
Abstract
A graphene oxide (GO) was produced for upgrading the thermal stability, flammability and decrease in the fire hazards of polystyrene (PS). Cone calorimeter and limiting oxygen index tests were utilized to assess the heat and flame tests of PS nanocomposites. The addition of GO in PS matrix efficiently improved the flame retardancy and reduced the density of carbon monoxide (CO) release, which attributed to the homogeneous dispersal of GO in the PS matrix upgraded barrier effect that reduced peak heat release rate, total heat release and toxic gas evolution during combustion. The characterization implied that the GO nanosheets were well distributed throughout the PS matrix without clear aggregates, leading to outstanding upgrading of thermal stability and fire safety properties. Dynamic mechanical analysis (DMA) showed that both the storage modulus and glass transition temperature (Tg) of PS/GO nanocomposites were significantly promoted related to that of pristine PS. Moreover, PS/GO nanocomposites clearly decreased the amount of flammable volatiles and CO evolved, suggesting suppressed fire hazards of the PS composites owing to proper distribution, physical barrier effect intumescent and loosely structure of char layers. This study provides useful insights into the flammability behavior of polystyrene compounds with layered fillers of GO.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polymeric materials have been widely consumed in our life owing to the incomparable advantages over the traditional materials. Though, many potential applications are restrained by serious fire hazards of the polymer materials, which affect the loss of life and property, attracting considerable attention from governments and society. It principally contains thermal hazards and nonthermal hazards. Thermal hazards are outlined as that polymer material produces a large amount of heat during combustion and directs to melt dripping which further encourages the spread of fire [1, 2]. The nonthermal hazards mean the releasing a great deal of toxic gases and smoke, such as CO, HCN, NOx, organic volatiles and smoke particles. The most fire deaths are toxic gases, oxygen deficiency and smoke breathing [3, 4]. The smoke produced in the fire is even the most important factor which straightforwardly puts people to death by poisoning and suffocation [5]. Furthermore, the impairing visibility and annoying effect of fire gases are regarded as the key factor which inhibits many fire victims to realize the possibilities of escape [6]. Some disadvantages of polystyrene (PS) are brittle, poor chemical resistance, susceptible to UV degradation and flammability. PS contains the toxic substances styrene and benzene, which are carcinogenic and harmful to humans. Extruded polystyrene is highly flammable and easily ignited. Meanwhile, the flame retardant used in all PS-building insulation is being prohibited by the European Union. PS, a general plastic, has been broadly used in automotive, thermal insulating materials and electrical application industries [7]. As a common law, polymers with aliphatic backbones are liable toward the low smoke generation, while polyene polymers and those with pendant aromatic groups create more smoke [8], which signifies that PS also generates huge smoke [8]. Though, the fire hazard of PS restricts its application in some fields, owing to the release of a large amount of heat and toxic smoke during combustion, which will direct to heavy casualties and property losses in case of fire accidents [9,10,11]. Therefore, decreasing the fire hazard of PS is an urgent requirement to arouse wide concerns. Heat is the direct hazard in fire accidents, which increases temperature and promotes fire spread. In order to decrease the peak heat release rate and the total heat release, flame retardants are broadly applied in the polymer composites [12, 13]. As far as we known, the excellent flame retardants usually contain some essential elements, such as halogen, phosphorus, nitrogen, boron and silicon. [14]. Owing to the superior requirement for environmental concerns, the halogen-free flame retardants have been widely used to inhibit the flammability of PS, especially phosphorus-containing flame retardants [15]. Up to now, two main flame-retardant mechanisms were proposed for the phosphorus-containing flame retardants [16, 17]. In the flame-retardant field of PS, the condensed phase mechanism presents higher efficiency and lower toxicity [18, 19]. Besides the heat hazard, there is also non-heat hazard in fire accidents, such as toxic smoke. Toxic smoke is composed of smoke particles, organic volatiles and carbon monoxide (CO) [20]. Inhalation of the toxic smoke is a major reason for the casualties in fire accidents, sometimes superior over the heat hazard. Therefore, an enormous amount of research and efforts have gone into the field of fire smoke toxicity suppression in the past few decades. Some findings implied that layered materials possess the property of decreasing the toxic smoke released in the fire condition, because of their peculiar two-dimensional (2D) structure which can form a barrier to restrain the spread of toxic escaping gas [21,22,23]. Among these layered materials, graphene has attracted considerable attention owing to its excellent physical and chemical functionalities [24, 25]. It has demonstrated that graphene is very thermally stable even after being exposed to a flame, representing the high intrinsic flame resistance of graphene [26]. Recently, graphene or its derivatives have been demonstrated as flame-retardant nanoadditives to decreased fire hazards of various polymers [27, 28]. Though, the problem of the nonuniform distribution of graphene in polymer matrix must be resolved urgently [29, 30], owing to that the reaggregation and incompatibility between the graphene and the polymer matrix may deteriorate the performance of the polymer nanocomposites. In general, there have been principally two strategies, namely covalent and non-covalent functionalization, to upgrade the distribution and compatibility of graphene or graphene-based materials with polymers. Direct covalent functionalization not only considerably upgrades the distribution, but also forms strong interfacial interactions with polymer materials via covalent linkages. Pristine graphene is liable to agglomerate and even restack in polymer matrices owing to the powerful van der Waals force and π–π interactions, which restrains the distribution of graphene in polymer matrix [31, 32]. Subsequently, GO was prepared from graphite. The morphology, thermal stability, flammability, flame-retardant mechanism and smoke toxicity characteristics of the PS/GO nanocomposites were studied and argued in this work. It is expected that GO will run a favorable solution to decrease the fire hazards of PS, thus promoting the development of flame-retardant additives for polymers. This study presents the influence of Gr to mitigate some PS drawbacks.
Experimental
Materials
All the commercial chemicals were used as received without further purification. Polystyrene (PS 158 K, melt index 3.00 g/10 min (200 °C/5 kg, ASTM D1238), density 1.04 g/cm3, ASTM D792, glass transition temperature 100 °C, ISO 11357) was supplied by BASF. Graphite flakes (RFL 99.5) were obtained from Kropfmühl AG. Graphene oxide (GO) single layers were produced from expanded graphite using a modified Hummers’method [33].
Characterization
The SEM images were obtained on a HITACHI S-4800 scanning electron microscope. SEM images were obtained on freeze fractured cross sections of the PS composite sample bars. Raman spectra of the samples were obtained by a micro Raman system (DXR, GX-PT-2412, Thermo, USA) with 532 nm lasers. The Raman detector was equipped with a charge-coupled device (CCD) multichannel detector and Olympus confocal microscope. The laser beam was focused on the sample surface and scanned for a 5 s exposure time for 180 times, meanwhile the powders were measured with extended range grating for 400–2000 cm−1. Dynamic mechanical properties were measured with a DMA/SDTA861e (Mettle, Switzerland) in the stretching mode on sample bars (9 × 4 × 0.5 mm3). A temperature ramp experiment (2 °C/min) was conducted under air from room temperature to 180 °C at a constant frequency of 1 Hz. X-ray diffraction (XRD) patterns were taken on a Japan Rigaku D/Max-Ra rotating anode X-ray diffractometer equipped with a Cu Ka tube and Ni filter (k = 0.1542 nm). Transmission electron microscopy (TEM) images were carried out to observe the morphology of GO, and the distribution of GO in PS matrix used a Hitachi model H-800 TEM. Fourier-transform infrared spectroscopy (FTIR) was performed on a Nicolet 6700 FTIR spectrophotometer to characterize the GO using a thin KBr disk with the scanning range of wave number from 4000 to 500 cm−1. Thermogravimetric analysis (TGA) was conducted using a Q5000 IR thermogravimetric analyzer (TA Instruments) at a linear heating rate of 20 °C/min under nitrogen or air atmosphere. The weights of all the samples were maintained within 5 ± 1 mg. Samples in an open Pt pan were examined in an air flow rate of 60 ml/min in the temperature ranging from room temperature to 700◦C. The cone calorimeter experiments were carried out using an ULTRAMAT 6 instrument from FIRE (35 kW/m2 heat flux with horizontal orientation of the samples) according to ASTM E 1354, on 10 × 10 cm2 plaques (4 mm thickness). All samples were tested in triplicate, and the recorded cone data were reproducible within ± 5%. Oxygen index tests were carried out using the FTT (Fire Testing Technology) oxygen index apparatus and samples of dimensions 120 × 10 × 4 mm3. Testing was done according to ISO 4589.
Results and discussion
Figure 1a, b displays SEM photographs of GO and PS/GO composite with 2.0 wt% GO content. The morphology of GO is observed to be a wrinkled and folded layered structure. The composite PS/GO 2.0 wt% displays an overlapped layered morphology with some GO layers interlocked together. The surface of PS/GO 2.0 wt% composite is shown in Fig. 1b which presents that the fractured surface of PS/GO 2.0 wt% is rough. Though, some scaly patterns are detected on the surface of PS/GO 2.0 wt% composite, which implies that the GO is strongly stuck to the matrix [1,2,3,4]. Many agglomerates can be detected in PS matrix. The PS/GO 2.0 wt% composite is arbitrarily distributed in the matrix. The distribution in PS/GO 2.0 wt% could be also associated with favorable π–π interactions among the graphene sheets and the phenyl rings of PS [5, 6]. The negative oxygen groups on GO sheets will disrupt the π–π interactions between GO and PS matrix.

a SEM photograph of GO, b SEM photograph of PS/GO 2.0 wt%
In order to further investigate the distribution of GO in the polymer matrix, TEM analysis is carried out. The morphology and microstructure of the GO are shown in Fig. 2a. Figure 2a shows that the exfoliated GO consists of large thin sheets with diameters up to several micrometers. Figure 2b shows the TEM image of PS/GO 2.0 wt% composite. In this figure, the dark lines denote the GO sheets, while the white area signifies the PS matrix. Figure 2b presents that the majority of GO sheets are dispersed homogeneously in the PS matrix and there are some aggregates (marked by the ellipse). The TEM image (Fig. 2b) displays that the GO sheets are well dispersed through the polymer matrix. The polymer matrix displays an intercalate-exfoliated composite structure, which is in consistent with the result achieved from the XRD analysis.

a TEM photograph of GO, b TEM photograph of PS/GO 2.0 wt%
DMA is used to assess the mechanical properties of the PS and PS/GO composite. From DMA, we can obtain the storage modulus (E′) corresponding to the elastic response to the deformation and the loss modulus [Tan(δ)] describing the plastic response to the deformation. Upon measuring varying temperature, these properties can be related to transitions involving polymer mobility or segmental dynamics within the sample, such as the glass transition. Figure 3a shows the storage E′ of PS and its nanocomposite. Associated with PS, a slight decrease in E′ is observed for PS/GO 2.0 wt% composite. The uniform distribution of GO layers can facilitate superior interfacial interaction between the sheet and polymer matrix and thus restrict the segmental movement of the polymer chains considerably [7], leading to a higher storage modulus. The glass transition temperature (Tg) corresponds to the maximum of loss factor (Fig. 3b) which expected to be higher for PS/GO 2.0 wt% composite in comparison with pure PS which implies that the motion of polymer chains is greatly affected by the addition of GO [8].

a Storage modulus for PS and PS/GO 2.0 wt% at a frequency of 1 Hz and various temperatures. b Loss factor for PS and PS/GO 2.0 wt% at a frequency of 1 Hz and various temperatures
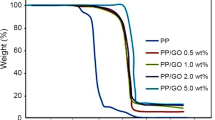
TGA was employed to examine the thermal degradation behaviors of GO and PS/GO composites. In the case of GO, the slight mass loss about 100 °C is ascribed to the release of adsorbed water. The maximum mass loss occurs in the temperature range of 180–250 °C, which is ascribed to the removal of unstable oxygen functional groups. Figure 4 shows the TGA curves of PS and the PS/GO nanocomposites under air and the relative data, including initial degradation temperature (T−0.5%), the temperature at a maximum mass loss rate (Tmax) and char yield at 700 °C. The inclusion of 0.5–2.0% GO considerably increases the T−5% of PS/GO nanocomposites associated with that of pure PS., which is ascribed to the combined effect of the physical hindrance and the capture of oxygen molecules and free radicals of GO layers under air atmosphere. As the GO content increases, the T−5% of the PS/GO nanocomposites is gradually decreased. Graphene could be easily burnt out under air atmosphere. Therefore, the earlier degradation of flame retardants on the surface of Gr protects properly the Gr against fire and thus reinforces the barrier effect of the GO sheets, which results in postponing thermal oxidation degradation process and upgrading the thermal stability of the PS nanocomposites. As shown in Fig. 6, the char residues at 700 °C of the PS nanocomposites under air increased with the inclusion of GO owing to the improved barrier effect of GOs, recommending the mass transfer deterring from nanocomposites to the flame region. Owing to the combined effect of the physical barrier of graphene and the upgraded char formation, the thermal stability of the PS/GO nanocomposites is significantly promoted. As can be observed, all the PS/GO nanocomposites present similar degradation behaviors to that of pure PS. Though, associated with that of the pristine PS, the thermal stability of the PS/GOs nanocomposites is improved clearly. It is summarized that the T−5%, T−10%, T−50% and Tmax of the PS nanocomposites are all higher than that of pure PS. The Tmax suggests GOs as an effective barrier to inhibit the mass loss during the thermal degradation process. These results suggest that including GOs into PS would retard the thermal degradation of PS molecular chains, which considerably developed the thermal stability of the PS nanocomposites which are ascribed to three aspects: (1) the strong interactions between graphene nanosheets and PS; (2) the physical barrier effect of the graphene nanosheets which deters the diffusion of volatile products; (3) the presence of GOs can promote char formation in the PS nanocomposites and the dense char layer will provide a good barrier to prevent the transfer of heat and volatiles, resulting in considerable upgrading of the thermal stability.

TGA curves of pure PS, GO and PS/GO nanocomposites
Figure 5a, b shows the digital photographs of the residual chars after cone calorimeter tests of PS and its nanocomposites. As can be seen, pristine PS almost does not form a char, while the addition of 2.0 wt% GO leads to the increase in char yield and forms a continuous and a compact char layer, which is good in agreement with the TGA results. The continuous and compact char surfaces are good barriers to protect the underlying polymers and inhibit the exchange of degradation products, combustible gases and oxygen. The mechanism of GO in decreasing the flammability of PS is probably ascribed to the higher char yield and the physical barrier effect of GO which inhibits the mass transfer and shield the underlying PS from the heat source. The flammability of the PS and PS/GO 2.0 wt% nanocomposite is performed by cone calorimetry, which can provide plenty of useful parameters including heat release rate (HRR), total heat release (THR), CO production and smoke production, etc. Figure 6a, b and Table 1 show the HRR and THR curves of PS and PS/GO nanocomposites versus time. It can be observed that pure PS burns up with a sharp peak heat release rate (PHRR) of 830 kW/m2 at 208 s after ignition. GO is usually used to impart flame-retardant properties of polymers owing to its unique 2D nanosheets structure [34]. As expected, the addition of 2.0 wt% GO gives rise to a 27% decrease in PHRR associated with that of pure PS and the time to PHRR is delayed to 233 s. These results imply that the barrier effect of graphene plays an important role in decreasing the heat release rate, which slows down the evolution of pyrolytic gases and transfer of radiant heat flux to the sample [9]. Moreover, including GO into PS also leads to the decrease in THR. The behavior in an ignition scenario was tested using the limiting oxygen index (LOI) test, where the minimum oxygen concentration necessary for combustion is obtained (Table 1). The LOI does not increase inconsiderably from 19% for the pristine polymer and PS/GOs nanocomposites. Table 1 presents the decreases in the PHRR of 30%, 44% and 53% obtained by PS/GO 0.5 wt%, PS/GO 1.0 wt% and PS/GO 2.0 wt%, respectively. Owing to the absence of diffraction peaks from GO in the composite, it suggested that GO was exfoliated during melt processing [10]. The distribution of GO in the polymer matrix is a key parameter for the decrease in PHRR. This behavior is best explained by the formation of a protective char layer on the sample surface [11]. This layer acts as a thermal insulator which slows down mass and heat transfer and separates the burning polymer from the flame [12]. Though, the massive decrease in the HRR at a later stage (after a char has formed) highlights the potential of this combination of fillers.

a Digital photographs of char for pure PS. b Digital photographs of char for PS/GO 2.0 wt%

Heat release rate curves for pure PS and PS/GO nanocomposites
Figures 6 and 7 present HRR and THR curves of PS and the PS/GO nanocomposites, and the detailed data are listed in Table 1. As shown in Fig. 6, the presence of GO markedly decreases the peak heat release rate (PHRR) values of the PS/GO nanocomposites. For example, including 2% GO into PS decreases the PHRR to 385 kW/m2 from 819 kW/m2 for pure PS, (approximately a 53% decrease). Moreover, GO increases the temperature at the maximum heat release rate, which is consistent with the superior thermal stability observed from TGA. The decreased heat release rate of PS/GO can be ascribed to two factors: on one hand, the GO layers, uniformly distributed in the PS matrix, play a good physical barrier role during combustion, which prevent fuels into the fire, capture oxygen free radicals and inhibit chain degradation of polymer matrix; on the other hand, the GO promotes the char formation on the surface of burning material and reinforces the barrier effect. Figure 7 shows that the addition of GO also decreased THR of PS/GO nanocomposites, and THR shows a similar trend to PHRR. Inclusion of GO decreased the PHRR and THR values of the PS nanocomposites, increased the temperature at the maximum heat release rate and thus upgrade the fire safety of PS. It can be observed that pure PS burns up with a sharp peak heat release rate (PHRR) of 819 kW/m2 at 208 s after ignition. GO is usually used to impart flame-retardant properties to polymers owing to its unique 2D nanosheets structure [13]. As expected, the addition of 2.0 wt% GO gives rise to a 53% decrease in PHRR associated with that of pure PS and the time to PHRR is delayed. These results imply that the barrier effect of graphene plays an important role in decreasing the heat release rate, which slows down the evolution of pyrolytic gases and transfer of radiant heat flux to the sample [14]. The ability of GO to decrease polymer flammability is probably ascribed to two factors: on one hand, the GO nanosheets, uniformly distributed in the PS matrix, play a good physical barrier role during combustion, which can retard the heat and mass transfer, and prevent the underlying material from further combustion. On the other hand, the presence of GO promotes the char formation on the surface of the burning material, reinforcing the barrier effect of the GO nanosheets, which is conducive to the upgrading of the flame retardancy. Overall, the introduction of GO into PS considerably decreases the PHRR and THR of the matrix, thereby upgrading the fire safety of PS nanocomposites.

Total heat release rate curves for pure PS and PS/GO nanocomposites
Figure 8 presents the smoke density of CO yield versus time of PS and PS/GO 2.0 wt% nanocomposite. Figure 8 shows the inclusion of GOs effectively decreases the CO yield and the smoke density associated with those of pure PS during combustion, which is principally ascribed to the adsorption effect of graphene [15].

Smoke density curves of pure PS and PS/GO 2.0 wt% nanocomposite
Figure 9 shows the FTIR spectra of PS, GO and PS/GO 2.0 wt%. In the FTIR spectrum of GO, peak at about 3410 cm−1 is assigned to the stretching vibration of CO–H. Peak at 1731 cm−1 is assigned to the C=O stretching, vibration; the absorption bands at 1624, 1052 and 1224 cm−1 are assigned to the stretching vibration of =C, C–O and C–OH, respectively [16]. In the FTIR spectrum of PS, bands at 3100–3000 cm−1 are assigned to =C–H aromatic stretching and vibration; peaks at 2920 and 2849 cm−1 present asymmetric and symmetric stretching vibration of CH2, respectively; peaks at 1600, 1580 and 1491 cm−1 are ascribed to stretching vibration of benzene ring; peaks at 753 and 697 cm−1 are related to the C–H out-of-plane bending vibration of the benzene ring [17]. For PS/GO composite, except the stretching vibration of C=C at about 1630 cm−1, the typical absorptions of GO are not detected because that is either too weak or overlap with the absorption peak of PS.

FTIR spectra of pure PS, GO and PS/GO 2.0 wt% nanocomposite
Figure 10 shows the Raman spectra of the residual char of the PS/GO nanocomposite. Raman scattering spectroscopy is an effective method to examine the microstructure of carbonaceous materials because of its sensitivity to these materials. Raman spectra were used to investigate the effect of GO on the condensed phase products of PS/GO nanocomposite after combustion. There is no result for PS because PS burns completely without char residues. The Raman spectra of PS/GO nanocomposite exhibit two strong peaks at approximately 1595 and 1360 cm−1,which are typical of graphitic phases [18]. The peak at 1595 cm−1 corresponds to the ordered carbon. The other peak at 1360 cm−1 is ascribed to the amorphous carbon. The relative intensity and width of the bands can be consistent with partially ordered carbon. Generally, the graphitization degree of residual char is assessed by the ratio of the intensity of ordering carbon to amorphous carbon. In brief, the relative content of graphitic carbon is increased, owing to the transformation from sp3 hybrid carbon to sp2 which implies a decrease in the size of the in-plane sp2 domains and higher exfoliation of graphene layers [19]. Higher ratio also suggests higher thermal stability of residual char which implies that the presence of GO upgrades the graphitization degree of char residues. Therefore, GO reinforces the char layers and retards the mass and heat transfer of PS/GO nanocomposites during the combustion process. These thermally stable char layers are responsible for the decreased heat release rate and suppressed smoke toxicity.

Raman spectra of the residual char of GO and PS/GO 0.5 wt%
XRD pattern of GO shows a sharp diffraction peak at about 11° corresponding to the (002) reflection of GO. The weak peak at about 42° is ascribed to the (100) reflection of GO [20]. It can be seen from Fig. 11 that the (002) peak intensity of the sample PS/GO 2.0 wt% considerably decreases and a new broad peak at about 22° appears. This may be owing to the partial decrease in GO sheets. The almost undetectable (002) peak and the new sharp peak at about 22° imply the further decrease in GO into graphene sheets.

XRD patterns of PS, GO and PS/GO 2.0 wt%
Conclusion
In this work, GO was successfully produced and well characterized by FTIR, XRD, SEM and TEM. TGA results denoted that the presence of GO promoted the thermal stability of PS. Then, the PS/GO nanocomposites with the different ratios of GO were prepared and the GO nanosheets were well distributed in the PS matrix confirmed by TEM and SEM. The PS/GO nanocomposites presented better thermal stability associated with pure PS, including higher char yields, higher initial and maximum decomposition temperature. The outstanding thermal stability of the PS/GO nanocomposites is credited to GO that reinforced the barrier effect and the char formation in the thermal degradation process. Furthermore, the flame retardancy and thermal stability of the PS/GO nanocomposites were promoted after GO addition, with clearly reduced PHRR and THR values, which is attributed to the good dispersal of nanosheets in the matrix and the high char yield during combustion. Similarly, the introduction of GO into PS reduced CO concentration during combustion. The inclusion of GO nanosheets reduced the decomposition rate of PS and augmented the amount of char residues. The development of a protective layer isolates the burning polymer from the flame and leads the combustion at much lower HRR, which consequently, delayed burn out time. DMA showed that both the storage modulus and Tg of PS/GO nanocomposites are significantly improved associated with that of pristine PS. In brief, GO delivers an efficient plan to minimize the fire hazards of the PS materials. Such a great advancement in thermal stability and decrease in fire hazards of PS were mainly attributed to the good dispersal, physical barrier effect and char development of GO nanosheets.
References
Sabet M, Hassan A, Ratnam CT (2013) Electron-beam irradiation of low density polyethylene/ethylene vinyl acetate blends. J Polym Eng 33(2):149–161
Sabet M, Savory RM, Hassan A, Ratnam CT (2013) The effect of TMPTMA addition on electron-beam irradiated LDPE, EVA and blend properties. Int Polym Process 28(4):386–392. https://doi.org/10.3139/217.2723
Sabet M, Anuwar MSB (2013) Calcium stearate and alumina trihydrate addition of irradiated LDPE, EVA and blends with electron beam. In: Applied mechanics and materials, vol 290, pp 31–37. https://doi.org/10.4028/www.scientific.net/AMM.290.31
Sabet M, Hassan A, Ratnam CT (2015) Properties of ethylene–vinyl acetate filled with metal hydroxide. J Elastom Plast 47(1):88–100
Sabet M, Hassan A, Ratnam CT (2012) Electron beam irradiation of low-density polyethylene filled with metal hydroxides for wire and cable applications. Polym Bull 69(9):1103–1114
Sabet M, Soleimani H, Hassan A, Ratnam CT (2014) Electron beam irradiation of LDPE filled with calcium carbonate and metal hydroxides. Polym Plast Technol Eng 53(13):1362–1366
Sabet M, Hassan A, Ratnam CT (2013) Flammability and thermal characterization of aluminum hydroxide filled with LDPE. Int Polym Process 28(4):393–397
Sabet M, Hassan A, Nooroldin S (2018) Effect of addition graphene to ethylene vinyl acetate and low-density polyethylene. 24:E177–E185. https://doi.org/10.1002/vnl.21628
Sabet M, Hassan A, Ratnam CT (2015) Properties of ethylene–vinyl acetate filled with metal hydroxide. Elastom Plast 47(1):88–100
Hosseini SN, Shuker MT, Sabet M, Zamani A, Hosseini Z, Shabib-Asl A (2015) Brine ions and mechanism of low salinity water injection in enhanced oil recovery: a review. Res J Appl Sci Eng Technol 11(11):1257–1264
Busahmin B, Maini B, Ramao RK, Sabet M (2016) Studies on the stability of the foamy oil in developing heavy oil reservoirs. Defect Diffus Forum 371:111–116
Huang G, Chen S, Liang H, Wang X, Gao J (2013) Combination of graphene and montmorillonite reduces the flammability of poly(vinyl alcohol) nanocomposites. Appl Clay Sci 80–81:433–437
Dittrich B, Wartig K-A, Hofmann D, Mülhaupt R, Schartel B (2013) Flame retardancy through carbon nanomaterials: carbon black, multi wall nanotubes, expanded graphite, multi-layer graphene and graphene in polypropylene. Polym Degrad Stab 98:1495–1505
Hong N, Song L, Richard Hull T, Stec AA, Wang B, Pan Y, Hu Y (2013) Facile preparation of graphene supported Co3O4 and NiO for reducing fire hazards of polyamide 6 composites. Mater Chem Phys 142:531–538
Han Z, Wang Y, Dong W, Wang P (2014) Enhanced fire retardancy of polyethylene/alumina trihydrate composites by graphene nanoplatelets. Mater Lett 128:275–278
Wang X, Xing W, Feng X, Yu B, Lu H, Song L, Hu Y (2014) The effect of metal oxide decorated graphene hybrids on the improved thermal stability and the reduced smoke toxicity in epoxy resins. Chem Eng J 250:214–221
Wang D, Zhang Q, Zhou K, Yang W, Hu Y, Gong X (2014) The influence of manganese–cobalt oxide/graphene on reducing fire hazards of poly(butylene terephthalate). J Hazard Mater 278:391–400
Huang G, Wang S, Song P, Wu C, Chen S, Wang X (2014) Combination effect of carbon nanotubes with graphene on intumescent flame-retardant polypropylene nanocomposites. Compos Part A 59:18–25
Huang G, Chen S, Song P, Lu P, Wu C, Liang H (2014) Combination effects of graphene and layered double hydroxides on intumescent flame-retardant poly(methyl methacrylate) nanocomposites. Appl Clay Sci 88–89:78–85
Li K-Y, Kuan C-F, Kuan H-C, Chen C-H, Shen M-Y, Yang J-M, Chiang C-L (2014) Preparation and properties of novel epoxy/graphene oxide nanosheets (GON) composites functionalized with flame retardant containing phosphorus and silicon. Mater Chem Phys 146:354–362
Wang Z, Wei P, Qian Y, Liu J (2014) The synthesis of a novel graphene-based inorganic–organic hybrid flame retardant and its application in epoxy resin. Compos Part B 60:341–349
Liu S, Yan H, Fang Z, Wang H (2014) Effect of graphene nanosheets on morphology, thermal stability and flame retardancy of epoxy resin. Compos Sci Technol 90:40–47
Hong N, Zhan J, Wang X, Stec AA, Richard Hull T, Ge H, Xing W, Song L, Hu Y (2014) Enhanced mechanical, thermal and flame retardant properties by combining graphene nanosheets and metal hydroxide nanorods for acrylonitrile–butadiene–styrene copolymer composite. Compos Part A 64:203–210
Hong N, Song L, Wang B, Stec AA, Richard Hull T, Zhan J, Hu Y (2014) Co-precipitation synthesis of reduced graphene oxide/NiAl-layered double hydroxide hybrid and its application in flame retarding poly(methyl methacrylate). Mater Res Bull 49:657–664
Hu W, Yu B, Jiang S-D, Song L, Hu Y, Wang B (2015) Hyper-branched polymer grafting graphene oxide as an effective flame retardant and smoke suppressant for polystyrene. J Hazard Mater 300:58–66
Feng Y, He C, Wen Y, Ye Y, Zhou X, Xie X, Mai Y-W (2018) Superior flame retardancy and smoke suppression of epoxy-basedcomposites with phosphorus/nitrogen co-doped graphene. J Hazard Mater 346:140–151
Zhou K, Gao R (2017) The influence of a novel two dimensional graphene-like nanomaterial on thermal stability and flammability of polystyrene. J Colloid Interface Sci 500:164–171
Ji Y, Li Y, Chen G, Xing T (2017) Fire-resistant and highly electrically conductive silk fabrics fabricated with reduced graphene oxide via dry-coating. Mater Des 133:528–535
Zhou K, Gui Z, Hu Y, Jiang S, Tang G (2016) The influence of cobalt oxide–graphene hybrids on thermal degradation, fire hazards and mechanical properties of thermoplastic polyurethane composites. Compos Part A 88:10–18
Zhou K, Gui Z, Hu Y (2016) The influence of graphene based smoke suppression agents on reduced fire hazards of polystyrene composites. Compos Part A 80:217–227
Chen X, Ma C, Jiao C (2016) Enhancement of flame-retardant performance of thermoplastic polyurethane with the incorporation of aluminum hypophosphite and iron-graphene. Polym Degrad Stab 129:275–285
Han Y, Wang T, Gao X, Li T, Zhang Q (2016) Preparation of thermally reduced graphene oxide and the influence of its reduction temperature on the thermal, mechanical, flame retardant performances of PS nanocomposites. Compos Part A 84:336–343
Edenharter A, Feicht P, Diar-Bakerly B, Beyer G, Breu J (2016) Superior flame retardant by combining high aspect ratio layered double hydroxide and graphene oxide. Polymer 91:41–49
Sun F, Yu T, Hu C, Li Y (2016) Influence of functionalized graphene by grafted phosphorus containing flame retardant on the flammability of carbon fiber/epoxy resin (CF/ER) composite. Compos Sci Technol 136:76–84
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sabet, M., Soleimani, H., Mohammadian, E. et al. Impact of inclusion graphene oxide nanosheets on polystyrene properties. Int J Plast Technol 23, 92–100 (2019). https://doi.org/10.1007/s12588-019-09236-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12588-019-09236-5