
Abstract
The formation of host–guest-complexes of Triton X-100 (1), a nonionic surfactant comprising of an oligo ethylene glycol chain and a bulky hydrophobic tail, with alpha-, beta- and gamma-Cyclodextrin (α-, β- and γCD) has been investigated by isothermal titration calorimetry (ITC). Especially, the interaction of βCD with Triton X-100 has been subject-matter of a series of analytical studies though the results are contradictorily. Equilibrium constants diversify in the range of 200–200000 M−1. Among these, even an isothermal titration calorimetric study is reported, indicating an association constant of 9100 M−1. In contrast, the findings we report in the present paper approve an exceptionally high association constant reported just recently. Moreover, the stoichiometry of the formed complexes and the binding sites were investigated. βCD and γCD interact with the octylphenyl residue of 1. In contrast, αCD forms pseudorotaxanes by threading onto the oligo ethylene glycol-part of the surfactant molecule.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cyclodextrins (CDs) are semi natural cyclic oligosaccharides based on α-(1,4)-linked glucopyranose units. The cone-shaped molecules are prefixed by α-, β- or γ-, due to their number of glucopyranose units—six, seven or eight. Their three-dimensional structure and some characteristic values are shown in Table 1. Besides α-, β- and γCD, CDs with more subunits are known but they are not of technical relevance.
Arising from its cone-shaped structure, CDs are hydrophilic molecules with a hydrophobic cavity. Hydrophobic molecules of adequate size are capable to penetrate into this cavity in polar media. Driving forces are both Van der Waals interactions as well as the increasing entropy of the system by liberation of the solvating molecules—known as hydrophobic effect.
In addition, hydrogen-bondings at the rim of the cavity may play a certain role [1–5].
Due to these rather weak interactions, equilibrium constants (K) for host–guest-complexes are rarely higher then 5 × 104 M−1 [1]. For comparison, a chelate-complex of calcium ions and EDTA (ethylenediaminetetraacetic acid) in MES-buffer has a value of K = 2 × 106 M−1 [6].
In order to assemble most stable supramolecular structures, complexes with high equilibrium constants are of great interest. Recent studies point out that the nonionic surfactant Triton X-100 (1, Fig. 1) is applicatively in this context. A photophysical study of inclusion of 1 into βCD by He et al. [7] confirmed the findings of Du et al. [8, 9] of an extraordinary high value of K > 1 × 105 M−1. Certainly, these results differ still in the stoichiometry of the formed complexes. He et al. report the coexistence of a 1:1 and 2:1 (host:guest) complex of βCD and 1, whereas Du et al. assume a 1:1 stoichiometry. Moreover, previous studies applying varying methods report values of K between 145 and 3 × 104 M−1 with stoichiometries of 1:1 or 2:1 respectively [10–14]. Among these, even an isothermal titration calorimetric study is reported, indicating an association constant of 9100 M−1 and a stoichiometry of 1:1 [15]. Against this background, the situation can be considered vaguely at most and needs clarification.

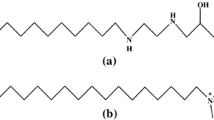
Chemical structure of Triton X-100
In the present study, we therefore revise the reported data by a new isothermal titration calorimetry (ITC) study. ITC is viewed as ‘method of choice’ for characterization thermodynamics and stoichiometry of inclusion complexes [16]. An important advantage is the direct determination of thermodynamic data without applying any auxiliary substances which may affect inaccuracies.
Experimental
Materials
Cyclodextrins were kindly provided by Wacker-Chemie, Germany and dried in a microwave drying system SAM 255 (CEM) prior to use. Triton X-100 was purchased from Sigma-Aldrich, Germany, and dried in vacuum for 24 h.
Instruments
Isothermal titration calorimetry experiments were performed on a VP-ITC Micro-Calorimeter from MicroCal (Northampton, MA) controlled by MicroCal’s VP viewer 2000 ITC software. 1H-NMR measurements were performed in D2O using a Bruker Advance DRX 500 spectrometer at 500.13 MHz. The δ scale was calibrated to the solvent signal at δ = 4.79 ppm. UV–visible-spectra were recorded on a Nicolet UV540 spectrometer in the range from 210 to 310 nm in a quartz glass cuvette with a layer thickness of 1 cm. The collection interval was set on 0.2 nm and the collection speed to 30 nm min−1.
Methods
ITC-titrations were performed at 25 °C. Against a 0.15 mM solution of Triton X-100, a 3 mM solution of βCD or γCD or a 40 mM solution of αCD was titrated. Titration was carried out by 40 injections of 7 μL with a spacing time of 300 s. The stirring speed was set on 300 min−1. The heat of dilution was determined in a separate experiment and was subtracted from the binding isotherm. Results were calculated by an iterative nonlinear regression (NLR) fit with Origin 7.0 software and the MicroCal LLC ITC add-on.
Job plots observed via 1H-NMR spectroscopy were prepared from 0.18 M stock solutions in D2O by simply mixing the appropriate ratios to a total concentration of 0.18 M. A measurement with the ratio 9:1 considering 1 vs. CD was left out to prevent formation of micelles. For the determination of the chemical shifts of 1, a 0.18 M solution of 1 in D2O was analyzed.
UV-titrations were performed by mixing equal volumes of a 0.19 M stock solution of 1 in water and CD solutions in water of varying concentrations. These concentrations were distributed in a pseudo logarithmic manner and ranged at least over three decades. Final concentrations of the solutions measured are declared in the correspondent figures.
Results and discussion
For all complexation studies of Triton X-100 (1) experiments should be carried out with special attention to the concentration of 1. As a nonionic detergent it forms micelles in water above its critical micelle concentration (CMC). Complex formation of CD with the hydrophobic residue of 1 thus would require a preceding dissociation of the micellar aggregates. To avoid such a competing effect, all measurements were carried out at a concentration of 0.15 mM of 1. This is definitely below the CMC, which is reported to be 0.22 mM [17].
Complexation study of Triton X-100 and βCD
Figure 2 shows the result of an ITC experiment of 1 and βCD. Although concentrations of solutions applied (0.15 mM for 1 and 3 mM for βCD) were exceptionally low, there was still a significant release of heat detectable. This effect alone points to a very high equilibrium constant. Curve-fitting of the ΔH plot revealed best accordance with a two sets of sites model with a stoichiometry of 1:1 each and values of K1 = 1.71 × 105 M−1 and K2 = 260 M−1. To ensure the accuracy of the results, we repeated this measurement five times from which a standard deviation for K1 of less then 3% emerged. K2 which is always less precisely, especially if it differs significantly from K1, had a standard deviation of 30%. Additionally, values for the entropy and enthalpy were iterated. They resulted in ΔH1 = −36.8 ± 0.1 kJ mol−1 and ΔS1 = −23.1 ± 0.6 J K−1 mol−1. Results for ΔH2 and ΔS2 are not presented, as they were too imprecisely.

ITC result for a titration of a 3 mM solution of βCD against 0.15 mM solution of 1 in water
Table 1 compares the equilibrium constants obtained with those published by other groups. Two things should be noted: First, the values of K, which differed greatly in the past, now level off at a value of K > 1 × 105 M−1 concerning more recent works. Thus, the remarkably high value of K can be considered to be confirmed. Second, in the past only complexes of 1 and βCD with a 1:1 stoichiometry were discussed. The present study included, there are only four works out of 11 reporting a 2:1 (host:guest) stoichiometry as well. This point is a little surprising, since if the dimensions of the involved molecules are regarded, it becomes apparent that a 2:1 stoichiometry is expectable. βCD has a total height of the cavity of 0.78 nm whereas the shortest length of the hydrophobic tail of 1 can be calculated to 0.85 nm. To confirm the expectation of a 2:1 stoichiometry, we performed 1H-NMR and UV-experiments (Table 2).
Figure 3 shows the result for a Job plot (method of continuous variation) observed via 1H-NMR-spectroscopy [19]. For a complex with 1:1 stoichiometry one would expect a triangular plot with a single maximum at a molar fraction of 0.5. In the case of other stoichiometric ratios, the maximum would be shifted to the appropriate molar fraction. However, if the equilibrium constants involved are not of the same magnitude, slopes become irregularly and the method no longer can give any information about the stoichiometric ratio [20]. This is clearly the case for the complex formation of 1 and βCD. Also a UV-titration approved a stoichiometry other then 1:1 as no isosbestic point could be observed (Fig. 4). Accordingly, the expectation of a 2:1 complex is most probable.

Job plot for 1 and βCD. Shifting of the proton signals at position C3 and C5 of the glucopyranose subunits of βCD is displayed depending on the molar fraction of βCD

UV-spectra for the titration of βCD (values are given in M) against 1 (concentration kept constant at 0.095 mM). Obviously, no distinct isosbestic point is observable
The question remains where the βCD molecules are located on 1. Three workgroups have investigated this topic by different NMR-methods and shared the same conclusion: Both binding sites for βCD are provided by the hydrophobic part of 1 [7, 18, 21]. However, none of these studies tried to determine, where the preferred binding site is. In the present work, we try to draw a conclusion from the method of continuous variation. Figure 5 displays the differences of the chemical shifts of proton signals of 1 depending on the molar fraction of βCD. While the signals of the aliphatic protons a and c (designation referred to Fig. 1) already shifted strongly upon small βCD addition, signals of the aromatic protons d and e changed when a greater amount of βCD was added. Consequently, the aliphatic residue of 1 seems to interact with βCD more favorably. Formation of a 2:1 complex occurs probably most likely when the first βCD molecule slides a bit in the direction of the oligo ethylene glycol (OEG) chain and thereby deblocks the terminal tert-butyl group. This group then can be included into a second βCD molecule (Fig. 6). This scenario is backed by the observation that hardly any shift of the OEG-proton signals appeared. Hence, the OEG chain is not involved in the complex formation of 1 and βCD in any way. Accordingly, the value K2 not alone describes the formation of the 2:1 complex but implies also a destructive fraction for the rearrangement of the 1:1 complex. That might be an explanation for the rather low value of K2 [1].

Differences of 1H-NMR shifts of 1 upon addition of βCD

Most likely scenario for the stepwise complex formation of 1 and βCD. In a first step mainly the aliphatic residue of 1 is included into the cavity of βCD (I). By sliding into the direction of the OEG chain βCD deblocks the terminal tert-butyl group which then can be included into a second βCD molecule (II)
Here it should be clarified that the previously described findings do not give any information about the orientation of the βCD molecules on 1. However, an intrusion of 1 through the secondary face of βCD as it is shown in Fig. 6 is most probable. A further head-to-head and tail-to-tail orientation is then favored, too [5].
Complexation study of Triton X-100 and γCD
Same analysis as explained in detail for βCD and 1 was performed for the system γCD and 1. In contrast to βCD, a Job plot via 1H-NMR (Fig. 7) and a UV-titration proved the formation of a 1:1 complex. Calculation of the ITC curve with a one set of site model gave a value of K1 = (2.16 ± 0.09) × 104 M−1. ΔH1 was estimated to −11.6 ± 0.21 kJ mol−1 and ΔS1 to 44.0 J K−1mol−1.

Job plot for 1 and γCD, showing for both curves a nearly perfect triangular shape with a maximum at a molar ratio of 1:1
Since the aromatic part of 1 is fully involved into the complex formation, it is possible to calculate K also by an iterative nonlinear regression (NLR) fit from the UV-dataset [22]. By Eq. 1, it is possible to get all relevant parameters:
Where A, A 0 and A 1 are the absorption intensities of the measurement mixture, uncomplexed 1 and the complex of γCD and 1 respectively. Applying this equation (Fig. 8) the following values are obtained: A 0 = 0.547 ± 0.002, A 1 = 0.697 ± 0.003, K1 = 21100 ± 16200 M−1 and n = 1.16 ± 0.09. Unfortunately, the margin of error is very high for K1. Nevertheless, it is worth noting that the estimated value is nearly the same as that from the ITC experiment.

NLR-fit for the UV-data set from the titration of γCD against 1
Table 3 compares the results for K1 with those from literature. In contrast to βCD the situation is clearer for the complex formation of γCD and 1. With the exception of the work of Nelson et al. the reported values of K are at least of the same magnitude.
Buschmann et al. are the only workgroup, who postulated also a 2:1 complex. This conclusion was drawn from the comparison of the margin of errors for a one set of site and a two sets of sides NLR-fit of the Fluorescence data set. However, such an evaluation is always limited to the accuracy of the data collection. Since the latter model has more free parameters to compensate inaccuracies of the data set, it usually will give the better fit [20]. So this work is a good example how valuable additional methods like the method of continuous variation are in order to get the right stoichiometric ratio. Although sometimes an additional experiment has to be carried out, results obtained normally are superior to a statistical evaluation.
As for βCD the binding site of γCD on 1 was investigated. In accordance to Saito et al. significant interactions were found for the proton signals c and d of 1, whereas the signals of a and e are only slightly shifted (Fig. 9) [18]. Hence, the former are located deep into the cavity and the latter can be found at the openings of γCD.

Differences of 1H-NMR shifts of 1 upon addition of γCD
Complexation study of Triton X-100 and αCD
Even the complex formation of αCD and 1 was investigated. In contrast to β- and γCD, αCD does not interact with the hydrophobic moiety of 1. A proof is given by the fact that no change of the UV-spectrum of 1 upon addition of αCD was observed. This coincides with the expectations, since the tert-octylgroup is too bulky for the small cavity of αCD. However, in an ITC experiment a small heat release could be detected. This exothermal effect can be led back on the formation of pseudorotaxane structures by threading from αCD onto the OEG chain of 1. The similar pseudopolyrotaxanes from poly(ethylene glycol) (PEG) and αCD are among the best studied polyrotaxane structures. They form spontaneously from a mixture of its components [5, 23]. Harada et al. also investigated the formation of inclusion complexes of monodisperse OEGs with αCD. Above a degree of polymerisation of three, pseudorotaxane structures are formed with a stoichiometry of 1:2 considering the proportion of αCD vs. the OEG repeating unit. If the degree of polymerisation exceeds 12 the solutions of the pseudorotaxanes have to be heated to prevent precipitation [24] The results of this study can be regarded as a model analysis for the complex formation of 1 and αCD. Indeed, the same group studied the binding behaviour of 1 and α- and βCD respectively. They identified as first group the site selective binding behaviour of 1. Though, they did not respond to the question how many αCD molecules are threaded onto the OEG chain [24]. With respect to their previous study, a maximum mean stoichiometric ratio of 5:1 (αCD:1) is thinkable, since the average degree of polymerisation of the OEG chain of 1 is 10. However, as a second group Saito et al. investigated the complex formation of αCD and 1 and examined also the binding stoichiometry. They prepared a precipitate from an aqueous αCD solution by addition of 1 which emerged to be a pseudorotaxane with a stoichiometry of 2:1 [18].
In contrast, under the conditions applied in the present study there was no precipitate formed. This is clearly supported by the result of the UV-titration where no decrease in the absorption intensity was observed upon αCD addition. However, we could not determine the complex stoichiometry by any of our methods either.
A stoichiometric ratio of 5:1 the pseudorotaxane is still thinkable though this question remains open. Consequently, it is not possible, to calculate reasonable complexation parameters from the ITC experiment, as the number of sequential binding sites is not known. Anyway, the ΔH plot is only of moderate quality, even after improvement of the measurement conditions. In order to detect a significant heat release, the concentration of the solution of αCD had to be raised to 40 mM (Fig. 10). Nevertheless, the maximal heat released upon addition of one equivalent of αCD was just −420 J mol−1. So at least it is possible to say that the pseudorotaxane formed from αCD and 1 is much weaker then those from β- or γCD and 1.

ITC result for a titration of a 40 mM solution of αCD against 0.15 mM solution of 1 in water
Conclusion
The present study reappraises the data reported so far on the complexation behaviour of α-, β- and γCD and Triton X-100 (1) by means of ITC experiments. As yet, these data were quite confusing for they differed in the complex stoichiometry and especially in the magnitude of the equilibrium constants. In order to get more reasonable results for the right complex stoichiometry, UV-titrations and Job plots were carried out in this work. Thus, the accuracy of the adopted stoichiometry for the fitting models applied in ITC experiments could be approved.
The results obtained confirm that βCD and 1 form a 1:1 and 2:1 complex with an exceptionally high K1 = 1.71 × 105 M−1 and a much lower K2 = 260 M−1. Both CD molecules are located on the hydrophobic residue of 1, as was shown by the shift on 1H-NMR signals. Thereby, the preferred binding site could be determined to be the tert-octylgroup. A second binding site is presumably provided by a sliding of the first βCD onto the phenylgroup, whereby the terminal tert-butyl group is deblocked. γCD forms with 1 a 1:1 complex with a value of K1 = 21600 M−1. γCD is located on the hydrophobic residue of 1, too. From ITC-experiments no equilibrium constants could be calculated for the pseudorotaxanes formed from αCD and 1 by threading of αCD onto the OEG chain. Also the complex stoichiometry could not be solved. From theoretical considerations, one might expect a maximum mean ratio of 5:1. At least it could be deduced that the equilibrium constant for these complexes are much weaker, then those from β- and γCD. The site selective complexation behaviour of α-, β- and γCD respectively and 1 as it was reworked in the present study is shown in Fig. 11.

Complexes of α-, β- and γCD with 1. While for the complexes of β- and γCD with 1 the maximum stoichiometric ratio is displayed, for the pseudorotaxanes of αCD and 1 stoichiometric ratios up to 5:1 are thinkable
References
Rekharsky, M.V., Inoue, Y.: Complexation thermodynamics of cyclodextrins. Chem. Rev. 98, 1875–1917 (1998)
Del Valle, E.M.M.: Cyclodextrins and their uses: A review. Process Biochem. 39, 1033–1046 (2004)
Szejtli, J., Osa, T.: Cyclodextrins. In: Atwood, J.L., Davies, J.E.D., Macnicol, D.D., Vögtle, F., Lehn, J.-M. (eds.) Comprehensive Supramolecular Chemistry. Elsevier Science Ltd, Oxford (1996)
Szejtli, J.: Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 98, 1743–1753 (1998)
Wenz, G., Han, B.H., Muller, A.: Cyclodextrin rotaxanes and polyrotaxanes. Chem. Rev. 106, 782–817 (2006)
Christensen, T., Gooden, D.M., Kung, J.E., Toone, E.J.: Additivity and the physical basis of multivalency effects: A thermodynamic investigation of the calcium EDTA interaction. J. Am. Chem. Soc. 125, 7357–7366 (2003)
He, Y.F., Shen, X.H., Gao, H.C., He, Y.K.: Spectral and photophysical studies on the inclusion complexation between Triton X-100 and beta-cyclodextrin: A competitive method using a substituted 3H-indole probe. J. Photochem. Photobiol. A 193, 178–186 (2008)
Du, X.Z., Zhang, Y., Huang, X.Z., Jiang, Y.B., Li, Y.Q., Chen, G.Z.: Intense room-temperature phosphorescence of 1-bromonaphthalene in organized media of beta-cyclodextrin and triton X-100. Appl. Spectrosc. 50, 1273–1276 (1996)
Du, X.Z., Lu, W.H., Ding, N., Dai, H.X., Teng, X.L., Deng, H.L.: Spectral properties and supramolecular inclusion complexes of beta-cyclodextrin with flexible amphiphilic and rigid compounds. J. Photochem. Photobiol. A 177, 76–82 (2006)
Nelson, G., Warner, I.M.: The Formation of Pyrene–Cyclomalto-Oligosaccharide Complexes in the Presence of Non-Ionic Surfactants. Carbohydr. Res. 192, 305–312 (1989)
Smith, V.K., Ndou, T.T., Delapena, A.M., Warner, I.M.: Spectral characterization of beta-cyclodextrin–Triton X-100 complexes. J. Incl. Phenom. Mol. Recogn. Chem. 10, 471–484 (1991)
Datta, A., Mandal, D., Pal, S.K., Das, S., Bhattacharyya, K.: Interaction of Triton X-100 with cyclodextrins. A fluorescence study. J. Chem. Soc. 94, 3471–3475 (1998)
Du, X.Z., Chen, X.G., Lu, W.H., Hou, J.G.: Spectroscopic study on binding behaviors of different structural nonionic surfactants to cyclodextrins. J. Colloid Interf. Sci. 274, 645–651 (2004)
Buschmann, H.J., Cleve, E., Schollmeyer, E.: The interactions between nonionic surfactants and cyclodextrins studied by fluorescence measurements. J. Incl. Phenom. Macrocycl. Chem. 33, 233–241 (1999)
Eli, W., Chen, W.H., Xue, Q.J.: The association of Triton X surfactants with beta-cyclodextrin. An isothermal titration calorimetry study. J. Incl. Phenom. Macrocycl. Chem. 36, 439–445 (2000)
Bouchemal, K.: New challenges for pharmaceutical formulations and drug delivery systems characterization using isothermal titration calorimetry. Drug Discov. Today 13, 960–972 (2008)
Tiller, G.E., Mueller, T.J., Dockter, M.E., Struve, W.G.: Hydrogenation of Triton X-100 eliminates its fluorescence and ultraviolet-light absorption while preserving its detergent properties. Anal. Biochem. 141, 262–266 (1984)
Saito, Y., Ueda, H., Abe, M., Sato, T., Christian, S.D.: Inclusion complexation of Triton X-100 with alpha-, beta- and gamma-cyclodextrins. Colloid Surf. A 135, 103–108 (1998)
Blanda, M.T., Horner, J.H., Newcomb, M.: Macrocycles containing tin—preparation of macrobicyclic lewis acidic hosts containing 2 tin atoms and Sn-119 NMR-studies of their chloride and bromide binding-properties in solution. J. Org. Chem. 54, 4626–4636 (1989)
Connors, K.A.: Binding Constants. Wiley-Interscience, New York (1987)
Harada, A., Okumura, H., Okada, M., Suzuki, S., Kamachi, M.: Site-selective complexation of amphiphilic compounds by cyclodextrins. Chem. Lett. 29, 548–549 (2000)
Abou-Zied, O.K.: Investigating 2, 2′-bipyridine-3, 3′-diol as a microenvironment-sensitive probe: Its binding to cyclodextrins and human serum albumin. J. Phys. Chem. B 111, 9879–9885 (2007)
Harada, A., Hashidzume, A., Yamaguchi, H., Takashima, Y.: Polymeric Rotaxanes. Chem. Rev. 109, 5974–6023 (2009)
Harada, A., Li, J., Kamachi, M.: Formation of inclusion complexes of monodisperse oligo(ethylene glycol)S with alpha-cyclodextrin. Macromolecules 27, 4538–4543 (1994)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Müller, BK., Ritter, H. Scrutinizing ITC-study on the formation of inclusion complexes of nonionic surfactant Triton X-100 and cyclodextrins. J Incl Phenom Macrocycl Chem 72, 157–164 (2012). https://doi.org/10.1007/s10847-011-9955-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-011-9955-0