
Abstract
A comparative study of thermal and thermal-oxidative degradation processes for polyethylene/organically modified montmorillonite (PE-MMT) nanocomposites, prepared by the ethylene intercalative polymerization in situ with or without subsequent addition of an antioxidant is reported. The results of TGA and time/temperature-dependent FTIR spectroscopy experiments have provided evidence for an accelerated formation and decomposition of hydroperoxides during the thermal oxidative degradation tests of PE-MMT nanocomposites in the range of 170–200 °C as compared to the unfilled PE, thus indicating to a catalytic action of MMT. It has been shown that effective formation of intermolecular chemical cross-links in the PE-MMT nanocomposite has ensued above 200 °C as the result of recombination reactions involving the radical products of hydroperoxides decomposition. Apparently, this process is induced by the oxygen deficiency in PE-MMT nanocomposite due to its lowered oxygen permeability. It is shown that the intermolecular cross-linking and dehydrogenation reactions followed by the shear carbonization lead to appreciable increase of thermal-oxidative stability of PE nanocomposite, as compared to that of pristine PE. Notably, the TGA traces for the antioxidant-stabilized PE-MMT nanocomposites recorded in air were quite similar to those obtainable for the non-stabilized PE-MMT nanocomposites in argon. The results of treatment of the experimentally acquired TGA data in frames of an advanced model kinetic analysis are reported and discussed.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyethylene (PE), being the most commercially important thermoplastic commodity, is heavily used for consumer products in many applications, but in a number of cases general applicability of PE turns out to be undermined by its relatively low thermal stability and flame resistance. The concept of compounding polymer matrices with nanoscale fillers (in particular, clays or layered silicates) has already been proved to be an effective method of preparing nanocomposites with excellent physical and mechanical properties [1−11]. It is believed that, in the course of high temperature pyrolysis and/or combustion, clay nanoparticles are capable of promoting formation of protective clay-reinforced carbonaceous char which is responsible for the reduced mass loss rates, and hence the lower flammability. Accordingly, considerable attention has been paid also to polyolefin/layered silicate nanocomposites. Reportedly, the latter have exhibited improved mechanical properties, gas impermeability, thermal stability, and flame retardancy as compared with corresponding pristine polymers [4, 5, 9, 10, 12].
This study deals with polyethylene/layered silicate nanocomposites prepared by intercalative polymerization route [12]. The method includes the intercalation of a metallorganic catalyst into the interlayer spacing of organically modified MMT and the subsequent polymerization of ethylene. In result of polymer chains growing within the interlayer spacing of montmorillonite (MMT) the exfoliation the original MMT particles to the nanoscale inorganic monolayers takes place.
In order to clarify the mechanisms of the carbonaceous char formation, that may be responsible for the reduced mass loss rates, and hence the lower flammability of the polymer matrices, a number of thermo-physical characteristics of the PE/MMT nanocomposites have been measured in comparison with those of the pristine PE (which, by itself is not a char former) in both inert and oxidizing atmospheres. The evolution of the thermal and thermal-oxidative degradation processes in these systems was followed dynamically with the aid of TGA and FTIR methods. The thermal-oxidative stability of PE nanocomposites in both the absence as well as in the presence of an antioxidant was investigated. Several sets of experimentally acquired TGA data have provided a basis for accomplishing thorough model-based kinetic analyses of thermal and thermal-oxidative degradation of both pristine PE and PE-n-MMT nanocomposites prepared in this work.
Experimental
Materials
A Cloisite 20A (purchased from Southern Clay Products, Inc.) has been used as the organically modified montmorillonite (MMT) to prepare PE/MMT nanocomposites throughout this study. The content of an organic cation-exchange modifier, N+2CH32HT (HT = hydrogenated tallow, C18 ≈ 65%; C16 ≈ 30%; C14 ≈ 5%; anion: Cl−), in the MMT was of 38 wt.%. VCl4 (vacuum distilled at 40 °С before use, TU 48-05-50-71) and Al(i-Bu)3 (Aldrich) have been used for catalytic activation of MMT. Ethylene monomer was of a standard polymerization grade.
Procedure of polyethylene nanocomposite synthesis
Intercalation of the catalyst has been accomplished by treating the freshly dehydrated MMT with Al(i-Bu)3 and then with VCl4. The polymerization reaction was started by admitting ethylene into the reactor and was carried out until of PE nanocomposite (PE-n-MMT) of adjusted composition was obtained. The polymerization reaction was stopped by adding ethanolic HCl solution (5 wt.% HCl) to the reactor. The polymer composite product was filtered off, washed several times with ethanol, and dried under vacuum at 60 °C. The weight loads of MMT in the resulting composites were calculated by neglecting the contribution of the organic modifier in MMT. The sample of unfilled polyethylene was prepared by ethylene polymerization on VCl3 activated with Al(i-Bu)3 at the same conditions as applied to the nanocomposite synthesis.
Stabilized samples of both the nanocomposites (st-PE-n-MMT) and pristine PE (st-PE) were prepared by treating them with synergetic composition of Topanol CA and di-lauryl-3,3′-thiodipropionate (DLTDP) [13] solutions in heptane at 70 °C, followed by drying in vacuum. The concentrations of Topanol and DLTDP in st-PE-n-MMT and st-PE comprised 0.3 and 0.5 wt.%, respectively. For further testing, the prepared materials were hot-pressed into films at applied pressure of 20 MPa and 160 °C.
Characterization of materials
Small-angle X-ray scattering (SAXS)
The structure of the composites was studied by SAXS using a KRM-1 camera (CuKα radiation, λ = 0.154 nm, Ni filter). The test samples were powders or films. The data collected were normalized with due regard to the concentration of MMT and the coefficients of attenuation.
Transmission electron microscopy (TEM)
Micrograph of PE nanocomposite sample was obtained on a JEM-100B transmission electron microscope at an accelerating voltage of 80 kV. The sample of 70 nm thickness was cut with the aid of LKV-III ultramicrotome from the composite plate prepared by hot pressing.
Thermogravimetric analysis (TGA)
A Perkin-Elmer TGA-7 instrument calibrated by Curie points of several metal standards has been employed for non-isothermal thermogravimetric analysis. The measurements were carried out at a desired heating rate (in the range of 3–40 K/min) in both inert (argon) and oxidizing (oxygen) atmospheres, as appropriate.
Fourier transform infrared spectroscopy (FTIR)
Infrared spectra of the investigated materials in their nascent form were acquired with the aid of a Perkin-Elmer 1725X FTIR instrument by using a Spectra-Tech “Collector” DRIFT accessory furnished with a heated sample post, embedded thermo couple and the corresponding external heater/controller providing temperature reading precision of ±1.0 °C. The series of FTIR spectra for the polymer samples have been recorded at systematically varied temperatures or over predetermined time intervals (in isothermal regimes) by employing a modified diffuse reflectance-absorbance Fourier Transform (DRAFT) spectroscopy technique published elsewhere [14]. All measurements were performed using the instrument DTGS detector and a 4 cm−1 resolution.
Kinetic analysis of PE compositions thermal degradation was carried out using Thermokinetics software by NETZSCH-Gerätebau GmbH.
Results and discussion
PE nanocomposite structure
Small-angle X-ray scattering (SAXS) has been used to evaluate the degree of exfoliation of the organoclay particles in the polymer matrix [12]. SAXS diffractograms of pristine C20A MMT and those of PE nanocomposites prepared by the intercalation polymerization route for MMT contents of 2.0 vol.% (2) and 6.5 vol.% (3) are shown in Fig. 1. The SAXS curve for C20A MMT shows a reflection at around of 3.6° corresponding to the interlayer mean distance of 2.46 nm (Fig. 1(1)). As can be seen from the same Fig. 1(2, 3), for the PE/clay nanocomposites having different MMT contents, the 3.6° reflection is absent. The full exfoliation of the MMT particles to the monolayers takes place under the action of PE forming in the course of polymerization in the interlayer spacing.

SAXS patterns for the original C20A MMT (1) and PE nanocomposites with MMT content of 2.0 vol.% (2) and 6.5 vol.% (3)
Figure 2 shows TEM image of the PE nanocomposite containing 1 vol.% of MMT. The dark features in the micrograph correspond to the exfoliated monolayers and nanostacks of MMT distributed throughout the PE matrix. It can be seen that the nanoscale MMT layers lack any sort of orientation in the matrix of the pressed composite. The exfoliated MMT particles are characterized by very high aspect ratio (longitudinal size:thickness).

TEM micrograph of the PE nanocomposite containing 1% by volume of MMT
It is worth noting here that the organic ammonium cations present in pristine C20A MMT are susceptible to washing out from the interlayer spacing of MMT under acidic treatment of the latter with ethanolic HCl (5 wt.%). The ammonium cations are substituted for protons, and this leads to a prominent decrease of the interlayer distances in the MMT structure (Fig. 3). If the intercalative polymerization of ethylene would not accomplish exfoliation of the MMT particles in the composite to the full extent, the diffractogram should contain a wide reflection positioned somewhere at greater angles than in the pristine C20A MMT. It is also obvious that the acidic after-treatment of the synthesized exfoliated nanocomposites should lead to removal of the major part of the original organic MMT modifier.

SAXS patterns for the original C20A MMT (1) and C20A MMT treated with HCl solution in ethanol (5 wt.% HCl) (2)
Thermal degradation of PE nanocomposite
It is accepted that thermal stability of polymer nanocomposites is higher than that of pristine polymers, and this gain is explained by the presence of anisotropic clay layers hindering diffusion of volatile products through the nanocomposite material. It is important to note that the exfoliated nanocomposites, prepared and investigated in this work, have much lower gas permeability in comparison with that of pristine unfilled PE [12]. Thus, the study of purely thermal degradation process of PE nanocomposite seemed to be of interest in terms of estimation of the nanoclay barrier effects on thermal stability of polyolefin/clay nanocomposites.
The radical mechanism of thermal degradation of pristine PE has been widely discussed in a framework of random scission type reactions [14–22]. It is known that PE decomposition products comprise a wide range of alkanes, alkenes, and dienes. The polymer matrix transformations, usually observed at lower temperatures and involving molecular weight alteration without formation of volatile products, are principally due to the scission of weak links, e.g., oxygen bridges, incorporated into the main chain as impurities. The kinetics of thermal degradation of PE is frequently described by a first-order model of mass conversion of the sample [22]. A broad variation in Arrhenius parameters can be found in literature, i.e., activation energy (E) ranging from 160 to 320 kJ/mol and pre-exponential factor (A) variations in the range of 1011 and 1021 s−1 [20–22] are not unusual. It is believed that the broad range of E values reported may be explained by the polymers molecular mass variations, by use of various additives, and by different experimental conditions [22] employed by different authors.
Previously Bockhorn et al. have reported that thermal degradation of PE leads to a large number of paraffins, dienes and olefins without a residue formation [20]. In order to formulate a simple kinetic model adequately explaining the isothermic global kinetic data of the authors, a mechanism embracing only the main reactions including initiation by random scission of the polymer chains into primary radicals, β-scission of these radicals, hydrogen transfer (intra- and inter-molecular) and recombination reactions has been proposed [20]. It is important that the change in the reaction order is dependent on the intermolecular hydrogen transfer in reaction leading to the alkanes. Thus, at high temperatures and at high degrees of conversion, the alkanes formation via reaction of intermolecular hydrogen transfer becomes favored and, therefore, the reaction order alters from 0.5 to 1.5 [20].
In the present work, the processes of thermal degradation of both unstabilized PE and PE-n-MMT nanocomposite with MMT content of 4.3 wt.% have been investigated by TGA in an inert (argon) atmosphere at the heating rates of 3, 5, 10, and 20 K/min. According to the dynamic TGA data, the polymer degradation starts at about 300 °C and then, through a complex radical chain process, the material totally destructs and completely volatilizes in the range of 500–550 °C (Fig. 4). It is obvious that, taken at the same heating rates in argon, the thermograms for pristine PE and PE-n-MMT are practically identical, except that the solid silicate residue amounting to 4–5 wt.% can be seen on the curves for the nanocomposites (Fig. 4). This result suggests that the mechanisms of thermal degradation of PE and PE-n-MMT nanocomposites, and hence the global kinetic parameters of their thermal degradation processes are rather similar.

TGA thermograms for PE (firm lines) and PE-n-MMT (dotted lines) taken in Ar at the heating rates of: 5 K/min—1, 2 and 20 K/min—3, 4
Kinetic analysis of PE nanocomposite thermal degradation based on TGA data
Kinetic studies of material degradation have long history, and there exists a long list of data analysis techniques employed for the purpose. Often, TGA is the method of choice for acquiring experimental data for subsequent kinetic calculations, and namely this technique was employed here.
It is commonly accepted that the degradation of materials follows the base Eq. 1 [14]
where: t—time, T—temperature, c o—initial concentration of the reactant, and c f—concentration of the final product. The right-hand part of the equation F(t,T,c o,c f) can be represented by the two separable functions, k(T) and f(c o,c f):
Arrhenius equation (4) will be assumed to be valid for the following:
Therefore,
All feasible reactions can be subdivided onto classic homogeneous reactions and typical solid-state reactions, which are listed in Table 1 [23]. The analytical output must provide good fit to measurements with different temperature profiles by means of a common kinetic model.
Kinetic analysis of PE and PE-n-MMT thermal degradation at heating rates of 3, 5, 10 and 20 K/min was accomplished by using a NETZSCH Thermokinetics software in accordance with a formalism we proposed earlier [7].
In order to assess the activation energy for development of a reasonable model for kinetic analysis of pristine PE and PE-n-MMT thermal degradation processes, a few evaluations by model-free Friedman analysis have been done as the starting point [24].
Further, nonlinear model fitting procedure for PE and PE-n-MMT TGA-curves has led to the following triple-stage model scheme of successive reactions (Fig. 5):
Taking this as a reasonable approximation for PE and PE-n-MMT, the fits with the aid of nonlinear regression were attempted by the model (5), where an one-dimensional diffusion type reaction was used for the first step and the nth-order (Fn) reaction—for the two subsequent steps of the overall thermal degradation process (Fig. 5; Table 1).

Outcome of multiple models-based nonlinear fitting for PE-n-MMT. The experimental TGA-data (dots) in comparison with the model calculations results (firm lines) are shown for different heating rates: 3 K/min—(1), 5 K/min—(2), 10 K/min—(3) and 20 K/min—(4)
Assuming a radical chain mechanism is operative in the process of PE and PE-n-MMT thermal degradation, the apparent activation energy and the pre-exponential factor values calculated in this work (Table 2) turned out to be in perfect match with the data from isothermal analysis and dynamic TGA published earlier (E a = 268 ± 3 kJ/mol, log A = 17.7 ± 0.01 min−1 and E a = 262.1 kJ/mol, log A = 18.09 ± 0.14 min−1 [25, 26]).
The TGA data acquired for PE and PE-n-MMT in argon has not provided any evidence in favor of the hypothesis that the barrier effect, being clearly manifested in the gas permeability experiments with the same PE-n-MMT at room temperature [12], is operative also during thermally stimulated degradation of PE-n-MMT. It should be noted that, in an inert atmosphere, degradation/volatilization of both PE and PE-n-MMT starts at about 350 °C and is totally completed up to 500–550 °C, not taking into account a solid silicate residue amounting to 4–5 wt.% which remains in the case of the nanocomposites (Figs. 4, 5). Based on TGA data, the first stage of the degradation process (1D-diffusion limiting stage) develops in the range of 350–410 °C corresponding to the overall mass loss of 5–7%. The subsequent steps of the thermal degradation processes (410–500 °C) for PE and PE-n-MMT proceed in the liquid melt of high molecular weight degradation products. In the light of the above findings, we believe that during the high-temperature degradation stages (above, e.g., 410 °C) in an inert atmosphere, the barrier diffusion restrictions can become insignificant since the viscosity of the pyrolyzed polymer melt at these temperatures is rather low and, because of the intensified mobility of the clay layers in such melt, the overall ‘labyrinth effect’ normally provided by the clay particles in more viscous matrices may be considerably diminished.

Differences in coloration of neat PE (1) and PE-n-MMT (2) samples—both heated for 2 min at 180 °C in air
Thermal-oxidative degradation of PE nanocomposite
Thermal-oxidative degradation of PE and PE nanocomposites has been extensively studied over the past decades [27–31]. It has been reported that the main oxidation products of PE are aldehydes, ketons, carboxylic acids, esters, and lactones [27, 28]. According to Lacoste and Carlsson [29], β-scission plays an important role in thermal oxidation of UHMWPE. Notably, the feasibility of intra-molecular hydrogen abstraction by the peroxy radicals for polyethylene has been questioned in frames of a thermal oxidation mechanism proposed by Gugumus [30, 31].
It is usually supposed that the reaction of hydrogen abstraction from an alkane molecule, R–H, may lead to hydroperoxides and alkyl radicals according to the overall reaction scheme (Scheme 1). A mechanism describing oxidation of organic molecules by virtue of complex chain reactions has been proposed earlier by Benson [32]. At temperature below 190 °C, oxidation of organic compounds involves free-radical chain initiation and the main products are hydroperoxides and oxygenated species indicated in the routes A1 and A2 of Scheme 1. At temperatures below 200 °C, the abstraction of H from \( {\text{R}}^{{\hbox{${\scriptscriptstyle \bullet}$}}} \) resulting to \( {\text{HO}}^{{\hbox{${\scriptscriptstyle \bullet}$}}}_{{\text{2}}} + {\text{olefin}} \) (routes B1 and B2) proceeds at least 200 times slower than the addition of O2 to \( {\text{R}}^{{\hbox{${\scriptscriptstyle \bullet}$}}} \) to give\( {\text{RO}}^{{\hbox{${\scriptscriptstyle \bullet}$}}}_{{\text{2}}} . \) Above 250 °C, the route A2 becomes reversible, and the very slow step B2 becomes rate determining. As a consequence, at temperatures above 300 °C, there is some retardation of the rate of oxidation of the polymer. The H2O2 can play the same role as ROOH in providing a secondary radical source just above 480 °C, where the rate of oxidation picks up again.

A flow-chart of elementary steps constituting PE thermal-oxidative degradation process
It is worth noting that a simple digital photo camera was of help for qualitative assessment of differences in the processes of thermal oxidation of neat PE as compared to PE-n-MMT. Figure 6 shows photograph of the neat PE (1) and PE-n-MMT (2) taken after both samples having been heated during 2 min at 180 °C in air. It can be seen that coloration of the PE-n-MMT sample (2) is much darker than that of the neat PE (1), thus evidencing that MMT promotes the carbonization of the polymer.
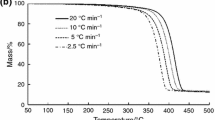
Figure 7 compares the TGA thermograms for neat PE and PE-n-MMT which has been acquired at a 10 K/min heating rate in air. Obviously, under the thermal-oxidative degradation conditions, these two materials demonstrate strikingly different behavior.

TGA curves for PE (1) and PE-n-MMT (2) recorded in air at the heating rates of 10 K/min
The earliest stage of thermal-oxidative degradation of unstabilized samples of PE-n-MMT and PE manifests itself as a clear weight gain feature emerging on the TGA curves well below 200 °C and is attributed to the oxygen absorption followed by the hydroperoxides formation (Fig. 7). Of importance, however, is the fact that for PE-n-MMT this process seems to be accelerated due to the presence of nanosilicate additive as compared with the pristine PE. Dependences of the hydroperoxides formation onset temperatures versus the heating rate, which have been derived from the TGA data for unstabilized PE-n-MMT and PE samples, are presented in Fig. 8.

The onset temperatures of hydroperoxides formation vs. heating rate for: PE (1), PE-n-MMT (2)
We believe that O2 molecules, being adsorbed on the defect centers of MMT represented by the impurities, transform into more active species which are able to react with PE at lower temperatures, thus inducing formation of active centers on the hydrocarbons chains (Scheme 2). Apparently, this chain of events should result to accelerated formation of PE hydroperoxides

The earliest stages of the process of PE thermal-oxidative degradation in the presence of exfoliated MMT nanoparticles
Notably, while the hydroperoxides accumulation starts at lower temperatures in PE-n-MMT than in the unfilled PE (cf., e.g., Fig. 8), the clearly visible mass loss of the nanocomposite (attributable solely to decomposition of the accumulated hydroperoxides) ensues at lower temperatures as well. It is reasonable to suggest that this effect is caused by a catalytic action of exfoliated MMT nanoparticles on the hydroperoxides decomposition. As it has been mentioned above, the treatment of PE-n-MMT with alcoholic HCl solution led to substitution of the major part of the organic modifier by acidic protonic centers. Moreover, it is widely accepted that MMT-type clay minerals always comprise a plenty of different catalytically capable sites, which may be represented by weakly acidic Brønsted-like Si–OH sites, by strongly acidic –OH groups localized at the edges of the silicate layers, by transition metal cations captured in the galleries, and by crystallographic defect sites within the layers [33, 34]. All these sites are able to trigger decomposition of hydroperoxides within the bulk of the PE-n-MMT. It has been shown that acid-catalyzed rearrangements of hydroperoxides can proceed in both polar and non-polar solvents. Hence, such rearrangements can be expected to occur also in PE-n-MMT. Acids can decompose primary and secondary hydroperoxides according to two different pathways [35]. Both these routes are depicted in Scheme 3 for the secondary hydroperoxides most probably present in PE. Since mobility of the methylene units in the PE backbone is rather limited, it is reasonable to assume that reaction (2) in Scheme 3 should be of minor importance. Then the main reaction (reaction (1) in Scheme 3) must lead to transformation of the hydroperoxide into the ketone group with elimination of water. The reaction might proceed according to the general mechanism or be simply dehydration [36].

Acid-catalyzed decomposition of PE hydroperoxides
At the same time, the acidic sites of MMT may turn out to be sufficiently active to abstract single electrons from donor molecules with formation of free radicals, the latter being capable of further accelerating the thermal oxidation of PE chains, e.g., by virtue of branching reactions. In addition to the accumulation and subsequent decomposition of the hydroperoxides on the earlier stage resulting to emergence of the oxygen-containing groupings, the process of thermal-oxidative degradation of the nanocomposite involves the reactions of oxidative dehydrogenation and intermolecular cross-linking. It seems reasonable to suggest that namely at this step the thermally stable carbonized charred layer on the nanocomposite surface is formed and starts to hinder the diffusion transport of both the volatile degradation products (out of the polymer melt into the gas phase) and the oxygen (from the gas phase into the polymer). The above set of events results in actual increase of the nanocomposite thermal stability in the temperature range of 350–500 °C, where normally a shear degradation of the main part of PE takes place. This point is illustrated by TGA and DTG plots presented in Figs. 7, 9, and 10.

Acquired at 10 K/min heating rate in air or argon TGA and DTG curves for: PE in air (1), st-PE in air (2), PE in Ar (3)

Acquired at 10 K/min heating rate in air or argon TGA and DTG curves for: PE-n-MMT in air (1), st-PE-n-MMT in air (2), PE-n-MMT in Ar (3)
The diverse behavior of stabilized and unstabilized samples (Figs. 9, 10, curves 1, 2 TG and DTG) shows that the addition of antioxidants has resulted to higher thermal-oxidative stability. It can be seen also that the overall thermal-oxidative stability of PE-n-MMT independently of the antioxidant presence was higher than that of the pristine PE. Moreover, incorporation of the antioxidants in PE-n-MMT has led to a notable change in the character of the mass loss process (Fig. 10, curves 2, 3 of TG and DTG). It is quite probable that the antioxidant is able to “deactivate” the sites of MMT that have been occupied earlier with absorbed oxygen. In the result, the MMT nanolayers could become chemically inert in respect to the hydroperoxides formation and hence to further accelerated PE oxidation. It may be seen as well that, with the exception of the first thermal oxidation step, the TGA and DTG curves for st-PE-n-MMT taken in air become closely resembling those characteristic for PE-n-MMT run in argon.
Having taken into account the above findings, it seems reasonable to explain the observed retardation of thermal-oxidative degradation of st-PE-n-MMT by the capability of exfoliated MMT nanolayers to hinder the diffusion of oxygen throughout the partly cross-linked and carbonized nanocomposite matrix.
Kinetic analysis of PE-n-MMT thermal-oxidative degradation
Kinetic analysis of thermal-oxidative degradation of unstabilized PE and PE-n-MMT at the heating rates of 3, 5, 10, and 20 K/min (Fig. 11(a, b)) (as well as of the same samples stabilized with antioxidants) has been accomplished by using the aforesaid interactive model based nonlinear fitting approach.

Nonlinear kinetic modeling of PE-n-MMT (a) and st-PE-n-MMT (b) in air. Comparison between experimental TGA data (dots) and the model results (firm lines) at several heating rates: 3 K/min (1), 5 K/min (2) and 10 K/min (3)
With best fidelity, the undertaken nonlinear model fitting for the stabilized samples of PE and PE-n-MMT has provided a triple-stage model scheme of successive reactions, wherein an nth-order autocatalysis reaction (Cn) was used at the first step, while a general nth-order (Fn) reaction was used for both the second and the third steps of the overall process of thermal-oxidative degradation (Table 1):
For unstabilized PE and PE-n-MMT at the beginning stage of degradation, the degree of conversion depends on the heating rate (Fig. 11(a)), such dependence being a strong evidence in favor of a branched reaction path. For this case, the same approach has provided a dissimilar triple-stage model scheme comprising two competitive reactions: an nth-order autocatalytic reaction (Cn), for the first competing path, and two nth-order (Fn) successive reactions, for the second competing path (7).

As data in Table 3 for the first stage of thermal-oxidative degradation reaction show, the activation energies values for st-PE and st-PE-n-MMT amount to 74 and 96 kJ/mol, while for unstabilized materials those values are of 65 and 51 kJ/mol, correspondingly, thus indicating that the degradation of these samples is initiated by the similar oxygen induced reactions. At this stage, the lower activation energy of PE-n-MMT compared to that of PE may be related to the catalysis exerted by the ММТ during formation and decomposition of hydroperoxides.
At the same time, the values of activation energy found at the second and the third stages of thermal-oxidative degradation for PE-n-MMT are higher than those for PE (Table 3). This difference might be attributed to a shift of the PE-n-MMT degradation process to a diffusion-limited mode owing to emergence in the system of a carbonized cross-linked material.
We explained the fact of the formation of chemical crosslinking among the polyethylene macromolecules in the nanocomposite by means of FTIR measurements. At the heating of non stabilized samples of PE and PE-n-MMT above 200 °C it is clearly seen a sharp increase of absorbance in a range of 1,700–1,800 cm−1, corresponding to the carbonyl-containing thermal degradation products (Fig. 12). In addition, Fig. 12 shows the medium absorption band at 1,162 cm−1 which can be attributed to intermolecular esters groups (>С–О–С<) [36]. At the same time this peak is absent at the spectra of the pristine PE. It is known [27] that the contribution of radical recombination reactions, which lead to the intermolecular crosslinking, increases under the oxygen deficiency conditions. Such conditions are realized at thermal-oxidative degradation of the PE nanocomposites due to the labyrinth effect of the silicate layers towards the diffusing gas. The formation of chemical crosslinking during the thermal-oxidative degradation of the PE nanocomposites is a necessary condition of PE carbonization process.

Dynamic FTIR analysis of PE (a) and PE-n-MMT (b)
The activation energy values of st-PE-n-MMT at the first and the last stages are higher than those of st-PE (Table 3). Actually, at the last stage, the activation energy of st-PE-n-MMT rises up to 274.8 kJ/mol, almost reaching the activation energy value found for degradation of PE-n-MMT in inert atmosphere (274.3 kJ/mol) (Table 2). This fact infers that the last stage of the st-PE-n-MMT degradation process is governed mainly by random scission of C–C bonds, rather than by an oxygen catalyzed reactions. On the other hand, these results are also consistent with the barrier model mechanism, which suggests that inorganic clay layers can play a role of barriers retarding the diffusion of oxygen from gas phase into the nanocomposite.
Thus, from the TGA data for both antioxidant-stabilized and unstabilized PE and PE-MMT nanocomposite it follows that the organoclay nanoparticles can exert two counteracting effects influencing the thermal-oxidative stability of the PE-MMT nanocomposite:
-
catalytically accelerated accumulation and decomposition of hydroperoxides observed at the earlier stages, thus in fact promoting degradation of the polymer matrix and hence impairing the thermal stability of PE-n-MMT;
-
promoting of the carbonization of the polymer matrix tending to improve the thermal-oxidative stability of the nanocomposite.
Dynamic FTIR analysis of PE-n-MMT thermal-oxidative degradation
Simultaneously to TGA measurements, thermal-oxidative degradation of PE and PE-n-MMT samples has been monitored in this work with the aid of a dynamic FTIR spectroscopy in a temperature range of 25–260 °С. The overall evolution of the dynamic FTIR spectra in the course of thermal-oxidative degradation of PE and PE-n-MMT in the condensed phase is shown in Fig. 12(a, b).
At room temperature (25 °C), the FTIR spectra of both materials are typical for PE. The absorption bands at 2,845–2,960 cm−1 are assigned to –CH–, –CH2–, or –CH3 stretching vibration [37]. The absorption at 1,472 cm−1 is due to the deformation vibration of –CH2– or –CH3 groups, while that at about 720 cm−1 is due to (CH2)n rock when n ≥ 4 [37, 38]. Beside these, the FTIR spectra of PE-n-MMT revealed the absorptions belonging to MMT (ν (Si–O) 1,047 cm−1) [38]. Another MMT absorption band at 3,630 cm−1 has been assigned to the structural hydroxyl groups, directed toward the vacant positions in the inner octahedral layer of montmorillonite. Else, a broad absorption band of hydroxyl groups was observed at 3,400 cm−1 [39].
During the dynamic recording of the PE and PE-n-MMT spectra under the step-wise heating a sharp growth of the absorption in the range of 1,700–1,800 cm−1 was noted at temperatures above 200 °C indicating the emergence and accumulation of carbonyl-containing products resulting from the thermal-oxidative degradation process (Figs. 12, 13).

FTIR spectra of PE (1) and of PE-n-MMT (2) taken at 220 °C in the course of the heating runs (excerpted from the corresponding dynamic FTIR spectra sets)
Obviously, the complex band in the range of 1,700–1,800 cm−1 (Fig. 13) comprises: a carbonyl absorption (shoulder at 1,717 cm−1) belonging to ketone groups embedded into the polymer chain [38, 40], which are known to be the main oxidation products for the neat PE [38]; a shoulder peaking close to 1,734 cm−1, which is attributed to aldehyde groups [41]; another shoulder with maxima in the vicinity of 1,746 cm−1, which is normally assigned to ester groups vibrations [38, 40], and an absorption at 1,790 cm−1 belonging to the carbonyl in γ-lactone moiety [42]. Gradual growth of concentration of the vinylene groups absorbing close to 1,600 cm−1 [38] has also been noted (Fig. 12). The dynamic FTIR spectra show that, while the fractional ratio of different carbonyl absorptions was almost the same for both PE and PE-n-MMT, the apparent concentration of the carbonyl-containing products (overall intensity of the complex absorption band) in PE-n-MMT is considerably higher than in pristine PE (Figs. 12, 13).
The temperature dependences of the carbonyl index (CI) for samples of PE and PE-n-MMT are shown in Fig. 14. The carbonyl index (CI) was defined to illustrate the formation of non-volatile carbonyl containing oxidation products:

The carbonyl index (CI) versus temperature dependences for: (1) PE; (2) PE-n-MMT
where \( {\text{S}}_{{{\text{C=O}}}} \) is peaks area of carbonyl containing groups at 1,800–1,700 cm−1, S2019 is internal reference at 2,019 cm−1, CIo is initial carbonyl index.
Along with the carbonyl absorptions growth, the FTIR spectra have revealed a clear decay of intensities of the bands belonging to –СH2– vibrations as the result of the PE chain scission. Both investigated materials experienced the chain scission during pyrolysis, but with drastically different rates. The relative rates of the decay are illustrated by the overlaid spectra in Fig. 15 showing evolution of the symmetrical and asymmetrical –CH2– stretch absorption bands with the pyrolysis temperature for pristine PE as compared to PE-n-MMT. At any given temperature above 200 °C, PE-n-MMT has higher content of intact CH2 units than the pristine PE sample, and the apparent rate of disappearance of the aliphatic units in the pyrolysis temperature range of 220–260 °C is much higher for pristine PE than for the corresponding nanocomposite. Thus, the observed evolution of the spectra is an extra proof of the fact that, at temperatures above 220 °C, PE-n-MMT nanocomposite undergoes thermal-oxidative degradation with a considerably slower rate than the neat PE does. The same conclusion has been derived in the preceding section based on the analysis of corresponding TGA data (Fig. 7).

Comparative evolution of the IR absorption bands attributed to –CH2– units in (a) neat PE, and (b) PE-n-MMT nanocomposite
We explained this fact by the formation of chemical crosslinking between the polyethylene macromolecules in the nanocomposite. Really, Figs. 12b and 13 show the medium absorption band at 1,162 cm−1 which can be attributed to intermolecular esters groups (>С–О–С<) [43]. At the same time this peak is absent at the spectra of the pristine PE (Figs. 12a, 13). It is known [27] that the contribution of radical recombination reactions, which lead to the intermolecular crosslinking, increases under the oxygen deficiency conditions (Scheme 4, reactions e, f). Such conditions are realized at thermal-oxidative degradation of the PE nanocomposites due to the labyrinth effect of the silicate layers towards the diffusing gas. This feature of thermal-oxidative degradation of the PE nanocomposites–the formation of chemical crosslinking is a necessary condition of PE carbonization.

Alternative reactions of alkyl radicals during thermal-oxidative degradation of polyethylene
Conclusion
It is shown that thermal degradation of PE in the inert environment proceeds identical with PE nanocomposite, which is the evidence against the MMT influence on this process.
On the other hand, during the thermal oxidation at 170–200 °C for inhibitor-free PE-MMT nanocomposites it was observed the accelerated formation and decomposition of hydroperoxides as compared with unfilled PE which is caused by catalytic action of montmorillonite (at the presence of oxygen). Cumulative action of chemical cross-linking and catalytic dehydrogenation presents a necessary and sufficient condition of carbonization, which is observed in the process of thermal-oxidative degradation of PE-MMT nanocomposites. Carbonized layer formation leads to appreciable increase of thermal stability of PE nanocomposite, owing to a hindrance of the mass transfer in the nanocomposite. An incorporation of the antioxidant in PE and its nanocomposite suppresses the formation and decomposition of hydroperoxides and reduces the catalytic action of MMT on these processes. These facts explain an increase of thermal-oxidative stability in both materials, at that the nanocomposite thermal stability exceeds PE and approaches the values obtained in inert atmosphere.
References
Messersmith PB, Giannelis EP (1993) Chem Mater 5:1064
Zanetti M, Lomakin S, Camino G (2000) Macromol Mater Eng 279:1
Alexandre M, Dubois P (2000) Mater Sci Eng R 28:1
Giannelis EP (1996) Adv Mater 8:29
Oya A (2000) In: Pinnavaia TJ, Beall GW (eds) Polymer clay nanocomposites. Wiley, London
Gilman JW, Kashiwagi T, Nyden M, Brown JET, Jackson CL, Lomakin SM, Gianellis EP, Manias E (1998) In: Al-Maliaka S, Golovoy A, Wilkie CA (eds) Chemistry and technology of polymer additives. Blackwell Scientific, London, p 249
Lomakin SM, Dubnikova IL, Berezina SM, Zaikov GE (2005) Polym Int 54(7):999
Lomakin SM, Zaikov GE (2003) Modern polymer flame retardancy. VSP Int. Sci. Publ. Utrecht, Boston, p 272
Gilman JW (1999) Appl Clay Sci 15:31
Gilman GW, Jackson CL, Morgan AB, Harris RH, Manias E, Giannelis EP, Wuthenow M, Hilton D, Phillips S (2000) Chem Mater 12:1866
Kashiwagi T, Harris RH Jr, Zhang X, Briber RM, Cipriano BH, Raghavan SR, Awad WH, Shields JR (2004) Polymer 45:881
Kovaleva NYu, Brevnov PN, Grinev VG, Kuznetsov SP, Pozdnyakova IV, Chvalun SN, Sinevich EA, Novokshonova LA (2004) Polym Sci Ser A 46(6):651
Voigt J (1966) Die Stabilisierung der Kunstoffe Gegen Licht und Wärme. Springer-Verlag, Berlin, p 542
Shchegolikhin AN, Lazareva OL (1997) Int J Vib Spect (www.ijvs.com) 1(4):95
Lacey DJ, Dudler V (1996) Polym Degrad Stab 51:1011
Paabo M, Levin BC (1987) Fire Mater 11:55
Lattimer RP (1995) J Anal Appl Pyrolysis 31:203
Kuroki T, Sawaguchi T, Niikuni S, Ikemura T (1982) Macromolecules 15:1460
Kiran E, Gillham JK (1976) J Anal Appl Pyrolysis 20:2045–2068
Blazso M (1993) J Anal Appl Pyrolysis 25:25
Hornung U, Hornung A, Bockhorn H (1998) Chem Ing Tech 70:45
Hornung U, Hornung A, Bockhorn H (1998) Chem Eng Technol 21:332–337
Bockhorn H, Hornung A, Horung U (1998) J Anal Appl Pyrolysis 46:1
Opfermann J (2000) J Thermal Anal Cal 60:641
Friedman HL (1965) J Polym Sci C6:175
Bockhorn H, Hornung A, Hornung U, Schawaller D (1999) J Anal Appl Pyrolysis 48:93
Grassie N, Scott G (1985) Polymer degradation and stabilization. Cambridge University Press, Cambridge, p 275
Gugumus F (2000) Polym Degrad Stab 69:23
Lacoste L, Carlsson DJ (1992) J Polym Sci Part A Polym Chem 30:493
Gugumus F (2002) Polym Degrad Stab 76(2):329
Gugumus F (2002) Polym Degrad Stab 77(1):147
Benson SW (1976) Thermochemical kinetics. Wiley, New York, p 114
Zaragoza DF (2000) Organic synthesis on solid phase. Wiley, New York
Xie W, Gao ZM, Pan WP, Hunter D, Singh A, Vaia R (2001) Chem Mater 13:2980
Yablokov VA (1980) Russ Chem Rev 49:833
Plesnicar B (1983) In: Patai S (ed) The chemistry of functional groups, peroxides. Wiley, New York, p 521
Bugajny M, Bourbigot S, Bras ML, Delobel R (1999) Polym Int 48:264
Xie RC, Qu BJ, Hu KL (2001) Polym Degrad Stab 72:313
Serratosa JM, Bradlay WF (1958) J Phys Chem 62:1164
Zanetti M, Bracco P, Costa L (2004) Polym Degrad Stab 85:657
Morlat S, Mailhot B, Gonzalez D, Gardette S (2004) J Chem Mater 16:377
Desai SM, Pandey JK, Singh RP (2001) Macromol Symp 169:121
Brown DW, Floyd AJ, Sainsbury M (1988) Organic spectroscopy. Wiley
Acknowledgements
This work is supported by Russian Foundation for Basic Research (Grant No. 06-03-08047) and Federal Agency on Science and Innovations (Contract No. 02.513.11.3161).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lomakin, S.M., Novokshonova, L.A., Brevnov, P.N. et al. Thermal properties of polyethylene/montmorillonite nanocomposites prepared by intercalative polymerization. J Mater Sci 43, 1340–1353 (2008). https://doi.org/10.1007/s10853-007-2295-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-007-2295-1