
Abstract
Understanding the interactions of cyclodextrins (CD) and cucurbit[n]uril (CB[n]) hosts with a variety of guest molecules following their encapsulation within the cavity of these macrocycles have become increasingly important in the recent years. The electronic charge distribution and the cavity dimension are some of the key factors those govern their interactions with cations or neutral guests. In the present work the molecular electrostatic potential (MESP) topography has been utilized to obtain the ‘effective’ cavity diameter and height of CB[n] (n = 6–8) homologues and 8 conformers each of α-, β- and γ-CD. It has been shown that the shape of CD cavity be it cone- or barrel-like stems from the hydrogen bonding patterns within primary hydroxyl groups. The width of CB[7] is comparable to the β-CD conformer that possesses either O6H–O5′ or intraglucose O6H–O5 interactions. The cavity diameters of α- and γ-CD are predicted to be respectively, 1.0 and 1.5 Å larger than CB[6] and CB[8] hosts. MESP topography reveals that the cavities of CB[n] are less hydrophilic with largely hydrophilic portals as compared to CD hosts. Cremer–Pople puckering parameters were derived for all the CD conformers and CB[n]. It has been demonstrated that the clockwise and anticlockwise hydrogen bonding patterns in the lower as well as upper rims of different CD conformers are less distorted and exhibit a little deviation from the °C3 chair conformation of α-d-glucopyranose constituting monomeric unit of CD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cyclodextrins (CD) are cyclic oligomers of glucose obtained from the enzymatic degradation of starch by cyclodextrin glucatranferase enzyme in intramolecular transglycosylation reaction [1]. The commercially available forms of CD are composed of α(1 → 4) linkage of 6–8 glucopyranoside units. CD are capable of encapsulating compounds such as straight or branched chain aliphatics, aldehydes, ketones, alcohols, organic acids, fatty acids, aromatic and polar compounds and form inclusion complexes in solid state [2, 3]. The guest molecules with varying size can be accommodated in the CD cavity in solution as well [4, 5]. Inclusion of guest molecule within the cavity of the host protects it from oxidation, reduction or hydrolysis, decreases sublimation as well as volatility [6] and thus alters its physical or chemical properties [7]. Along with this the enhanced stability and water solubility of a drug molecule confined within the CD cavity leads to a controlled releasing ability [8, 9], improved bioavailability [10] and reduces its toxicity [11, 12].
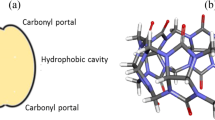
It should be remarked here that the cavity size and the binding ability of CD towards different guests are often compared [12–15] with CB[n], cyclic oligomers of glycouril units linked by a pair of methylene group [16], which represent a different family of hosts [17]. Like CDs, the CB[n] macrocycles offer varying cavity size and regio-selectivity toward binding of variety of guests with the enhanced solubility in different solvents. Hydrophilic portals of CB[n] bind to metal ions [18, 19], inorganic salts [20], whereas its hydrophobic cavity accommodates organic dyes [21], protonated alkyl/aryl [22] or platinum amine [23]. Host–guest interactions in such systems led to a wide range of applications in the recent years that include drug delivery [24, 25], separation technology [26], catalysis [27], rotaxanes [28] and nanotechnology [29].
To understand the host–guest interactions of α-, β- and γ-CD and their inclusion complexes calculations at different levels of theory, that include molecular mechanics [30–34], semi-empirical methods [35–39], Hatree–Fock, density functional methods [40–43] and simulations based on molecular dynamics [44] have been carried out. Topography of scalar fields such as molecular electron density (MED) or molecular electrostatic potential (MESP) have successfully been utilized to analyze hydrogen bonded interactions from the primary as well as secondary hydroxyl groups of α-, β- and γ-CD [42, 43]. Theoretical investigations on CB[n] homologues along with their inclusion complexes are also reported [45–49]. These results have shown that CB[6] and CB[7] hosts are largely stabilized over its acyclic polymer devoid of ring strain and large electrostatic repulsion due to ureido oxygens. It has been well known that matching of size and shape complimentarity of host and guest [50, 51] leads to substantial binding affinity toward the guest. Recent density functional calculations [45] have shown that the interaction of ferrocene with the CB[n] hosts tends to form inclusion complexes only with the CB[7] and CB[8] homologues while interacts laterally in case of CB[6]. Several molecular recognition theories have been put forth which highlights on the complementarity of guests in terms of size and shape for hosts [52]. This further necessitates quantifying the dimensions (diameter and height or depth) of the host cavity. As a pursuance to this we present a systematic MESP based approach to estimate “effective cavity dimensions” of CB[n] as well as those of CD hosts exhibiting different hydrogen bonding patterns in their conformations. The shape of host cavity can be further also be gauged from the charge distribution in terms of the MESP topography. To this end 24 different conformers of α-, β- and γ-CD, including all the 3 CD conformers reported earlier in Ref. [42], have been considered. This method enables one to compare directly the cavity dimension of different host families and therefore, should serve as an initial step toward the molecular level understanding of the host–guest interactions.
Computational method
CB[n] (n = 6–8) homologues and 8 conformers each of α-, β- and γ-CD were optimized employing the density functional theory incorporating Becke’s three-parameter exchange with Lee, Yang, and Parr’s (B3LYP) correlation functional [53, 54]. The internally stored 6-31G(d) basis set was used. Optimizations (without any symmetry constraint) were performed using the GAUSSIAN 03 program [55]. The molecular electrostatic potential (MESP), V(r) in these hosts have been calculated. The V(r) essentially comprised of two terms with one representing the repulsion between the positive probe and nuclei and the second term is its attraction to the electrons; the latter essentially represents the columbic interactions between the charge and the electrons. Substantial effective negative values in V(r) arising from the net balance of aforementioned electronic and the bare nuclear contributions reveal regions conducive to electrophilic interactions. The MESP exhibits rich topographical features, which has subsequently been mapped by examining eigenvalues of the Hessian matrix at the point where gradient V(r) vanishes and thus the critical points (CPs) were located using the code written in our laboratory [56]. The CPs are characterized in terms of an ordered pair (R,σ), where R and σ denote the rank and signature (i.e. the sum of algebraic signs of the eigen values) of the Hessian matrix, respectively. These CPs can further be grouped into three sets: (3,+3) as local minima and (3,+1) or (3,−1) which correspond to saddle points. Interestingly lone pairs on electronegative atoms in molecules can be inferred from (3,+3) CPs in the MESP topography. Furthermore the (3,+3) CPs (local minima) herein represent the potential binding sites of the host toward binding of an electrophile or the neutral guest [57, 58]. Thus the MESP topography has proven useful for modeling an electrophilic attack on a molecule. UNIVIS-2000 was utilized for visualization of the MESP along with its isosurface [59].
Results and discussion
Atomic numbering scheme and optimized geometry of a glucose residue cut out of CD is shown in Fig. 1a. Likewise Fig. 1b displays the glycouril monomer in CB[n]. Conformers possessing primary as well as secondary hydroxyl groups oriented in clockwise as well as anticlockwise fashion and combinations therein of α-, β- and γ-CD hosts were considered. CB[n] homologues yield only one conformer. Hydrogen bonding patterns at the primary hydroxyl groups allow CD conformers to be classified in three different classes with those comprising of (i) O6H–O6′ interactions referred as “A”, (ii) O6H–O5′ interactions which are denoted by “B” and (iii) intra-glucose O6H–O5 interactions (involving ether oxygens) called as “C”. The single prime here refers to atom from the adjacent glucose unit. These conformers of α-CD, as a prototype example, are displayed in Fig. 2 along with the CB[n] (n = 5–8) homologues. Different patterns of these hydrogen bonded interactions from primary and secondary hydroxyl functionalities yield four conformers of “A” denoted by A1, A2, A3, and A4. Here, 1 refers to the pattern where the hydroxyl protons oriented anticlockwise in top rim and clockwise in bottom rim; the conformer designated as 2 has exactly reverse orientations in both these rims. Furthermore, in the conformer labeled as A3, 3 denotes the hydroxyl protons arranged clockwise in both top and bottom rims and finally the conformer possessing the hydrogen bonding pattern opposite to that of conformer 3 are denoted by 4; that is hydroxyl groups in both these rims orient in anticlockwise manner. With these notations only B2, B3 conformers were derived in class “B”. Likewise C1, and C4 conformers in class “C” can be characterized. Thus O2H–O3′ interactions were noticed only in A2, A4, B2 and C4 conformers whereas rest of the conformers facilitate O3H–O2′ interactions from the secondary hydroxyl groups of the bottom rim. It may further be stated here A1, B3, and C1 conformers possess the O3H–O2′ hydrogen bonded interactions and turn out to be of lowest energy in the respective class.

Atomic numbering scheme in a the glucose unit cut from CD and b glycouril unit in CB[n]

B3LYP optimized structures of “A”, “B” and “C” conformers of α-CD and CB[n] (n = 5–8) homologues
The Cremer–Pople puckering parameters [60] viz. phase angle (Φ), magnitude of puckering (θ), and puckering amplitude (Q) which engender various possible conformations on the surface of sphere were obtained by using the Hyperchem macro and are given in Table 1. The Θ values in the A1–A4 conformers increase from α-, to γ-CD are all near zero or show a small deviation from it. This suggests the chair form of glucose. The distortion magnitude, Θ, for A1 or A3 is larger than those possessing anticlockwise orientation in the lower rim of CD hosts. Thus it should be remarked here that the A2 conformer in α-CD and similarly B2 and C1 of β-CD match well (the maximal deviation being 0.5°) with the ideal value of the chair conformation of α-d-glucopyranose (2.7°) reported in the literature [61]. Furthermore as may be noticed Q, turns out to be the same for all the CD conformers and agree well with that obtained for the pyranoid ring (0.58 Ǻ). Thus ring conformation (°C3) for glucose, monomer of CD, has been predicted for these conformers. As opposed to this the phase angle (Φ) changes significantly with the relative orientations of primary hydroxyl groups as in the “A”, “B” and “C” conformers. Relatively large values can be noticed for “A” conformers. Accordingly for the α-CD conformers these value turn out to be from 332.6° to 354.5° as opposed to 57.8° and 79.1° for the B2 and B3 conformers, respectively. For C1 and C4 conformers the corresponding values are predicted to be 39.8°, and 63.5°. It may also be noticed that the phase angle, Φ, decreases steadily in the “A” conformers with an increasing size of the macrocycle (α- to γ-CD) and thus in the A1 conformers of α-CD to γ-CD these values decrease from 354.5° to 297.1°. Further the A2 and A4 conformers possessing anticlockwise orientation of the secondary hydroxyl groups exhibit lower Φ values than those in the conformers A1 or A3 wherein the clockwise (O3H–O2′) interglucose hydrogen bonding pattern can be noticed. It was pointed out earlier [42] that the strength of O6H–O6′ interactions from the primary hydroxyls decreases steadily on going from α-CD to γ-CD. A similar trend was shown in the calculated Φ values of these conformers. The corresponding Φ values are quite different in the B2 and B3 conformers, those possess the anticlockwise and clockwise hydrogen bonding patterns in its bottom rim, respectively. Thus it may be inferred that the phase angle, (Φ) has largely been influenced by the hydrogen bonded interactions. The Φ values for the C1 and C4 displayed in Table 1 also support these conclusions. Cremer–Pople parameters of CB[n] (n = 5–8) macrocycles are presented in Table 1. Here the puckering amplitude, (Q), turns out to be 0.05 ± 1 along CB[5] to CB[8] series which stems from the planar five-membered rings of CB[n]. These inferences are in consonant with those reported for acylated glycourils in the literature [62].
It was pointed out in the preceding section that the MESP brings about the effective localization of electron-rich regions and further yield insights for interaction of the host with electrophilic functionalities of the guest. An electrostatic potential in the CD compared with CB[n] (cf. Fig. 3) shows large electron-rich regions near glycosidic oxygens in CD, whereas its cavity turns out to be less electron-rich. An MESP isosurface (V = −105.0 kJ mol−1) in different α-, β- and γ-CD conformers and that in the CB[n] (n = 6–8) hosts are depicted in Fig. 1S. The electron-rich regions within the top rim of “A” conformers project outward whereas within the “B” and “C” conformers those direct toward the cavity concomitantly encompassing the openings of the cavity partially at the primary hydroxyl functionalities. As shown in Fig. 1S the lowest energy conformer in each of the aforementioned “A”, “B” and “C” class emerge with small electron-rich regions near O2 which predict that the O3H–O2′ hydrogen bonded interactions are relatively strong and follow the rank order “C” > “B” > “A”. As may further be inferred within the CB[n] homologues MESP isosurface extends within the cavity. This partly explains their large affinity toward cationic guests. The (3,+3) MESP minima in the α-, β- and γ-CD conformers and CB[n] homologues were identified. A lateral view of CPs in the lowest energy A1, B3, and C1 conformers of α-, β- as well as γ-CD and in the CB[n] macrocycles are displayed in Fig. 2S. The minima were assigned notations namely ‘v’ (shown in aqua) near ring oxygen O5, ‘w’ (green) and ‘w*’(olive green) those near primary hydroxyl oxygen O6 within and outside the cavity, ‘x’ (blue) near the glycosidic oxygen O4 and finally ‘y’ (black) and ‘z’ (pink) that corresponding to O2 and O3 atoms from the secondary hydroxyl group, respectively. MESP value at these minima in α-, β- and γ-CD conformers are given in Table 2. The A1, B3, and C1 conformers engender deepest minima (‘w’) for the primary hydroxyl oxygen in the respective class. Accordingly the B3 conformer of α-CD yields a minimum with V = −240.4 kJ mol−1 compared to a marginally (5.9 kJ mol−1) shallow minimum for B2. On the other hand for α-CD the minima (‘w’) near primary hydroxyl oxygens in conformer “A” turns out to be shallow relative to those in β- and γ-CD. The “B” and “C” conformers render O6 to be the electron-rich. Furthermore, when O3 is hydrogen bond acceptor (conformer 2 and 4 having O2H–O3′ interactions) it renders electron rich nature in the α-CD. For example, in C4 conformer of α-CD the ‘z’ minimum near O3 turns out to be −124.2 kJ mol−1 whereas shallow minima with V = −122.9 and −120.2 kJ mol−1 were located in the C4 conformers of β- and γ-CD, respectively. Thus MESP topography suggests relatively strong O2H–O3′ hydrogen bonded interactions in γ-CD. Similarly the deeper minima near O2 are located in α-CD. These conclusions are in line with those drawn earlier from the calculated NMR chemical shifts and from the MED topography [43]. A large downshifted signal of hydroxyl (O3H) proton relative to those bonded to O2 was also observed in the measured NMR spectra [63]. The experimental observations are accordingly in line with these conclusions. Interestingly the semi-empirical AM1 quantum chemical calculations pointed out that the secondary hydroxyl functions (O2H, O3H) of saccharides are more acidic than those from the primary hydroxyl functions [64]. It has also been noticed that the ‘w’ and ‘x’ CPs in CD cavity are sensitive to the cavity size, and become deeper in γ-CD.

MESP mapped (from −131.28 to 266 kJ mol−1) on CD and CB[n] conformers
MESP minima (in kJ mol−1) both at the portals (‘w’) and within the cavity (‘x’) of CB[n] hosts are reported in Table 3. The minima (‘w’) near portal oxygens (V = −277.5 kJ mol−1) in the CB[6] were noticed to be deeper than those within the cavity (V = −68.5 kJ mol−1). As opposed to this, the CB[8] exhibits a deeper minimum (−85.1 kJ mol−1) within the cavity and renders it to be largely electron-rich. It may, therefore, be inferred that unlike in CB[8], the cavity of its lower homologue CB[6] is less hydrophilic with its portals exhibiting large hydrophilicity. The MESP minima near portal oxygens (‘w’) in CB[n] homologues are deeper than those identified near primary hydroxyl oxygens (O6) of CD. The shallow minima were located within the cavity of CB[n] hosts. It may therefore, be concluded that CB[n] cavity possess less hydrophilic character than CD hosts, whereas its portals are predicted to be largely hydrophilic compared to those of CD.
In the following we outline how the MESP minima yield a measure of the effective cavity dimensions of CB[n] or CD. Here we gauge the diameter and the height of the cavity in a variety of α-, β- and γ-CD conformers (aforementioned “A”, “B” and “C” classes) and further analyze how the cavity shape be it cone or barrel, can be governed by hydrogen bonded interactions from the upper and lower rims. For CD hosts, the ‘w’ CPs near primary hydroxyl functionalities and ‘x’ CPs near glycosidic oxygens (O4) impart the characteristic shape to its cavity. Top view of these CPs (‘w’ and ‘x’) in the lowest energy A1, B3, and C1 conformers of β-CD and those within the CB[7] host, as prototype examples, are displayed in Fig. 4. A side view showing the separation of planes containing ‘w’ and ‘x’ CPs in figure yields the ‘effective’ cavity height within the host. A cone shaped cavity of β-CD is clearly discernable with the ‘w’ CPs being relatively nearer compared to the separation of ‘x’ CPs in its “A” type conformers. It is further evident that the separation of two planes containing ‘w’ and other with ‘x’ CPs for “B” and “C” type conformers is nearly the same and concomitantly led to a barrel shape cavity. In other words the barrel shaped cavity can be noticed not only for β- and γ-CD but also for the α-CD in the “B” and “C” conformers. A top view containing the array of ‘w’ and ‘x’ CP in α- and γ-CD and those in CB[6] and CB[8] are compared in Fig. 3S of the supporting information. As is evident the cavity shape whether cone or barrel is not dependent on varying number of glucose units in CD macrocycles but is primarily governed by hydrogen bonding patterns at the primary hydroxyl groups. As shown in Fig. 4 the CB[n] homologues exhibit barrel-shaped cavity and unlike in CD two planes associated with different sets of ‘x’ CPs may be noticed for the CB[n] hosts. Hence the CD and CB[n] macrocycles herein show distinct electron distribution characteristics within the cavity.

Top view of array of MESP CP (‘w’ and ‘x’) in β-CD and CB[7]. Planes comprising of CPs ‘w’ and ‘x’ in β-CD and CB[7] are shown. See text for details
The cavity diameter and the cavity height can be estimated by exploring the MESP topography. The cavity diameters of CB[n] portals and the CD rims were calculated from the separation between the radially opposite critical points in the MESP topography. An average separation of the radially opposite CPs yields the cavity diameter. The cavity height can be derived as follows: the x, y, z coordinates of all the (3,+3) CPs have been transformed to a new coordinate system, wherein the Z-coordinates of ‘w’ CPs in the CD hosts (or one of the portals in case of CB[n] homologues) were set to zero. Consequently the cavity has been set parallel to the Z-axis and thus the planes formed by different set of CPs become perpendicular to the axis. The z-coordinate of the CPs in a given plane turn out to be nearly identical in the coordinate framework defined here. Accordingly the cavity height in the α-, β-, and γ-CD can be calculated from the separation between two planes, viz., one formed by the CPs in the top rim (‘w’) and the other resulting from those within the cavity (‘x’).
The cavity diameter and the cavity height of α-, β- and γ-CD conformers thus obtained are given in Table 4. For the “A” conformers of α-CD the upper and lower rim diameters (dw and dx) turn out to be 4.38 and 7.01 Å, respectively. The upper rim diameter has been noticed to be less sensitive to hydrogen bonding patterns in the secondary hydroxyl groups; the diameter of lower rim on the other hand, varies from 6.39 to 7.01 Å in the A1–A4 conformers. The lower rim diameters in β- and γ-CD conformers are nearly 3.0 and 3.5 Å larger than those calculated for the upper rim. In “B” and “C” conformers both dw and dx estimated to be nearly the same. The cavity-height, separation of planes formed by ‘x’ and ‘w’ CPs, reported in Table 4 turn out to be largest for the “C” conformers (8.6–8.8 Å) than those for “B” or “A” conformers of CD hosts. Thus the cavity height does not vary significantly within the given class of CD conformers.
The diameter and height of the CB[n] homologues can also be derived using the methodology similar to that for CD hosts outlined earlier. Here the effective cavity height (hww) for CB[n] can be obtained from the separation of planes constituting the ‘w’ CPs one each at both the portals of host. The separation of two planes, viz., one containing the ‘w’ CPs at the portal and the other defined by ‘x’ CPs (midway between the two portals shown in Fig. 4) provides the estimate of hwx. The diameter (dw, and dx), and heights (hxx, hwx and hww) thus obtained are presented in Table 5. As may readily be noticed along CB[n] series, hww turns out to be ~14.0 Å and remains nearly unchanged. The distinct electronic distribution within the CB[n] cavity can readily be noticed from the different values of hwx and hww. As displayed, the cavity diameter of the upper rim of CB[5] turns out to be nearly less than one-half as compared to that for CB[8] homologue. Thus it may be inferred that penetration of the large guest within the CB[5] cavity will be relatively difficult and accordingly the CB[5] host does not conduce the inclusion complex. The earlier observation that ferrocene can not be encapsulated within the CB[5] cavity [45] can also be explained on similar lines.
A comparison of cavity dimensions in CB[n] and CD hosts reveals that both upper and lower rim diameters of “B” and “C” conformers in γ-CD (exhibiting barrel-like cavity) are 0.5–1.0 Å larger than those of CB[8]. Similar inferences may be drawn in case of α-CD and CB[6] hosts. On the other hand ‘effective cavity diameter’ of β-CD and CB[7] are comparable. It may be worth noting that cavity-height of CB[n] homologues are 5–6 Å larger compared to the “C” conformer of CD hosts exhibiting larger height.
Conclusions
This work presents a systematic investigations on the cavity size and molecular electrostatic potential topography in CB[n] (n = 5–8) and different (“A”, “B” and “C”) α-, β- and γ-CD conformers. It has been demonstrated how the ‘effective’ cavity height and diameter in different CD conformers can be gauged from the MESP topography. In case of α-, β- and γ-CD, the cavity shape is dependent on hydrogen bonding patterns from primary hydroxyl groups and not on the number of repeating sugar units in the macrocycle. Thus, “A” type conformers of α-, β- and γ-CD yield “cone-like cavity” while “B” and “C” conformers possesses a “barrel shaped cavity”. The largest cavity height (8.6–8.8 Å) was noticed within the “C” conformers (possessing O6H–O5 interactions) of α-, β- and γ-CD. Cavity width (diameter) in the conformers facilitating either O6H–O5′ or O6H–O5 interactions (as in the “B” and “C” conformers) turns out to be nearly the same. Furthermore the cavity diameter estimated from the MESP topography reveal that the width of CB[7] is comparable to “B” or “C” conformers of β-CD whereas that of α- and γ-CD are 1.0–1.5 Å larger compared to the CB[6] and CB[8], respectively. The distinct electronic charge distribution within the CB[n] (n = 6–8) cavity may also be noticed from the separation of planes constituting the MESP minima. On the other hand, the portals of CB[n] cavity turn out to be hydrophilic. This partly explains large affinity of CB[n] hosts toward the cations or metal ions. The cavity-height of CB[n] homologues are relatively large (5–6 Å) compared to “C” type conformers of CD hosts. Furthermore the calculated phase angle Φ in the Cremer–Pople parameters depends on the type of hydrogen bonded interaction as well as hydrogen bond strength. In brief, the host–guest interactions at the molecular level can be analyzed by exploring the MESP topography.
Supporting information
The lateral view of (3,+3) CPs, top view of array of CP ‘w’ and ‘x’ in the electrostatic potential topography, MESP isosurface (V = −105.0 kJ mol−1) for the conformers of α-, β- and γ-CD as well as CB[n] (n = 6–8) homologues.
References
Szejtli, J.: Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 98, 1743–1753 (1998). doi:10.1021/cr970022c
Cunha-Silva, L., Teixeira-Dias, J.J.C.: How humidity affects the solid-state inclusion of 2-phenoxyethanol in β-cyclodextrin: a comparison with β-cyclodextrin. New J. Chem. 28, 200–206 (2004). doi:10.1039/b309491j
Vrielynck, L., Lapouge, C., Marquis, S., Kister, J., Dupuy, N.: Theoretical and experimental vibrational study of phenylurea: structure, solvent effect and inclusion process with the β-cyclodextrin in the solid state. Spectrochim. Acta A 60, 2553–2559 (2004). doi:10.1016/j.saa.2003.12.035
Al-Shihry, S.S.: Spectroscopic studies of inclusion complexes of 1-naphthol-4-sulfonate with β-cyclodextrin in aqueous solution. Spectrochim. Acta A 61, 2439–2443 (2005). doi:10.1016/j.saa.2004.09.006
Buchanan, C.M., Alderson, S.R., Cleven, C.D., Dixon, D.W., Ivanyi, R., Lambert, J.L., Lowman, D.W., Offerman, R.J., Szejtli, J., Szente, L.: Synthesis and characterization of water-soluble hydroxybutenyl cyclomaltooligosaccharides (cyclodextrins). Carbohydr. Res. 337, 493–507 (2002). doi:10.1016/S0008-6215(01)00328-7
Mosinger, J., Tomankova, V., Nemacova, I., Zyka, J.: Cyclodextrins in analytical chemistry. Anal. Lett. 34(12), 1979–2004 (2001). doi:10.1081/AL-100106834
Szejtli, J., Szente, L.: Elimination of bitter, disgusting tastes of drugs and foods by cyclodextrins. Eur. J. Pharm. Biopharm. 61, 115–125 (2005). doi:10.1016/j.ejpb.2005.05.006
Redenti, E., Szente, L., Szejtli, J.: Cyclodextrin complexes of salts of acidic drugs. Thermodynamic properties, structural features, and pharmaceutical applications. J. Pharm. Sci. 90, 979–986 (2001). doi:10.1002/jps.1050
Redenti, E., Szente, L., Szejtli, J.: Drug/cyclodextrin/hydroxy acid multicomponent systems. Properties and pharmaceutical applications. J. Pharm. Sci. 89, 1–8 (2000). doi:10.1002/(SICI)1520-6017(200001)
Géczy, J., Bruhwyler, J., Scuvée-Moreau, J., Seutin, V., Masset, H., Van Heugen, J.C., Dresse, A., Lejeune, C., Decamp, E., Szente, L., Szejtli, J., Liégeois, J.-F.: The inclusion of fluoxetine into γ-cyclodextrin increases its bioavailability: behavioural, electrophysiological and pharmacokinetic studies. Psychopharmacology 151, 328–334 (2000). doi:10.1007/s002130000512
Loftsson, T., Jarvinen, T.: Cyclodextrins in ophthalmic drug delivery. Adv. Drug Deliv. Rev. 36, 59–79 (1999). doi:10.1016/S0169-409X(98)00055-6
Jeon, W.S., Moon, K., Park, S.H., Chun, H., Ko, Y.H., Lee, J.Y., Lee, E.S., Samal, S., Selvapalam, N., Rekharsky, M.V., Sindelar, V., Sobransingh, D., Inoue, Y., Kaifer, A.E., Kim, K.: Complexation of ferrocene derivatives by the cucurbit[7]uril host: a comparative study of the cucurbituril and cyclodextrin host families. J. Am. Chem. Soc. 127, 12984–12989 (2005). doi:10.1021/ja052912c
Buschmann, H.-J., Schollmeyer, E., Mutihac, L.: The formation of amino acid and dipeptide complexes with α-cyclodextrin and cucurbit[6]uril in aqueous solutions studied by titration calorimetry. Thermochim. Acta 399, 203–208 (2003). doi:10.1016/S0040-6031(02)00462-8
Mohanty, J., Bhasikuttan, A.C., Nau, W.M., Pal, H.: Host–guest complexation of neutral red with macrocyclic host molecules: contrasting pK a shifts and binding affinities for cucurbit[7]uril and β-cyclodextrin. J. Phys. Chem. B 110(10), 5132–5138 (2006). doi:10.1021/jp056411p
Moghaddam, S., Inoue, Y., Gilson, M.K.: Host–guest complexes with protein–ligand-like affinities: computational analysis and design. J. Am. Chem. Soc. 131(11), 4012–4021 (2009). doi:10.1021/ja808175m
Kim, J., Jung, I.-S., Kim, S.-Y., Lee, E., Kang, J.-K., Sakamoto, S., Yamaguchi, K., Kim, K.: New cucurbituril homologues: syntheses, isolation, characterization, and X-ray crystal structures of cucurbit[n]uril (n = 5, 7 and 8). J. Am. Chem. Soc. 122, 540–541 (2000). doi:10.1021/ja993376p
Koner, A.L., Nau, W.M.: Cucurbituril encapsulation of fluorescent dyes. Supramol. Chem. 19(1–2), 55–66 (2007). doi:10.1080/10610270600910749
Liu, J.-X., Long, L.-S., Huaang, R.-B.L., Zheng, L.-S.: Interesting anion-inclusion behavior of cucurbit[5]uril and its lanthanide-capped molecular capsule. Inorg. Chem. 46, 10168–10173 (2007). doi:10.1021/ic701236v
Osaka, I., Kondou, M., Selvapalam, N., Samal, S., Kim, K., Rekharsky, M.V., Inoue, Y., Arakawa, R.: Characterization of host–guest complexes of cucurbit[n]uril (n = 6, 7) by electrospray ionization mass spectrometry. J. Mass Spectrom. 41, 202–207 (2006). doi:10.1002/jms.978
Buschmann, H.-J., Cleve, E., Jansen, K., Wego, A., Schollmeyer, E.: Complex formation between cucurbit[n]urils and alkali, alkaline earth and ammonium ions in aqueous solution. J. Incl. Phenom. Macrocycl. Chem. 40, 117–120 (2001). doi:10.1023/A:1011159119554
Bhasikuttan, A.C., Mohanty, J., Nau, W.M., Pal, H.: Efficient fluorescence enhancement and cooperative binding of an organic dye in a supra-biomolecular host-protein assembly. Angew. Chem. Int. Ed. Engl. 46, 4120–4122 (2007). doi:10.1002/anie.200604757
Marquez, C., Hudgins, R.R., Nau, W.M.: Mechanism of host–guest complexation by cucurbituril. J. Am. Chem. Soc. 126(18), 5806–5816 (2004). doi:10.1021/ja0319846
Jeon, Y.J., Kim, S.-Y., Ko, Y.H., Sakamoto, S., Yamaguchi, K., Kim, K.: Novel molecular drug carrier: encapsulation of oxaliplatin in cucurbit[7]uril and its effects on stability and reactivity of the drug. Org. Biomol. Chem. 3, 2122–2125 (2005). doi:10.1039/b504487a
Kemp, S., Wheate, N.J., Stooman, F.H., Aldrich-Wright, J.R.: The host-guest chemistry of proflavine with cucurbit[6, 7, 8]urils. Supramol. Chem. 19, 475–484 (2007). doi:10.1080/10610270601124019
Kim, J., Ahn, Y., Park, K.M., Kim, Y., Ko, Y.H., Oh, D.H., Kim, K.: Carbohydrate wheels: cucurbituril-based carbohydrate clusters. Angew. Chem. Int. Ed. Engl. 119, 7537–7539 (2007). doi:10.1002/ange.200702540
Wei, F., Liu, S.-M., Xu, L., Cheng, G.-Z., Wu, C.-T., Feng, Y.-Q.: The formation of cucurbit[n]uril (n = 6, 7) complexes with amino compounds in aqueous formic acid studied by capillary electrophoresis. Electrophoresis 26, 2214–2224 (2005). doi:10.1002/elps.200410260
Tuncel, D., Steinke, J.H.G.: Catalytic self-threading: a new route for the synthesis of polyrotaxanes. Macromolecules 37, 288–302 (2004). doi:10.1021/ma034294v
Liu, S.-M., Wu, X., Huang, Z., Yao, J., Liang, F., Wu, C.: Construction of pseudorotaxanes and rotaxanes based on cucurbit[n]uril. J. Incl. Phenom. Macrocycl. Chem. 50, 203–207 (2004). doi:10.1007/s10847-004-6472-4
Corma, A., Garcıa, H., Montes-Navajas, P.A., Primo, J.J., Calvino, S., Trasobares, S.: Gold nanoparticles in organic capsules: a supramolecular assembly of gold nanoparticles and cucurbituril. Chemistry 13, 6359–6364 (2007). doi:10.1002/chem.200601900
Cervello, E., Jaime, C.: β-cyclodextrin bimodal complexes with n-alkylbenzenes and n-alkylcyclohexanes A molecular mechanics study. J. Mol. Struct. 428, 195–201 (1998). doi:10.1016/S0166-1280(97)00279-0
Madrid, J., Paozuelo, J., Mendicuti, F., Mattice, W.L.: Molecular mechanics study of the inclusion complexes of 2-methyl naphthoate with α- and β-cyclodextrins. J. Colloid Interface Sci. 193, 112–120 (1997). doi:10.1006/jcis.1997.5061
Margheritis, C., Sinistri, C.: β-cyclodextrin in aqueous solution: MM and semiempirical calculations. Farmaco 52, 435–438 (1997)
Casadesus, R., Moreno, M., Gonzalez-Lafont, A., Lluch, J.M., Repasky, M.P.: Testing electronic structure methods for describing intermolecular H···H interactions in supramolecular chemistry. J. Comput. Chem. 25, 99–105 (2004). doi:10.1002/jcc.10371
Britto, M.A.F.O., Nascimnento, C.S., Jr., Dos Santos, H.F.: Structural analysis of cyclodextrins: a comparative study of classical and quantum mechanical methods. Quim. Nova. 27(6), 882–888 (Portuguese) (2004); Sociedade Brasileira de Quimica (2004). doi:10.1590/S0100-40422004000600008
Nascimento Jr., C.S., Dos Santos, H.F., De Almedia, W.B.: Theoretical study of the formation of the α-cyclodextrin hexahydrate. Chem. Phys. Lett. 397, 422–428 (2004). doi:10.1016/j.cplett.2004.09.026
Nascimento Jr., C.S., Cleber, P.A., Dos Santos, H.F., De Almedia, W.B.: Theoretical study of the α-cyclodextrin dimer. J. Phys. Chem. A 109, 3209–3219 (2005). doi:10.1021/jp044490j
Bako, I., Jicsinszky, L.: Semiempirical calculations on cyclodextrins. J. Incl. Phenom. Mol. Recognit. Chem. 18(3), 275–289 (1994). doi:10.1007/BF00708734
Margheritis, C., Sinistri, C.: β-cyclodextrin and water semiempirical calculations. Z. Naturforsch. A 51(8), 950–956 (1996)
Avakyan, V.G., Nazarov, V.B., Alfimov, M.V., Bagatur’yants, A.A., Voronezheva, N.I.: The role of intra- and intermolecular hydrogen bonds in the formation of b-cyclodextrin head-to-head and head-to-tail dimers. The results of ab initio and semiempirical quantum- chemical calculations. Russ. Chem. Bull. 50(2), 206–216 (2001). doi:10.1023/A:1009557729668
Anconi, C.P.A., Nascimento Jr., C.S., Fedoce-Lopes, J., Dos Santos, H.F., De Almeida, W.B.: Ab initio calculations on low-energy conformers of α-cyclodextrin. J. Phys. Chem. A 111, 12127–12135 (2007). doi:10.1021/jp0762424
Karpfen, A., Liedl, E., Snor, W., Weiss-Greiler, P., Viernstein, H., Wolschann, P.: Homodromic hydrogen bonds in low-energy conformations of single molecule cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 57, 35–38 (2007). doi:10.1007/s10847-006-9166-2
Pinjari, R.V., Joshi, K.A., Gejji, S.P.: Molecular electrostatic potentials and hydrogen bonding in α-, β-, and γ-cyclodextrins. J. Phys. Chem. A 110, 13073–13080 (2006). doi:10.1021/jp065169z
Pinjari, R.V., Joshi, K.A., Gejji, S.P.: Theoretical studies on hydrogen bonding, NMR chemical shifts and electron density topography in α-, β-, and γ-cyclodextrins conformers. J. Phys. Chem. A 111, 13583–13589 (2007). doi:10.1021/jp074539w
El-Barghouthi, M.I., Jaime, C., Al-Sakhen, N.A., Issa, A.A., Adboth, A.A., Al Omari, M.M., Badwan, A.A., Zughul, M.B.: Molecular dynamics simulations and MM–PBSA calculations of the cyclodextrin inclusion complexes with 1-alkanols, para-substituted phenols and substituted imidazoles. J. Mol. Struct. 853, 45–52 (2008). doi:10.1016/j.theochem.2007.12.005
Pinjari, R.V., Gejji, S.P.: Electronic structure, molecular electrostatic potential, and NMR chemical shifts in cucurbit[n]urils (n = 5–8), ferrocene, and their complexes. J. Phys. Chem. A 112, 12679–12686 (2008). doi:10.1021/jp807268v
Carlqvist, P., Maseras, F.: A theoretical analysis of a classic example of supramolecular catalysis. Chem. Commun. 7, 748–750 (2007). doi:10.1039/b613434c
Buschmann, H.-J., Wego, A., Zielesny, A., Schollmeyer, E.: Structure, stability, electronic properties and NMR-shielding of the cucurbit[6]uril–spermine-complex. J. Incl. Phenom. Macrocycl. Chem. 54, 241–246 (2006). doi:10.1007/s10847-005-8140-8
Pichierri, F.: DFT study of cucurbit[n]uril, n = 5–10. J. Mol. Struct. 765, 151–152 (2006). doi:10.1016/j.theochem.2006.03.039
Oh, S.K., Yoon, J., Kim, K.S.: Structural stabilities and self-assembly of cucurbit[n]uril (n = 4–7) and decamethylcucurbit[n]uril (n = 4–6): a theoretical study. J. Phys. Chem. B 105, 9726–9731 (2001). doi:10.1021/jp011919n
Kaanumalle, L.S., Gibb, C.L.D., Gibb, B.C., Ramamurthy, V.: A hydrophobic nanocapsule controls the photophysics of aromatic molecules by suppressing their favored solution pathways. J. Am. Chem. Soc. 127, 3674–3675 (2005). doi:10.1021/ja0425381
Smit, B., Maesen, T.L.M.: Towards a molecular understanding of shape selectivity. Nature 451, 671–678 (2008). doi:10.1038/nature06552
Steed, J.W., Atwood, J.L. (eds.): Encyclopedia of Supramolecular Chemistry. Marcel Dekker, New York (2004)
Becke, A.D.: Density-functional thermochemistry. III. The role of exact exchanges. J. Chem. Phys. 98, 5648–5652 (1993). doi:10.1063/1.464913
Lee, C., Yang, W., Parr, R.G.: Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988). doi:10.1103/PhysRevB.37.785
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery, J.A. Jr., Vreven, T., Kudin, K.N., Burant. J.C., Millam, J.M., Iyengar, S.S., Tomasi, J., Barone, V., Mennucci, B., Cossi, M., Scalmani, G., Rega, N., Petersson, G.A., Nakatsuji, H., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Klene, M., Li, X., Knox, J.E., Hratchian, H.P., Cross, J.B., Bakken, V., Adamo, C., Aramillo, J.J., Gomperts, R., Stratmann, R.E., Yazyev O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Ayala, P.Y., Morokuma, K., Voth, G.A., Salvador, P., Dannenberg, J.J., Zakrzewski, V.G., Dapprich, S., Daniels, A.D., Strain, M.C., Farkas, O., Malick, D.K., Rabuck, A.D., Raghavachari, K., Foresman J.B., Ortiz, J.V., Cui, Q., Baboul, A.G., Clifford, S.J., Cioslowski, B.B., Stefanov, G., Liu, A., Liashenko, P., Piskorz, I., Komaromi, R.L., Martin, D.J., Fox, T.M.A., Keith, C.Y., Al-Laham, A., Peng, M., Nanayakkara, M., Challacombe, P.M.W., Gill, B., Johnson, W., Chen, M.W., Wong, C., Gonzalez, J. Pople, A.: Gaussian 03, Revision C.02. Gaussian, Wallingford (2004)
Balanarayan, P., Gadre, S.R.: Topography of molecular scalar fields. I. Algorithm and Poincaré–Hopf relation. J. Chem. Phys. 119, 5037–5043 (2003). doi:10.1063/1.1597652
Politzer, P., Trulhar, D.G. (eds.): Chemical Applications of Atomic and Molecular Electrostatic Potentials. Plenum, New York (1981)
Naray-Szabo, G., Ferenczy, G.G.: Molecular electrostatics. Chem. Rev. 4, 829–847 (1995). doi:10.1021/cr00036a002
Limaye, A.C., Gadre, S.R.: UNIVIS-2000: an indigenously developed comprehensive visualization package. Curr. Sci. 80, 1298–1301 (2001)
Cremer, D., Pople, J.A.: A general definition of ring puckering coordinates. J. Am. Chem. Soc. 97, 1354–1358 (1975). doi:10.1021/ja00839a011
Gessler, K., Krauss, N., Steiner, T., Betzel, C., Sarko, A., Saenger, W.: β-D-cellotetraose hemihydrate as a structural model for cellulose II. An X-ray diffraction study. J. Am. Chem. Soc. 117(46), 11397–11406 (1995). doi:10.1021/ja00151a003
Matta, C.F., Cow, C.N., Harrison, P.H.M.: Twisted amides: X-ray crystallographic and theoretical study of two acylated glycourils with aromatic substituents. J. Mol. Struct. 660, 81–97 (2003). doi:10.1016/j.molstruc.2003.08.005
Bekiroglu, S., Kenne, L., Sandstrom, C.: 1H NMR studies of maltose, maltoheptaose α-, β-, and γ-cyclodextrins, and complexes in aqueous solutions with hydroxy protons as structural probes. J. Org. Chem. 68, 1671 (2003). doi:10.1021/jo0262154
Brewster, M.E., Huang, M., Pop, E., Pitha, J., Dewar, M.J.S., Kaminski, J.J., Bodor, N.: An AM1 molecular orbital study of a-d-glucopyranose and β-maltose: evaluation and implications. Carbohydr. Res. 242, 53–67 (1993). doi:0008-6215/93/
Acknowledgements
SPG acknowledges support from the University Grants Commission (UGC), New Delhi, India [Research Project F34-370/2008(SR)] and University of Pune. RVP is grateful to Council of Scientific and Industrial Research, New Delhi, India for a Senior Research fellowship. JKK thanks UGC for the award of meritorious student fellowship. Authors thank the Center for Network Computing, University of Pune, for providing computational facilities.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Pinjari, R.V., Khedkar, J.K. & Gejji, S.P. Cavity diameter and height of cyclodextrins and cucurbit[n]urils from the molecular electrostatic potential topography. J Incl Phenom Macrocycl Chem 66, 371–380 (2010). https://doi.org/10.1007/s10847-009-9657-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-009-9657-z