J 1:26
Lindström T, O’Brian H (1986) Nord Pulp Paper Res J 2:31
Garnier G, Bertin M, Smrckova M (1999) Langmuir 15:7863
Seppänen R, Tiberg F, Valignat MP (2000) Nord Pulp Paper Res J 15:452
Karademir A (2002) Turk J Agric For 26:253
Von Bahr M, Seppänen R, Tiberg F, Zhmud B (2004) J Pulp Paper Sci 30:74
Quanxiao L, Wencai X, Yubin I (2011) Adv Mater Res 332–334:1872
Shen W, Filonanko Y, Truong Y, Parker IH, Brack N, Pigram P, Liesegang J (2000) Colloids Surf A 173:117
Shen W, Parker IH, Brack N, Pigram PJ (2001) Appita J 54:352
Seppänen R (2009) J Dispers Sci Technol 30:937
Hiroshi O, Takanori M (2000) Tappi J 54:812
Kilpeläinen T, Manner H (2000) In: Proceedings international printing & graphic arts conference 2000. Tappi Press, Atlanta, p 1
Yu L, Garnier G (2002) J Pulp Paper Sci 28:327
Fernandes S, Duarte AP (2006) Tappi J 5:17
Quan C, Werner O, Wagberg L, Turner C (2009) J Supercrit Fluids 49:117
Werner O, Can Q, Turner C, Pettersson B, Wagberg L (2010) Cellulose 17:187
Trombetta T, Iengo P, Turri S (2005) J Appl Polym Sci 98:1364
Shimada K, Dumas D, Biermann CJ (1997) Tappi J 80:171
Koskela JP, Hormi OE (2002) JAOCS 79:921
Asakura K, Iwamoto M, Isogai A (2006) Appita J 59:285
Jing Q, Chen M, Biermann CJ (1998) Tappi J 81:193
Finlayson MF, Cooper JL, Springs KE, Gathers JJ, Hodgson KT (1996) In: Proceedings of papermakers conference. Tappi Press, Atlanta, p 309
Ibrahim MM, Mobarak F, El-Din EIS, Ebaid A, Youssef MA (2009) Carbohydr Polym 75:130
Ishida Y, Ohtani H, Tsuge S (1994) Anal Chem 66:1444
Yang N, Deng Y (2000) J Appl Polym Sci 77:2067
Wang T, Simonsen J, Biermann CJ (1997) Tappi J 80:77
Valton E, Schmidhauser J, Sain M (2004) Tappi J 3:25
Alince B (2004) Tappi J 3:16
Hossain HM, Uddin MK, Saifullah K, Rashid MM, Mollah MM (2010) Daffodil Int Univ J Sci Technol 5:48
Andersson C, Ernstsson M, Järnström L (2002) Packag Technol Sci 15:209
Järnström J, Väisänen M, Letho R, Jäsberg A, Timonen J, Peltonen J (2010) Colloids Surf A 353:104
Krook M, Gällstedt M, Hedenqvist MS (2005) Packag Technol Sci 18:11
Lacroix M, Le Thien C (2005) In: Han JH (ed) Innovations in food packaging. Elsevier Academic Press, Amsterdam, p 338
Fortuny RS, Rojas-Grau MA, Belloso OM (2011) In: Bai J (ed) Edible coatings and films to improve food quality. CRC Press, Boca Raton, p 103
Gonzalez I, Boufi S, Pelach MA, Alcala M, Vilaseca F, Mutje P (2012) Bioresources 7:5167
Syverud K, Stenius P (2009) Cellulose 16:75
Lavoine N, Desloges I, Dufresne A, Bras J (2012) Carbohydr Polym 90:735
Hult EL, Iotti M, Lenes M (2010) Cellulose 17:575
Aulin C, Gällstedt M, Lindström T (2010) Cellulose 17:559
Rodinova G, Roudot S, Erikson A, Männle F, Gregersen O (2012) Bioresources 7:3690
Minelli M, Baschetti MG, Doghieri F, Ankerfors M, Lindström T, Siró I, Plackett D (2010) J Membr Sci 358:67
Aulin C, Ahola S, Josefsson P, Nishino T, Hirose Y, Osterberg M, Wagberg L (2009) Langmuir 25:7675
Belkekhouche S, Bras J, Siqueira G, Chappey C, Lebrun L, Khelfi B, Marais S, Dufresne A (2011) Carbohydr Polym 83:1740
Spence KL, Venditti RA, Rojas OJ, Habibi Y, Pawlak JJ (2010) Cellulose 17:835
Rodionova G, Lenes M, Eriksen O, Gregersen O (2011) Cellulose 18:127
Andresen M, Johansson LS, Tanem BS, Stenius P (2006) Cellulose 13:665
Chinga-Carrasco G, Kuznetsova N, Garaeva M, Leirset I, Galiullina G, Kostochko A, Syverud K (2012) J Nanopart Res 14:1280
Stenstad P, Andresen M, Tanem BS, Stenius P (2008) Cellulose 15:35
Xhanari K, Syverud K, Chinga-Carrasco G, Paso K, Stenius P (2011) Cellulose 18:257
Chinga-Carrasco G, Syverud K (2012) Nanoscale Res Lett 7:192
Missoum K, Belgacem MN, Bras J (2013) Materials 6:1745
Shen J, Qian X (2012) Bioresources 7:4495
Hu Z, Zen X, Gong J, Deng Y (2009) Colloids Surf A 351:65
Määttänen A, Ihalainen P, Bollström R, Toivakka M, Peltonen J (2010) Colloids Surf A 367:76
Ihalainen P, Määttänen A, Järnström J, Tobjörk D, Österbacka R, Peltonen J (2012) Ind Eng Chem Res 51:6025
Mesic B, Kugge C, Järnström L (2010) Tappi J 9:33
Arbatan T, Zhang L, Fang XY, Shen W (2012) Chem Eng J 210:74
Mertaniemi H, Laukkanen A, Teirfolk JE, Ikkala O, Ras RH (2012) RSC Adv 2:2882
Gil G, Marques PAAP, Trindade T, Neto CP, Gandini A (2008) J Colloid Interface Sci 324:42
Kalia S, Dufresne A, Cherian BM, Kaith BS, Avérous L, Njuguna J, Nassiopoulos E (2011) Int J Polym Sci 837875:35 pp
Roy D, Semsarilar M, Guthrie JT, Perrier S (2009) Chem Soc Rev 38:1825
Belgacem MN, Gandini A (2005) Compos Interfaces 12:41
Roy D, Guthrie JT, Perrier S (2005) Macromolecules 38:10363
Takacs E, Wojnarovits L, Borsa J, Racz I (2010) Radiat Phys Chem 79:467
Dahou W, Ghemati D, Oudia A, Aliouche D (2010) Biochem Eng J 48:187
Paquet O, Krouit M, Bras J, Thielemans W, Belgacem MN (2010) Acta Mater 58:792
Song D, Zhao Y, Dong C, Deng Y (2009) J Appl Polym Sci 113:3019
Morandi G, Health L, Thielemans W (2009) Langmuir 25:8280
Zampano G, Bertolde M, Bronco S (2009) Carbohydr Polym 75:22
Xiao MM, Li S, Chanklin W, Zhen A, Xiao H (2011) Carbohydr Polym 82:512
Carlmark A, Malmström E (2002) J Am Chem Soc 124:900
Nyström D, Lindqvist J, Östmark E, Hult A, Malmström E (2006) Chem Commun 34:3594
Nyström D, Lindqvist J, Östmark E, Antoni P, Carlmark A, Hult A, Malmström E (2009) Appl Mater Interfaces 1:816
Baiardo M, Frisoni G, Scandola M, Licciardello A (2001) J Appl Polym Sci 83:38
Vuoti S, Laatikainen E, Heikkinen H, Johansson LS, Saharinen E, Retulainen E (2013) Carbohydr Polym 96:549
Cunha AG, Freire CS, Silvestre AJ, Neto CP, Gandini A, Orblin E, Fardim P (2007) Biomacromolecules 8:1347
Cunha AG, Freire CS, Silvestre AJ, Neto CP, Gandini A, Orblin E, Fardim P (2007) Langmuir 23:10801
Cunha AG, Freire CS, Silvestre AJ, Neto CP, Gandini A (2006) J Colloid Interface Sci 301:333
Cunha AG, Freire CS, Silvestre AJ, Neto CP, Gandini A, Orblin E, Fardim P (2007) J Colloid Interface Sci 316:360
Ly B, Belgacem MN, Bras J, Brochier Salon MC (2010) Mater Sci Eng C 30:343
Cunha AG, Freire CSR, Silvestre AJD, Neto CP, Gandini A (2010) Carbohydr Polym 80:1048
Cunha AG, Freire C, Silvestre A, Neto CP, Gandini A, Belgacem MN, Chaussy D, Beneventi D (2010) J Colloid Interface Sci 344:55
Oh MJ, Lee SY, Paik PH (2011) J Ind Eng Chem 17:149
Aulin C, Yun SH, Wagberg L, Lindstrom T (2009) Appl Mater Interfaces 1:2443
Li S, Xie H, Zhang S, Wang X (2007) Chem Commun 45:4857
Shang SM, Li Z, Xing Y, Xin JH, Tao XM (2010) Appl Surf Sci 257:1495
Sayyah SM, Khaliel AB, Mohamed EH (2013) J Appl Polym Sci 127:4446
Jandura P, Riedl B, Kokta BV (2002) J Chromatogr A 969:301
Freire CSR, Silvestre AJD, Neto CP, Gandini A, Fardim P, Holmbom B (2006) J Colloid Interface Sci 301:205
Freire CSR, Silvestre AJD, Neto CP, Rocha RM (2005) Cellulose 12:449
Chuan-Fu L, Ai-Ping Z, Wei-Ying L, Feng-Xia Y, Run-Cang S (2009) J Agric Food Chem 57:1814
Wagberg L, Forsberg S, Johansson A, Juntti P (2002) J Pulp Paper Sci 28:222
Eriksson M, Torgnysdotter AS, Wagberg L (2006) Ind Eng Chem Res 45:5279
Lingström R, Notley SM, Wagberg L (2007) J Colloid Interface Sci 314:1
Wang L, Wei J, Su Z (2011) Langmuir 27:15299
Nurmi L, Kontturi K, Houbenov N, Laine J, Ruokolainen J, Seppälä J (2010) Langmuir 26:15325
Ogawa T, Ding B, Sone Y, Shiratori S (2007) Nanotechnology 18:165607 (8 pp)
Yang H, Deng Y (2008) J Colloid Interface Sci 325:588
Gustafsson E, Larsson PA, Wagberg L (2012) Colloid Surf A 414:415
Ou R, Zhang J, Deng Y, Ragauskas AJ (2007) J Appl Polym Sci 105:1987
Findenig G, Leimgruber S, Kargl R, Spirk S, Stana-Kleinschek K, Ribitsch V (2012) Appl Mater Interfaces 4:3199
Sahin HT, Arslan MB (2008) Int J Mol Sci 9:78
Bayer IS, Fragouli D, Attanasio A, Sorce B, Bertoni G, Brescia R, Di Corato R, Pellegrino T, Kalyva M, Sabella S, Pompa P, Cingolani R, Athanassiou A (2011) Appl Mater Interfaces 3:4024
Koga H, Kitaoka T, Isogai A (2012) J Mater Chem 22:11591
Koga H, Kitaoka T, Isogai J (2011) J Mater Chem 21:9356
Abdelmouleh M, Boufi S, Belgacem MN, Duarte AP, Ben Salah A, Gandini A (2004) Int J Adhes Adhes 24:43
Huang X, Wen X, Cheng J, Yang Z (2012) Appl Surf Sci 258:8739
Maity J, Kothary P, O’Rear EA, Jacob C (2010) Ind Eng Chem Res 49:6075
Denes F, Hua ZQ, Barrios E, Young RA, Evans J (1995) J Macromol Sci Pure Appl Chem 32:1405
Carlsson CMG, Strom G (1991) Langmuir 7:2492
Nithya E, Radhai R, Rajendran R, Shalini S, Rajendran V, Jayakumar S (2011) Carbohydr Polym 83:1652
Vander Wielen LC, Östenson M, Gatenholm P, Ragauskas AJ (2006) Carbohydr Polym 65:179
Sepieha S, Wrobel AM, Werthmeier MR (1988) Plasma Chem Plasma Process 8:331
Balu B, Berry D, Hess DW, Breedveld V (2009) Lab Chip 9:3066
Balu B, Berry AD, Patel KT, Breedveld V, Hess DW (2011) J Adhes Sci Technol 25:627
Li R, Ye L, Mai YW (1997) Composites A 28:73
Navarro E, Da’valos F, Denes F, Cruz LE, Young RA, Ramos J (2003) Cellulose 10:411
Vaswani S, Koskinen J, Hess DW (2005) Surf Coat Technol 195:121
Mukhopadhyay SM, Joshi P, Datta S, Zhao JG, France P (2002) J Phys D Appl Phys 35:1927
Sahin HT, Manolache S, Young RA, Denes F (2002) Cellulose 9:171
Mukhopadhyay SM, Joshi P, Datta S, Macdaniel J (2002) Appl Surf Sci 201:219
Sahin HT (2007) Appl Surf Sci 253:4367
Yasuda T, Okuno T, Tsuji K, Yasuda H (1996) Langmuir 12:1391
Song Z, Tang J, Li J, Xiao H (2013) Carbohydr Polym 92:928
Li X, Tian J, Garnier G, Shen W (2010) Colloids Surf B 76:564
Li X, Tian J, Ngyuen T, Shen W (2008) Anal Chem 80:9131
Li X, Tian J, Shen W (2010) Anal Bioanal Chem 396:495
Chitnis G, Ding Z, Chang CL, Chang CA, Savran CA, Ziaie B (2011) Lab Chip 11:1161
Martinez AW, Phillips ST, Butte MJ, Whitesides GM (2007) Angew Chem Int Ed 46:1318
Carrilho E, Philips ST, Vella SJ, Martinez AW, Whitesides GM (2009) Anal Chem 81:5990
Martinez AW, Phillips ST, Carrilho E, Thomaz SW, Sindi H, Whitesides GM (2008) Anal Chem 80:3699
Carrilho E, Martinez AW, Whitesides GM (2009) Anal Chem 81:7091
Shiroma LY, Santhiago M, Gobbi AL, Kubota LT (2012) Anal Chim Acta 725:44
Lu Y, Shi W, Qin J, Lin B (2010) Anal Chem 82:329
Wang S, Ge L, Song X, Yan M, Ge S, Yu J, Zheng F (2012) Analyst 137:3821
Määttänen A, Fors D, Wang S, Valtakari D, Ihalainen P, Peltonen J (2011) Sens Actuators B 160:1404
Sarkar MK, He F, Fan J (2010) Thin Solid Films 518:5033
Hsieh CT, Chen JM, Kuo RR, Lin TS, Wu CF (2005) Appl Surf Sci 240:318
Kim DY, Steckl AJ (2010) Appl Mater Interfaces 2:3318
You H, Steckl AJ (2010) Appl Phys Lett 97:023514
Jin C, Yan R, Huang J (2011) J Mater Chem 21:17519
Mahltig B, Haufe H, Böttcher H (2005) J Mater Chem 15:4385
Daoud WA, Xin JH, Tao X (2004) J Am Ceram Soc 87:1782
Taurino R, Messori EFM, Pilati F, Pospiech D, Synytska A (2008) J Colloid Interface Sci 325:149
Kiuberis J, Kazlauskas R, Grabauskaite L, Tautkus S, Kareiva A (2003) Environ Chem Phys 25:81
Yoldas BE (1998) J Sol-Gel Sci Technol 13:147
Trepte J, Bötther H (2000) J Sol-Gel Sci Technol 19:691
Yagi O, Iwamiya Y, Suzuki K (2005) J Sol-Gel Sci Technol 36:69
Sequeira S, Evtuguin DV, Portugal I, Esculcas AP (2007) Mater Sci Eng C 27:172
March J (1977) Advanced organic chemistry—reactions, mechanisms and structure. McGraw Hill, New York
Mahltig B, Bötther H (2003) J Sol-Gel Sci Technol 27:43
Li Z, Xing Y, Dai J (2008) Appl Surf Sci 254:2131
Tan B, Rankin SE (2006) J Phys Chem B 110:22353
Yeh J, Chen C, Huang K (2007) J Appl Polym Sci 103:3019
Vince J, Orel B, Vilcenik A, Fir M, Vuk AS, Jovanovski V, Simoneie B (2006) Langmuir 22:6489
Roe B, Zhang X (2009) Text Res J 79:1115
Huang W, Song Y, Xing Y, Dai J (2010) Ind Eng Chem Res 49:9135
Huang W, Song Y, Xing Y, Shang S, Dai J (2011) Appl Surf Sci 257:4443
Textor T, Mahltig B (2010) Appl Surf Sci 256:1668
Zhu Q, Gao Q, Guo Y, Yang CQ, Shen L (2011) Ind Eng Chem Res 50:5881
Huang J, Kunitake T (2003) J Am Chem Soc 125:11834
Li S, Wei Y, Huang J (2010) Chem Lett 39:20
Lee K, Jur JS, Kim DH, Parsons GN (2012) J Vac Sci Technol A 30:01A163 (7 pp)
Zhang Y, Ji Q, Qi H, Liu Z (2012) Phys Proced 32:706
Ngo YH, Li D, Simon GP, Garnier G (2011) Adv Colloid Interface Sci 163:23
Yuen CW, Li Y, Ku SK, Mak CM, Kan CW (2005) AATCC Rev 5:41
Wang T, Hu X, Dong S (2007) Chem Commun 18:1849
Fragouli D, Bayer IS, Di Corato R, Brescia R, Bertoni G, Innocenti C, Gatteschi D, Pellegrino T, Cingolani R, Athanassiou A (2012) J Mater Chem 22:1662
Daoud WA, Xin JH (2004) J Am Ceram Soc 87:953
Huang L, Chen K, Lin C, Yang R, Gerhardt R (2011) J Mater Sci 46:2600. doi:10.1007/s10853-010-5112-1
Su C, Li J (2010) Appl Surf Sci 256:4220
Gashti MP, Alimohammadi F, Shamei A (2012) Surf Coat Technol 206:3208
Nallathambi G, Ramachandran T, Rajendran V, Palanivelu R (2011) Mater Res 14:552
Hsieh CT, Chen WY, Wu FL, Shen YS (2008) J Adhes Sci Technol 22:265
Wang H, Fang J, Cheng T, Ding J, Qu L, Dai L, Wang X, Lin T (2008) Chem Commun 7:877
Yu MH, Gu GT, Meng WD, Qing FL (2007) Appl Surf Sci 253:3669
Ramaratnam K, Tsyalkovsky V, Klep V, Luzinov I (2007) Chem Commun 49:4510
Hoefnagels HF, Wu D, De With G, Ming W (2007) Langmuir 23:13158
Pereira C, Alves C, Monteiro A, Magén C, Pereira AM, Ibarra A, Ibarra MR, Tavares PB, Araújo JB, Blanco G, Pintado JM, Carvalho AP, Pires J, Pereira MFR, Freire C (2011) Appl Mater Interfaces 3:2289
Gao Q, Zhu Q, Guo Y (2009) Ind Eng Chem Res 48:9797
Chen X, Liu Y, Lu H, Yang H, Zhou X, Xin JH (2010) Cellulose 17:1103
Abidi N, Aminayi P, Cabrales L, Hequet E (2012) Funct Mater Renew Source 1107:149
Zhang J, France P, Radomyselskiy A, Datta S, Zhao J, van Ooij W (2003) J Appl Polym Sci 88:1473
Li S, Zhang S, Wang X (2008) Langmuir 24:5585
Shirgholami MA, Shateri-Khalilabad M, Yazdanshenas ME (2013) Text Res J 83:100
Zimmermann J, Reifler FA, Fortunato G, Gerhardt L, Seeger SA (2008) Adv Funct Mater 18:3662
Ogihara H, Xie J, Okagaki J, Saji T (2012) Langmuir 28:4605
Li J, Wan H, Ye Y, Zhou H, Chen J (2012) Appl Surf Sci 261:470
Barona D, Amirfazli A (2011) Lab Chip 11:936
Stepien M, Saarinen JJ, Teisala H, Tuominen M, Aromaa M, Kuusipalo J, Makela JM, Toivakka M (2011) Appl Surf Sci 257:1911
Teisala H, Tuominen M, Aromaa M, Mäkelä JM, Stepien M, Saarinen JJ, Toivakka M, Kuusipalo J (2010) Surf Coat Technol 205:436
Teisala H, Tuominen M, Aromaa M, Mäkelä JM, Stepien M, Saarinen JJ, Toivakka M, Kuusipalo J (2013) Colloid Polym Sci 291:447
Teisala H, Tuominen M, Aromaa M, Mäkelä JM, Stepien M, Saarinen JJ, Toivakka M, Kuusipalo J (2013) Cellulose 20:391
Liu Y, Tang J, Wang R, Lu H, Li L, Kong Y, Qi K, Xin JH (2007) J Mater Chem 17:1071
Liu YY, Tang J, Xin JH (2004) Chem Commun 24:2828
Laird ED, Wang W, Cheng S, Li B, Presser V, Dyatkin B, Gogotsi Y, Li CY (2012) ACS Nano 6:1204
Samyn P, Deconinck M, Schoukens G, Stanssens D, Vonck L, Van den Abbeele H (2012) Polym Adv Technol 23:11
Samyn P, Deconinck M, Schoukens G, Stanssens D, Vonck L, Van den Abbeele H (2010) Prog Org Coat 69:442
Samyn P, Schoukens G, Vonck L, Stanssens D, Van den Abbeele H (2011) Langmuir 27:8509
Stanssens D, Van den Abbeele H, Vonck L, Schoukens G, Deconinck M, Samyn P (2011) Mater Lett 65:1781
Samyn P, Schoukens G, Van den Abbeele H, Vonck L, Stanssens D (2011) J Coat Technol Res 8:363
Schuman T, Adolfson B, Wikström M, Rigdahl M (2005) Prog Org Coat 54:188
Stauffer TC, Venditti RA, Gilbert RD, Kadla JF, Montero G (2001) J Appl Polym Sci 81:1107
Food and Drug administration (2003) Department of Health and Human Services, chap 1, title 21 (food and drugs), 21 CFR—Code of Federal Regulations Register
Rom M, Dutkiewicz J, Fryczkowka B, Fryczkowka R (2007) Fibres Text East Eur 15:141
Dankovich TA, Hsieh YL (2007) Cellulose 14:469
Shang W, Huang J, Luo H, Chang PR, Feng J, Xie G (2013) Cellulose 20:179
Samyn P, Schoukens G, Stanssens D, Vonck L, Van den Abbeele H (2012) J Nanopart Res 14:1075
Khwaldia K, Tehrany AM, Desobry S (2010) Compr Rev Food Sci Food Saf 9:82
Andersson C (2008) Packag Technol Sci 21:339
Bordenave N, Grelier S, Pichavant F, Coma V (2007) J Agric Food Chem 55:9479
Bordenave N, Grelier S, Coma V (2010) Biomacromolecules 11:88
Reis AB, Yoshida CM, Reis AP, Franco TT (2011) Polym Int 60:963
Fernandes SC, Freire CS, Silvestre AJ, Desbrières J, Gandini A, Neto CP (2010) Ind Eng Chem Res 49:6432
Rhim JW, Kim JH (2009) J Food Sci 74:105
Rhim JW, Lee JH, Hong S (2007) Packag Technol Sci 20:393
Bourbonnais R, Marchessault RH (2010) Biomacromolecules 11:989
Cyras VP, Soledad CM, Analía V (2009) Polymer 50:6274
Thellen C, Coyne M, Froio D, Auerbach M, Wirsen C, Ratto JA (2008) J Polym Environ 16:1
Obeso CG, Sousa MP, Song W, Rodriguez-Perez MA, Bhushan B, Mano JF (2013) Colloids Surf A 416:51
Han J, Salmieri S, Le Tien C, Lacroix M (2010) J Agric Food Chem 58:3125
Olabarrieta I, Forsström D, Gedde U, Hedenqvist M (2001) Polymer 42:4401
Garcia-Ubasart J, Colom JF, Vila C, Hernández NG, Blanca Roncero M, Vidal T (2012) Bioresour Technol 112:341
Maximova N, Österberg M, Laine J, Stenius P (2004) Colloids Surf A 239:65
Alves NM, Shi J, Oramas E, Santos JL, Tomas H, Mano JF (2009) J Biomed Mater Res A 91:480
Shi J, Alves MN, Mano JF (2008) Bioinspir Biomimetics 3:034003 (6 pp)
Song W, Gaware VS, Runarsson OV, Marsson M, Mano JF (2010) Carbohydr Polym 81:140
Acknowledgements
P.S. acknowledges the Robert Bosch Foundation for support as Juniorprofessor in Sustainable Use of Natural Materials (Foresnab-Project), and the State of Baden-Württemberg for financial support in the Juniorprofessorenprogramm (NaCoPa-Project).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Samyn, P. Wetting and hydrophobic modification of cellulose surfaces for paper applications. J Mater Sci 48, 6455–6498 (2013). https://doi.org/10.1007/s10853-013-7519-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-013-7519-y