
Abstract
In this study, graphite oxides (GOs) with different oxidation degrees and graphene nanosheets were prepared by a modified Hummers method and thermal exfoliation of the prepared GO, respectively. Polystyrene (PS)/GO and PS/graphene nanocomposites were prepared via melt blending. X-ray diffraction results showed that GOs and graphene were exfoliated in the PS composites. It could be observed from the scanning electron microscope images that GOs and graphene were well dispersed throughout the matrix without obvious aggregates. Dynamic mechanical thermal analysis suggested that the storage modulus for the PS/GO1 and PS/graphene nanocomposites was efficiently improved due to the low oxygen content of GO1 and the elimination of the oxygen groups from GO. The flammability of nanocomposites was evaluated by thermal gravimetric analysis and cone calorimetry. The results suggested that both the thermal stability and the reduction in peak heat release rate (PHRR) decreased with the increasing of the oxygen groups in GOs or graphene. The optimal flammability was obtained with the graphene (5 wt%), in which case the reduction in the PHRR is almost 50 % as compared to PS.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polystyrene (PS) is a widely used general polymer due to its low density, good chemical resistance, high performance price ratio, and convenience of processing and molding. However, its high flammability and severe dripping during combustion greatly limit its application in buildings, transportation, and electrical appliances. Flame-retardant treatment of organic polymer materials is essential from the viewpoint of disaster prevention because their heat release occurs rapidly after ignition. In recent years, the development of halogen-free flame-retardant polymeric materials has become a potential trend [1–3]. Conventional flame-retardant additives containing halogen elements are currently being forced by legal obligations to be replaced by halogen-free counterparts such as phosphorus and/or nitrogen-based compounds and mineral-type metal hydroxides. Very recently, carbon nanoadditives, including expanded graphite [4, 5], carbon nanotubes [6–8], graphite oxide (GO) [9–15], and graphene [16–18], have been extensively used into various polymer systems for improving their flame-retardant properties.
GO, with a low fabrication cost and an environmentally friendly nature, can be obtained on a large scale by the chemical oxidation of graphite and possesses a number of hydroxyl and epoxide functional groups anchored on sp 3-hybridized carbon atoms on both surfaces of each sheet and considerable amounts of sp 2-hybridized carbon atom-containing carboxyl and carbonyl groups at the sheet edges [19–21]. These functional groups make it easy for GO to absorb polar molecules and polar polymers by different means to form polymer/GO nanocomposites, which have some physical and chemical properties, including fire retardancy, that are dramatically different from that of their bulk counterparts. Many studies show that the composites consisting of GO and a polymer matrix, such as epoxy resin [9, 13], polyacrylate ester [10], polycarbonate [11], acrylonitrile butadiene styrene [11], high-impact PS [11], PS [14], polyamide 6 [15], and styrene-butyl acrylate copolymer [17, 22], exhibit enhanced thermal stability and flame retardancy compared with that of pristine polymers. The efficiency of GO on flame retardancy of polymers depends primarily on two factors: the oxidation extent of natural graphite and the dispersion quality of the GO [13].
The reduction of GO sheets produces reduced graphene oxide (r-GO), also known as chemically modified graphene. Extensive research into the application of graphene including its potential application as a flame-retardant additive has been carried out due to a unique two-dimensional (2D) atomic carbon sheet structure. It is reported that the combination of graphene and intumescent flame retardant can be used as flame-retardant systems [17]. Previous research on graphene or functional graphene into polymer has been primarily focused on the improvement of the thermal stability, melt flow index, or anti-dripping properties [16, 17]. The incorporation of graphene and metal compound-loaded graphene into PS by a novel masterbatch-melt blending technique for improved thermal stability and fire safety properties was investigated in a recent publication [18]. It was stated that the physical barrier effect of graphene, the interaction between graphene and PS, and the synergistic effect of the metal compounds are the causes for the improvements. However, little attention has been paid to the comparison of different oxidation degrees of GO with graphene on both flame retardancy and dynamic viscoelastic properties of the polymer composites.
In this work, GO with different oxidation degrees and graphene have been prepared. PS/GO and PS/graphene nanocomposites have also been prepared by melt blending. The structure, morphology, dynamic mechanical properties, thermal stability, and flame retardancy of the composites were studied.
Experiments
Materials
GO was synthesized from natural graphite (1000 mesh) that was kindly supplied by Shandong Pingdu Graphite Company. Concentrated sulfuric acid (H2SO4) (A.R), concentrated hydrochloride acid (HCl) (A.R), and potassium permanganate (KMnO4) (A.R) were purchased from Nanjing Chemical Reagent Company. Sodium nitrate (A.R, NaNO3) and H2O2 (A.R, 30 %) were purchased from Shanghai Renyu Chemicals Company. All these commercial chemicals were used as received without further purification.
Preparation of GO and Graphene
GO was synthesized from graphite powder by a modified Hummers method [23]. According to the different reaction times at 10–15 °C, GO was designated as GO1, GO2, and GO3. Natural graphite powder (10 g) was added to 230 mL of cooled (0 °C) H2SO4 (98 %). KMnO4 (30 g) and NaNO3 (5 g) were added gradually with stirring and cooling, so that the temperature of the mixture was maintained at 10–15 °C for a desired time (1, 2, and 4 h for GO1, GO2, and GO3, respectively). The mixture was then stirred at 35 °C for 30 min. 250 mL deionized water was slowly added to increase temperature to 98 °C (60 °C for GO1) and the mixture was stirred at that temperature for 15 min. The reaction was terminated by adding 1 L of deionized water followed by 100 mL of 30 % H2O2 solution. The solid product was separated by centrifugation, washed repeatedly with 5 % HCl solution, then washed 3–4 times with acetone, and dried in a vacuum oven at 60 °C for 24 h. The elemental analysis data (see Table 1) showed that the composition of GO1, GO2, and GO3 is C8O2.1H1.2, C8O2.8H1.5, and C8O3.7H1.9, respectively.
The dried GO3 powder was placed in a muffle furnace at ~1050 °C for ~30 s under N2 atmosphere to get graphene nanosheets.
Preparation of PS/GO and PS/graphene composites
The PS/GO and PS/graphene composite samples were obtained by melt mixing the GO and graphene with the desired amount of PS, using a HAAKE Rheocord 90 internal mixer at the rotor rate of 45 rpm at 180 °C for 12 min. The weight ratio of GO or graphene to PS is 5:95. Sample bars suitable for cone calorimeter testing and dynamic mechanical thermal analysis (DMTA) were prepared via compression molding at a processing temperature of 180 °C.
Characterization
Cone calorimeter test
The combustion properties of PS/GO and PS/graphene nanocomposites were evaluated using a cone calorimetry experiment. All samples (100 × 100 × 3 mm3) were exposed to a FTT 0007 cone calorimeter (FTT Company, England) under a heat flux of 50 kW/m2 according to ISO-5660 standard procedures.
Thermal gravimetric analysis (TGA)
TGA was carried out in air at a heating rate of 10 °C/min using a NETZSCH STA409 PC thermoanalyzer instrument.
Dynamic mechanical thermal analysis (DMTA)
Dynamic mechanical properties were measured with a DMA + 450 (MetraviB, France) in the stretching mode on sample bars (10 × 20 × 1 mm3). A temperature ramp experiment (2 °C/min) was conducted under air from room temperature to 160 °C at a constant frequency of 1 Hz.
Scanning electron microscopy (SEM)
The SEM images were obtained on a HITACHI S-4800 scanning electron microscope. SEM images were obtained on freeze-fractured cross sections of the PS composite sample bar.
Fourier-transform infrared spectroscopy (FTIR)
Samples for FTIR measurements were mixed with KBr powders and pressed into a tablet. The FTIR spectra were obtained using a Bruker VECTOR22 FTIR spectrophotometer in the range from 400 to 4000 cm−1.
X-ray diffraction (XRD)
XRD patterns were obtained with a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation (λ = 0.1541 nm).
Results and discussion
Structure and morphology
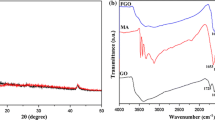
Figure 1 shows the FTIR spectra of pristine GO1, GO2, and GO3. The spectrum of GO3 displays a strong –OH peak at 3417 cm−1, a C=C peak at 1619 cm−1 corresponding to the remaining sp 2 character [24], the band at 1384 cm−1 attributed to deformations of the CO–H group, the band at 1720 cm−1 associated with stretching of the C=O bond of carbonyl or carboxyl groups, and the intense band at 1051 cm−1 assigned to the deformation of the C–O bond. For GO3, bands located at 3417, 1714, and 1384 cm−1 attributed to the stretching of –OH, C=O bond, and CO–H groups provided a stronger intensity compared with that of GO1 and GO2, indicating that more oxygen-containing functional groups were introduced after the oxidation of natural graphite. As a result, lower oxidation degrees of GO1 and GO2 were obtained compared with GO3. The red shift of C=C peak at 1619 cm−1 of GO1 and GO2 compared with GO3 can be attributed to the π–π interactions between C=C induced from the different oxidation structure of GO1 and GO2. It can be observed from Fig. 1d that after thermal reduction, most of the oxygen functional groups of GO are removed from the GO layers. FTIR spectra of PS/GO and PS/graphene composites are shown in Fig. 2. In the FTIR spectrum of pure PS, characteristic peaks at 3100–3000 cm−1 represent =C–H aromatic stretching vibration; peaks at 2920 and 2849 cm−1 demonstrate asymmetric and symmetric stretching vibration of CH2, respectively; peaks at 1600, 1580, and 1491 cm−1 pertain to stretching vibration of benzene ring; and peaks at 753 and 697 cm−1 are related to the C–H out-of-plane bending vibration of the benzene ring [25]. For PS/GO and PS/graphene composites, the typical absorption of GO is not detected because it is either too weak or overlaps with the absorption peak of PS.

FTIR spectra of GO1; GO2; GO3; and graphene

FTIR spectra of a PS; b PS/GO1; c PS/GO2; d PS/GO3; e PS/graphene
XRD is a powerful tool for the structure analysis of layered materials like GO. XRD patterns of GO1, GO2, GO3, and graphene are shown in Fig. 3. In the case of GO1, the diffraction (002) peak of graphite at 2θ = 25° vanished, which indicates that the crystal structure of graphite was reduced. In addition, reflection at around 2θ = 11.9°, corresponding to the (001) plane of GO, is weak, which may be because of the low content of oxygen groups in GO1. In GO2 and GO3 diffractograms (Fig. 3b, c), this peak became sharp and moved to a slightly lower angle (2θ = 11.3° and 10.6°). The interlayer spacing of GO1, GO2, and GO3 calculated from the Bragg’s equation (2d sinθ = nλ, λ = 0.1541 nm) is 0.74, 0.78 and 0.83 nm, respectively. The increase of the GO (001) diffraction peak and interlayer spacing indicated the increasing oxidation degree from GO1, GO2 to GO3. These XRD results related to the degree of oxidation correspond to that of the elemental analysis described in Table 1. As shown in Fig. 3d, compared with the XRD pattern of GO3, the disappearance of the diffraction peak at 10.6° gives evidence that the oxygen functional groups of GO3 are eliminated. A broad and weak peak centered at ca. 25° appears, corresponding to the graphitic (002) face, which is characteristic of disordered, exfoliated, and loose-packed graphene sheets, indicating successful exfoliation [26, 27]. The XRD patterns of PS/GO and PS/graphene composites are shown in Fig. 4. It can be seen from Fig. 4a that a wide peak from 2θ = 15° to 25° belongs to amorphous PS. However, after GO or graphene was dispersed into the PS matrix, the XRD pattern of the PS/GO and PS/graphene nanocomposites (Fig. 4b–e) only showed the PS diffraction peak from PS; the diffraction peak of GO or graphene was hard to be detected. The XRD results demonstrate that GO was exfoliated in the polymer matrix and that the regular and periodic structure of GO or graphene disappeared [20, 22, 28, 29].

XRD patterns of GO1; GO2; GO3; and graphene

XRD patterns of a PS; b PS/GO1; c PS/GO2; d PS/GO3; e PS/graphene nanocomposites
Figure 5 shows the SEM images of GO, graphene, and the nanocomposites. The morphology of all GOs in Fig. 5a–c is observed to be of a wrinkled and folded texture, reflecting its layered microstructure. It can be seen from the SEM image (Fig. 5d) of the as-exfoliated graphene after thermal shock on GO3 that the exfoliated graphene sheets were several micrometers in size, the delicate graphene could be folded over repeatedly without breaking, and the graphene nanoflakes tended to organize to an expanded layered structure. In the transmission electron microscopy (TEM) image (insert image of Fig. 5d) of the thermally exfoliated graphene, the graphene nanosheet looks like a transparent ultrathin film with ripples within the plane. Figure 6 shows SEM micrographs of the fractured surfaces of PS/GO and PS/graphene nanocomposites, revealing that the compact layers of pristine GO1 and GO2 (Fig. 5a, b) with a micrometer size have been exfoliated into irregular flakes in the nanocomposites (Fig. 6a, b) and dispersed well in the PS matrix with a few restacks. However, it is hard to a distinguish GO flake from PS matrix in PS/GO3 nanocomposites, probably due to the agglomeration of GO3 in PS matrix. Noticeably, graphene flakes tend to organize inside the polymer matrix and are well dispersed in the PS (Fig. 6d).

SEM images of a GO1; b GO2; c GO3; and d graphene

SEM images a PS/GO1; b PS/GO2; c PS/GO3; d PS/graphene nanocomposites
Dynamic mechanical thermal analysis
DMTA was performed at various temperatures to evaluate the dynamic viscoelastic properties. Figure 7a shows the temperature dependence of storage modulus (E′) obtained from DMTA of the PS and its nanocomposites. It can be seen from Fig. 7a that a decrease of the storage modulus was present for the PS/GO3 nanocomposites. For the PS/GO2 nanocomposites, the difference of the storage modulus was not remarkable. The storage modulus for the PS/GO1 and PS/graphene sample was obviously improved. From the above results, it can be deduced that high oxidation degree of GO is not beneficial for the enhancement of storage modulus. One possible explanation for these observations is that the storage modulus values are highly influenced by the interfacial interactions between the GO or graphene nanosheets and the PS matrix [30]. The π–π interfacial interactions between GO and PS chain could be decreased by the unstable oxygen-containing groups. A high number of oxygen groups (for example, GO3) might lead to the poor compatibility between the matrix polymer and the inorganic layers.

a Storage modulus and b Tan curves for PS; PS/GO1; PS/GO2; PS/GO3; PS/graphene nanocomposites at a frequency of 1 Hz at various temperatures
Figure 7b shows the temperature dependence of tanδ for neat PS and its nanocomposites. A variation of T g of nanocomposites relative to the virgin polymers can provide evidence of specific interactions between the inorganic particle and polymer matrix. The T g data of the samples in our studies are presented in Table 2. Compared with pure PS, the T g for the PS/GOs and PS/graphene nanocomposites increases obviously. The T g of the nanocomposites is about 15–20 °C, higher than that of the neat PS. It was believed that the efficient dispersion of exfoliated GOs and graphene layers in the nanocomposites results in the restricted segmental motions at the organic–inorganic interface and stronger interactions between the inorganic layers and the polymer chains.
Thermal gravimetric analysis
TGA curves and the detailed data for neat PS and its nanocomposites in air are presented in Fig. 8 and Table 2, respectively. The data include the temperature at which 10 % weight loss occurs (T 0.1, a measure of onset temperature of degradation), the temperature for 50 % degradation (T 0.5) as the mid-point of degradation, and the amount of char at 600 °C. Interestingly, the T 0.1 of PS is 320 °C, while the T 0.1 values of the PS/GO1, PS/GO2, PS/GO3, and PS/graphene nanocomposites are 370, 363, 336, and 400 °C, respectively. T 0.1 of PS/GO1, PS/GO2, PS/GO3, and PS/graphene is 50, 43, 16, and 80 °C, higher than that of pure PS. T 0.5 of the PS nanocomposites is also higher than that of neat PS. These results prove that the high oxidation degree of GO (GO3) is not beneficial for the enhancement of the thermal stability of the nanocomposites. Particularly, among all the nanocomposites, PS/graphene nanocomposites provide the optimal thermal stability compared with pure PS. This improvement might be attributed to the elimination of the oxygen functional groups from GO3 and the dispersion of graphene nanosheets in the PS matrix. The well-dispersed and special structure of graphene in the polymer matrix is expected to be an effective barrier to the permeation of flammable gases. A similar result was also found in previous graphene-based nanocomposites [17, 31, 32]. In the case of char residues at 600 °C, virgin PS left little residue. The char yield of the nanocomposites increased in comparison to that of pure PS, possibly because of the GOs or graphene in PS matrix promoting carbonization on the polymer surface. Furthermore, unburned filler and high heat resistance exerted by the filler itself additionally contributed to the higher char residues.

TGA curves of PS; PS/GO1; PS/GO2; PS/GO3; and PS/graphene nanocomposites
Flame-retardant properties
It is well known that cone calorimetry is one of the most effective bench-scale methods for studying the flammability properties of materials. The heat release rate (HRR), in particular the peak HRR (PHRR) value, proves to be the most important parameter to evaluate fire safety. The HRR plots for pure PS and its nanocomposites are shown in Fig. 9. It was found that the nanocomposites had a much lower PHRR than that of the virgin polymer. The PHRR values of PS/GO1, PS/GO2, PS/GO3, and PS/graphene nanocomposites are 39, 38, 32, and 47 %, respectively, lower than that of virgin PS (as shown in Table 3). Among GOs, GO3 with the highest oxidation degree attained by elemental analysis, FTIR, and XRD results showed the highest PHRR. These results indicated that the high oxidation degree of GO does not contribute to the enhancement of the flame-retardant properties. It is clear that the lowest PHRR was obtained when the graphene was incorporated in neat PS. The reduction of HRR values was accompanied by a pronounced prolongation of burning time with a flat curve, while it presents a very sharp and short HRR curve for pure PS as shown in Fig. 9, which can be ascribed to the degradation as well as the stabilization of the char formation [10]. The addition of GO or graphene to PS matrix tends to cause a decrease in the time to ignition. At first, GO or graphene absorbs and re-irradiates the heat to cause a rapid charring of the polymer, resulting in the rapid rise seen in the HRR curve, followed by the thermal degradation of the charred polymer, which is albeit slower than that of the virgin polymer, since the GO or graphene now serves as a barrier to mass transport and somewhat insulates the underlying polymer from the heat source.

Heat release rates versus burning time for PS; PS/GO1; PS/GO2; PS/GO3; and PS/graphene nanocomposites
Figure 10 shows the macromorphologies of the final chars after cone calorimeter tests of pure PS and its nanocomposites using a digital camera. The pure PS matrix has been burnt out (Fig. 10a) and all the aluminum foil has become visual, whereas the other samples for PS nanocomposites remain more or less residues (Fig. 10b–e) and incumbent nearly or completely on the aluminum foil. For the PS/graphene samples, intact and uniform chars are left at the end of the test as shown in Fig. 10e. As compared with PS/GOs nanocomposite, the char of PS/graphene nanocomposites is obviously larger, more swollen, and compact, in accordance with its high stability and flammability.

Residues at the end of cone calorimeter tests a PS; b PS/GO1; c PS/GO2; d PS/GO3; e PS/graphene nanocomposites
The GO and graphene layers consequently formed a charred layer, which acts as a heat insulation barrier, preventing the heat transfer and transportation of degraded products between the melting polymer and surface, thus reducing the HRR and related parameters. In this research, for different oxidation degrees of GO, the number of oxygen functional groups is different. It is known that the deflation of GO mainly depends on the number of oxygen functional groups and the temperature increasing rate [33]. For GO3, the unstable oxygen-containing groups on the GO sheet could probably lead to the deflation of GO3 in the PS matrix during the combustion. This deflation behavior of GO3 might make the GO layer become damaged or accumulated together. Eventually, the thermal stability and the flame retardancy of GO3 were decreased. As a comparison, GO1 or GO2 with relatively fewer oxygen functional groups might avoid the deflation during the combustion process. For graphene, the thermal stability and the flame retardancy are increased due to the elimination of oxygen-containing groups on the GO sheets and the dispersion of graphene sheets in the nanocomposites.
Conclusions
In our study, GOs with different oxidation degrees or graphene were blended at 5 wt% into PS to serve as flame-retarding nanoadditives. Structural characterization by elemental analysis, FTIR, and XRD illustrated that GO1, GO2, and GO3 possess different oxidation degrees. SEM analysis showed that except for GO3 with a high oxidation degree, GO1, GO2, and graphene all formed a well-dispersed morphology in the PS matrix. XRD results demonstrated that GOs and graphene were exfoliated in the PS composites. DMTA revealed that a high oxidation degree is not beneficial for the enhancement of storage modulus. PS/graphene provided the highest storage modulus due to the elimination of the functional groups from GO. The T g of the nanocomposites is about 15–20 °C higher than that of the neat PS. The TGA and cone calorimetry results suggested that both the thermal stability and the reduction in PHRR decreased with the increasing content of the oxygen groups in GOs or graphene. The optimal flammability is obtained with the graphene (5 wt%), in which case the reduction in the peak PHRR is almost 50 % as compared to PS.
References
Sudhakara P, Kannan P, Obireddy K, Rajulu AV (2011) J Mater Sci 46:5176. doi:10.1007/s10853-011-5451-6
Konnicke D, Kuhn A, Mahrholz T, Sinapius M (2011) J Mater Sci 46:7046. doi:10.1007/s10853-011-5673-7
He QL, Song L, Hu Y, Zhou S (2009) J Mater Sci 44:1308. doi:10.1007/s10853-009-3266-5
Murariu M, Dechief AL, Bonnaud L, Paint Y, Gallos A, Fontaine G, Bourbigot S, Dubois P (2010) Polym Degrad Stab 95:889
Bian XC, Tang JH, Li ZM, Lu ZY, Lu A (2007) J Appl Polym Sci 104:3347
Peeterbroeck S, Laoutid F, Taulemesse JM, Monteverde F, Lopez-Cuesta J, Nagy JB, Alexandre M, Dubois P (2007) Adv Funct Mater 17:2787
Peeterbroeck S, Laoutid F, Swoboda B, Lopez-Cuesta J, Moreau N, Nagy JB, Alexandre M, Dubois P (2007) Macromol Rapid Commun 28:260
Isitman NA, Kaynak C (2010) Polym Degrad Stab 95:1523
Lee YR, Kim SC, Lee H, Jeong HM, Raghu AV, Reddy KR, Kim BK (2011) Macromol Res 19:66
Wang JQ, Han ZD (2006) Polym Adv Technol 7:335
Higginbotham AL, Lomeda JR, Morgan AB, Tour JM (2009) ACS Appl Mater Interfaces 1:2256
Zhang R, Hu Y, Xu JY, Fan WC, Chen ZY, Wang QN (2004) Macromol Mater Eng 289:355
Wang Z, Tang XZ, Yu ZZ, Guo P, Song HH, Du XS (2011) Chin J Polym Sci 29:368
Kedzierski M, Jankowski P, Jaworska G, Niska A (2012) Polimery 57:347
Dasari A, Yu ZZ, Mai YW, Cai GP, Song HH (2009) Polymer 50:1577
Shi Y, Li LJ (2011) J Mater Chem 21:3277
Wang X, Song L, Yang H, Lu H, Hu Y (2011) Ind Eng Chem Res 50:5376
Bao CL, Song L, Wilkie CA, Yuan BH, Guo YQ, Hu Y, Gong XL (2012) J Mater Chem 22:16399
Rourke JP, Pandey PA, Moore JJ, Bates M, Kinloch LA, Young RJ, Wilson NR (2011) Angew Chem Int Ed 50:3173
Wilson NR, Pandey PA, Beanland R, Young RJ, Kinloch IA, Gong L et al (2009) ACS Nano 3:2547
Lee DW, De Los Santos LV, Seo JW, Felix LL, Bustamante DA, Cole JM, Barnes CHW (2010) J Phys Chem B 114:5723
Hussein MT (2012) New approaches to the synthesis and exfoliation of polymer/functional graphene nanocomposites by miniemulsion polymerization. Dissertation, University of Stellenbosch
Hummers WS, Offeman RE (1958) J Am Chem Soc 80:1339
Bissesur R, Liu PKY, White W, Scully SF (2006) Langmuir 22:1729
Bhutto AA, Vesely D, Gabrys BJ (2003) Polymer 44:6627
Zhang DS, Yan TT, Shi LY, Zheng P, Wen XR, Zhang JP (2012) J Mater Chem 22:14696
Yang S, Chang K, Tien H, Lee Y, Wang Y, Wang J, Ma CM, Hu C (2011) J Mater Chem 21:2374
Du XS, Yu ZZ, Dasari A, Ma J, Mo MS, Meng YZ, Mai YW (2008) Chem Mater 20:2066
Du XS, Xiao M, Meng YZ, Hay AS (2005) Carbon 43:195
Vadukumpully S, Paul J, Mahanta N, Valiyaveettil S (2011) Carbon 49:198
Liang JJ, Huang Y, Zhang L, Wang Y, Ma YF, Guo TY, Chen YS (2009) Adv Funct Mater 19:2297
Kim H, Miura Y, Macosko CW (2010) Chem Mater 22:3441
Han YQ, Lu Y (2007) Carbon 45:2394
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21173120), the Postdoctoral Science Foundation of China (2012M511184), and the Postdoctoral Science Foundation of Jiangsu Province (No. 1102075C).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Han, Y., Wu, Y., Shen, M. et al. Preparation and properties of polystyrene nanocomposites with graphite oxide and graphene as flame retardants. J Mater Sci 48, 4214–4222 (2013). https://doi.org/10.1007/s10853-013-7234-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-013-7234-8