
Abstract
The genetic structure of lineages can provide important information for delineating “evolutionarily significant units” (ESUs) for conservation, and for planning actions to protect and restore taxa threatened with extinction. Speyeria zerene hippolyta, the Oregon silverspot butterfly, is a U.S.A. federally threatened subspecies that is the focus of considerable conservation effort, but whose evolutionary relationships with other Speyeria taxa are not well-understood. We conducted a genetic analysis of nine Speyeria species and 25 subspecies from western U.S.A., using both mitochondrial and nuclear markers. Our goal was to determine whether such data supported (a) S. z. hippolyta’s designation as an ESU, and (b) the current morphologically-based taxonomy of Speyeria spp. Our data for S. z. hippolyta were equivocal; while nuclear markers resolved all these individuals into a single clade, mtDNA data suggested the existence of two clades. Aside from S. cybele, which was consistently supported as monophyletic, our data provided little support for most of the species currently recognized for western U.S. Speyeria, including S. zerene, and even less for the many subspecies designations. These genetic findings stand in contrast to the morphological differences recognized by experts, and suggest a relatively recent origin for many of these taxa. Two of 66 individuals screened for Wolbachia infection tested positive for this symbiont. Our results provide no persuasive evidence that S. z. hippolyta should lose its status as an ESU, but they have important implications for ongoing management actions such as population augmentation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Developing effective strategies for protecting and restoring sensitive taxa requires that we be able to define and identify those units in need of protection. It is widely recognized that protected groups should represent distinctive evolutionary histories and potentials (Moritz 1994), i.e. that they be “evolutionarily significant units” (ESUs) (Waples 1991; Crandall et al. 2000). Knowledge of the genetic structure of lineages is an important complement to behavioral, morphological, and ecological information in determining the distinctiveness of groups (DeSalle and Amato 2004), the recognition of which can help guide management decisions and set conservation priorities (Dayrat 2005).
The Oregon Silverspot Butterfly (Speyeria zerene hippolyta) provides one example of a group for which ambitious conservation activities are underway even though little is known about the population processes and historical biogeography that underlie its current distribution. Listed in the U.S. as a threatened subspecies in 1980 (45 FR 44935–44939), S. z. hippolyta has been the focus of considerable management efforts, including habitat improvement as well as augmentation of low and/or declining populations with captively reared larvae (Crone et al. 2007). Some sites of extirpated populations are undergoing habitat restoration in preparation for re-introduction of individuals from other locations (U.S. Fish and Wildlife Service 2001). But little is known about the genetic relationships among the five extant populations of S. z. hippolyta in Oregon and northern California, nor is there much information about their genetic relationships to other subspecies of S. zerene or other species of Speyeria.
Speyeria are a North American group of Nymphalid butterflies. The 16 recognized species (Pelham 2008; Dunford 2009) were defined on morphological grounds by dos Passos and Grey (1947), and are mostly distributed in western North America. The larvae feed exclusively on Viola spp. (Brittnacher et al. 1978; Hammond 1981), and the ranges of various taxa are limited, in part, by the distribution of these plants. The group is notoriously variable in wing pattern and color (Pyle 2002; Dunford 2009), and this variation has formed the basis of numerous subspecies designations. Adding to the complexity is the parallel nature of the morphological variation observed among sympatric taxa. Within the same region, species tend to be similar in color and size. Differences between subspecies from different regions sometimes exceed differences observed among sympatric species (Hovanitz 1943). Convergence in form within a region may be influenced by selection favoring an advantageous phenotype, developmental response to a common environment, a more recent shared ancestry than the species designations indicate, or to hybridization. While Brittnacher et al. (1978) report that naturally occurring hybrids are rare, most subspecies and even many species have successfully interbred under laboratory conditions (Paul Hammond and David McCorkle, personal communication, Feb 25, 2012). No key to the group has been published (but see keys in Hammond 1978 and Dunford 2007), and only experts are able to reliably identify individuals (Pyle 2002), even then requiring knowledge of the geographic region from which they were collected. Though a preliminary investigation of mitochondrial DNA variation was conducted by Dunford (2007), Brittnacher et al.’s 1978 allozyme study of California Speyeria remains the only published investigation of genetic relationships among western North American Speyeria to date.

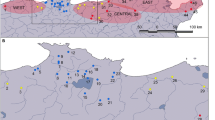
Map of collection locations. Three-letter geographical codes correspond to those used in the trees and in “Appendix 1”
In addition to the threatened S. z. hippolyta, Speyeria includes three U.S. “endangered” subspecies, S. z. behrensii, S. z. myrtleae, and S. callippe callippe, and two other taxa regarded as vulnerable, S. z. bremnerii and S. idalia (Xerces Society 2012). The taxonomic confusion characterizing Speyeria raises the question of whether these taxa are as genetically distinct as their protected status implies, and suggests that a genetic analysis of Speyeria could provide important information to help guide conservation efforts.
Many recent attempts to use genetic data to identify ESUs have focused on mitochondrial DNA (mtDNA) markers, because the rapid rate at which variation accumulates in the mitochondrial genome makes mtDNA useful for assessing differences among closely related taxa. In particular, much attention has been given to the potential of a fragment of the mitochondrial cytochrome c oxidase subunit one (COI) gene to serve as a universal “barcode” for identifying and delineating taxa (Hebert et al. 2003).
However, there are many reasons why variation within a single locus, especially one from the mitochondrial genome, might be a poor indicator of evolutionary patterns and processes, both for organisms in general and for Lepidoptera in particular (Wahlberg et al. 2003a, b; Gompert et al. 2006; Forister et al. 2008; Wahlberg et al. 2009). For this reason our genetic analysis employs both a mtDNA marker and several nuclear markers. Because infection by the endosymbiont Wolbachia has been known to complicate the interpretation of patterns of mtDNA and to pose a threat to the persistence of arthropod populations (Nice et al. 2009), we screened a subset of our samples for evidence of Wolbachia infection.
Our goal is to develop a molecular phylogeny for the Speyeria taxa in the western U.S., with a particular focus on S. z. hippolyta and other subspecies of S. zerene, in order to determine whether molecular phylogenetic data support S. z. hippolyta’s designation as a distinctive evolutionary lineage. This analysis also provides an opportunity to determine whether the phylogenetic patterns we discover are coincident with current taxonomy for other taxa in this group.
Methods
Taxon sampling
We attempted to include a representative sample of individuals of S. z. hippolyta, from both extant and extirpated populations, a representative sample of most of the other subspecies of S. zerene, and a sampling of other species of Speyeria from across a wide geographic distribution. We sampled a total of 121 Speyeria individuals from the western United States (Fig. 1) and two outgroup specimens, Brenthis daphne and Argynnis aglaja (“Appendix 1”). Our sampling structure for individuals and populations of species and subspecies is summarized in Table 1. Eighty of the Speyeria individuals were collected for or donated to this project by Paul C. Hammond, David McCorkle and Anne McHugh. Thirty-two specimens were obtained from the Arthropod Collection at Oregon State University (OSU). Nine S. zerene specimens were provided by the McGuire Center for Lepidoptera and Biodiversity at the Florida Museum of Natural History in Gainesville, Florida. In addition to these 121 samples, mitochondrial sequence data from 6 haplotypes representing 67 individuals of S. zerene hippolyta were provided by Richard Van Buskirk (RVB), either from individuals or tissue collected in 1995–1996 under USFWS permit number PRT-806058 or from additional specimens from OSU. All collections of S. z. hippolyta pre-dated the augmentations from other populations currently taking place. RVB also provided mitochondrial sequence data from 45 additional haplotypes representing 81 individuals of other Speyeria species and subspecies. DNA vouchers of all specimens and DNA samples (where available) have been archived at OSU.
DNA isolation
All genomic DNA isolated at Lewis & Clark College (LC) was extracted from one leg using QIAgen’s DNEasy extraction kit, according to manufacturer’s instructions, except eluted in 30 μl water. DNA was stored at −20 °C. Genomic DNA data provided by RVB came from wing tissue non-destructively sampled from live individuals (for S. z. hippolyta), leg tissue (from museum specimens), or thorax muscle (for all other live-caught specimens). This genomic DNA was isolated using a proteinase digest followed by phenol–chloroform extraction (for details see Van Buskirk 2000).
Gene selection
At LC, we amplified a single 1,410-base pair (bp) mtDNA fragment that included two genes, cytochrome c oxidase subunit I (COI) and cytochrome c oxidase subunit II (COII). RVB amplified a 613 bp region of the COII subunit for some individuals, and a 456 bp region for others. To optimize taxon inclusion while minimizing missing data, our phylogenetic analysis used the 554 base pair region of COII that allowed maximal overlap among these three datasets (see sequence assembly and alignment section below). At LC we also amplified four nuclear markers: glyceraldehyde 3-phosphate dehydrogenase (GAPDH), ribosomal protein subunit 5 (RpS5), triosephosphate isomerase (TPI), and wingless. We also amplified an 850 bp fragment of carbamoyl phosphate synthetase 2, aspartate carbamyltransferase, dihydrorotase (CAD) from a small but diverse subset of taxa, but did not analyze this fragment because it lacked variability. All these nuclear markers have proven to be informative at the species level for Lepidoptera (Brower and DeSalle 1998; Beltrán et al. 2002; Wahlberg et al. 2003b; Regier et al. 2008; Wahlberg and Wheat 2008; Wahlberg et al. 2009).
Molecular data acquisition
We used polymerase chain reaction (PCR) protocols and primers from several sources (“Appendix 2”). We purified post-PCR products using QIAgen PCR purification kits (LC) or Millipore filtration tubes with double-distilled water as a rinsing agent (RVB). We analyzed LC samples with a Nanodrop 1000 Spectrophotometer for DNA concentration and sent them to the University of Arizona Genetics Core for sequencing in two directions. RVB samples were sequenced with an ABI377 Perkin-Elmer automated sequencer.
Sequence assembly and alignment
We assembled the two strands for each fragment and checked sequence quality using Sequencher 4.6. To confirm amplification of the intended gene fragments, we subjected a subset of assembled sequences to homology searches in GenBank using BLASTn. We aligned sequences using the online server for MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/; Edgar 2004) and used default alignments for phylogenetic analyses. We viewed alignments, trimmed ragged ends, and concatenated our multigene datasets (see below) using Mesquite version 2.75 (Maddison and Maddison 2011). The COI/COII fragments were trimmed after alignment to the 554 nucleotides of COII that maximized overlap between the regions amplified at LC and by RVB. Preliminary analyses of the entire 1,410 base pair region of COI/II amplified at LC resulted in nearly identical tree topologies to those created using the shorter fragment; minor discrepancies between the analyses do not affect our conclusions. Sequences were deposited in GenBank (Accession Numbers available from the authors).
Phylogenetic analyses
We reconstructed separate phylogenetic hypotheses (a) for individual genes, (b) for a concatenated dataset of all nuclear genes, and (c) for a concatenation of all genes, nuclear and mitochondrial. Though our goal was to amplify four nuclear genes, there were some taxa for which we succeeded in amplifying only one (either wingless or RpS5, “Appendix 1”). Preliminary analyses of concatenated datasets that included taxa with single gene representation yielded trees that placed these taxa in unresolved basal polytomies. Therefore, our concatenated analyses only include taxa for which we had sequences for two or more genes. Most of the excluded taxa were S. zerene specimens from OSU (“Appendix 1”) that were represented only by wingless sequences, which were nearly invariant (Table 2).
We used jModelTest version 0.1.1 with the Bayesian Information Criterion (BIC) to determine the optimal model of evolution for each gene dataset. All phylogenetic models were constructed using Bayesian inference as implemented in MrBayes version 3.1.2 (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003), with model parameters optimized from the results of BIC. For concatenated analyses we used separate model parameters for each gene partition. All analyses were done with 10,000,000 Markov Chain Monte Carlo generations, saving every 100th tree, with two iterations of four chains for each analysis. We used Tracer version 1.5 (Rambaut and Drummond 2007) to determine the appropriate burn-in value, and in all cases discarded the first 10 % of saved trees as burn-in. We assessed confidence in particular clades using posterior probabilities.
In addition, we analyzed the data using parsimony, neighbor joining, and maximum likelihood methods. These resulted in either similar tree topologies as Bayesian analyses or reduced resolution; therefore, for simplicity, we only report the Bayesian results.
Pairwise genetic distances
We calculated uncorrected p-distances (i.e. the proportion of nucleotide sites at which two sequences differ, with no correction for multiple substitutions at the same site) within and among resolved clades for the COII dataset and for the concatenated nuclear dataset using MEGA version 5.1 (Tamura et al. 2011) with pairwise deletion of gaps and missing data.
Wolbachia screening
We screened a subset of the LC individuals for Wolbachia infection by amplifying a Wolbachia-specific 16S gene from genomic DNA isolated from Speyeria tissue (“Appendix 1”). As a positive control for PCR amplification, we used a genomic DNA template from a spider previously determined by GJB to be infected with Wolbachia. This template yielded a positive band in every PCR reaction we attempted with Speyeria gDNA. Positive bands were subjected to the same protocols for molecular data acquisition, sequence assembly and alignment as other LC samples (“Appendix 2”). Sequences were identified using homology searching with BLASTn.
Results
Data characteristics and model choice
Of the 121 Speyeria sampled at Lewis & Clark, we obtained quality sequences from 90 individuals; the taxa included, collection locations, and other information are summarized in “Appendix 1”. With the addition of RVB sequence data (see “Methods” section and Table 1), our dataset included data from nine of the 16 recognized species of Speyeria in the western U.S. and from 25 of the 104 described subspecies of these nine species, including eight of the 15–16 described subspecies of S. zerene (Pelham 2008; Dunford 2009; Table 1). In addition to S. zerene, five other species in our analysis (S. atlantis, S. callippe, S. egleis, S. hydaspe, and S. mormonia) were represented by more than one subspecies (Table 1).
DNA from different taxa and different markers amplified with varying success; our final datasets were most complete for COII and wingless (“Appendix 1”). We had particular difficulty amplifying genes from museum specimens (Watts et al. 2007), which constituted most of our samples of S. z. hippolyta; as a result, these museum specimens are represented only in the wingless and COII datasets. The numbers of bases in final alignments, and model characteristics, are summarized in Table 2. Models selected by jModelTest for all individual gene partitions indicate that two-rate parameters provided the best models of substitution patterns for both nuclear and mitochondrial datasets. Of the 2,624 nucleotides in our full-concatenated dataset, the mitochondrial gene constituted 21 % of the dataset, and 43 % of the parsimony-informative sites (Table 2).
Phylogenetic analyses
The degree to which relationships were resolved varied among markers, with mitochondrial COII resolving a higher proportion of nodes than any analyses of the nuclear sequences. More nodes were resolved in the concatenated nuclear gene analysis than in individual analyses of nuclear genes; however, analyses of nuclear data resolved fewer nodes, even when concatenated, than did analyses of mitochondrial data (Fig. 2).

Mirrored COII (left) and nuclear concatenated (right) 50 % majority rule consensus phylogenies from Bayesian analyses. Branch width is proportional to posterior probabilities of clades, with the widest branches equivalent to probabilities >.95. Scale bar on tree represents numbers of nucleotide substitutions per site. S. zerene in blue; S. z. hippolyta in red. Photos display ventral morphology on the left and dorsal morphology on the right. They are scaled for relative size; scale bars in photos represent 1 cm. (Color figure online)
Support of monophyly of nominal species
Speyeria cybele was the only nominal species that was monophyletic in all of our analyses (Figs. 2, 3, 4). S. hydaspe and S. mormonia were supported as monophyletic by the full concatenated analysis, which contained the most complete dataset (Fig. 4), as well as by either the COII (in the case of S. hydaspe) or the nuclear (for S. mormonia) analyses, but not by all of the analyses. Many species were present in unresolved polytomies that included other taxa, or were resolved into clades that were not monophyletic. Some species were more solidly supported than others as not monophyletic; most notable were S. atlantis and S. zerene, each consistently supported as polyphyletic, or included in multiple clades that were not resolved by our analyses.

50 % majority rule consensus phylogeny from Bayesian analysis of the wingless dataset. Branch width is proportional to posterior probabilities of clades, with the widest branches equivalent to probabilities >.95. Scale bar represents numbers of nucleotide substitutions per site. S. zerene in blue; S. z. hippolyta in red. (Color figure online)

50 % majority rule consensus phylogeny from Bayesian analysis for the full concatenated dataset. Branch width is proportional to posterior probabilities of clades, with the widest branches equivalent to probabilities >.95. Scale bar represents numbers of nucleotide substitutions per site. S. zerene in blue; S. z. hippolyta in red. (Color figure online)
S. zerene is not supported as monophyletic
None of our analyses supported the monophyly of the focal species, S. zerene. In the COII analyses, S. zerene was strongly supported as polyphyletic, with two clades (M1 and M2) that were themselves paraphyletic (Fig. 2, left). In clade M1 the paraphyly was caused by the inclusion of a single haplotype of S. callippe. Clade M1 was contained in another clade that also included S. hollandi, S. atlantis sorocko, S. atlantis dodgei, other S. callippe individuals, and S. egleis (Fig. 2, left). Another S. zerene (spp. hippolyta from Westport, WA) resolved with S. callippe. Most of our S. zerene specimens were resolved in clade M2, which contained two clades that we refer to as M3 and M4 (Fig. 2, left). In clade M3, a subset of S. zerene would be monophyletic except for the inclusion of five individuals of S. coronis snyderi. Clade M4 consisted primarily of S. zerene but also included S. atlantis subspecies and S. egleis.
The concatenated nuclear analysis (Fig. 2, right) had a large polytomy that did not resolve all S. zerene taxa. However, it did resolve two clades that were predominantly S. zerene. One of these, N1, while weakly supported (posterior probability = 0.52), contained individuals of all S. zerene subspecies in our dataset except S. z. hippolyta. (S. z. bremnerii is not included because this taxon was represented by only a single nuclear gene.) Clade N1 also included one S. callippe (of three in the dataset) and two S. atlantis hesperis individuals. The second clade, N2, included all five S. z. hippolyta for which we amplified more than one nuclear marker. However, this clade of S. z. hippolyta was paraphyletic; it included a monophyletic clade of seven S. cybele. Nine of the S. zerene in this dataset were in neither clade N1 nor N2, but were in an unresolved polytomy that contained clades N1 and N2 (Fig. 2, right).
In the full concatenated analysis (Fig. 4), relationships among S. zerene individuals reflected the influence of signals from both the nuclear and mitochondrial data (Fig. 2). There was one strongly supported monophyletic clade of S. zerene, A1 (Fig. 4), that included all taxa resolved in clade M1 of the COII analysis (Fig. 2, left). A second strongly supported clade, A2 (Fig. 4), included all S. zerene taxa in clade M3 (Fig. 2, left), including a S. coronis snyderi individual that rendered S. zerene paraphyletic. A third clade, A3 (Fig. 4), contained taxa that corresponded to clade M4 (Fig. 2, left) and was paraphyletic, including the two S. atlantis hesperis individuals in the analysis.
Mitochondrial and nuclear data differ in support of monophyly of S. zerene hippolyta
There was a striking disparity between the mitochondrial and nuclear analyses in their degree of support for the monophyly of S. z. hippolyta (Fig. 2). In the COII analysis, S. z. hippolyta was strongly supported as polyphyletic, with some individuals falling into clade M1 (those from populations at Rock Creek, Bray Point, Lake Earl, and Boiler Bay), and others falling into clade M3 (from populations at Cascade Head, Mt. Hebo, Cape Meares, and other individuals from the Boiler Bay and Lake Earl populations). In neither clade M1 nor M3 did S. z. hippolyta resolve as monophyletic. Moreover, a single individual from Westport, WA, fell outside of both clades. The average p-distances between this individual’s sequence and those of S. z. hippolyta in clades M1 and M3 were 2.8 and 1.6 %, respectively. In the COII analysis, this individual paired with S. callippe; it was not represented in our nuclear analysis.
In contrast, in the analysis of concatenated nuclear data (Fig. 2, right), all S. z. hippolyta resolved into a single clade (N2); this clade included S. cybele as well. We analyzed the wingless dataset independently, because it included the full set of taxa, including many museum specimens of S. z. hippolyta. This analysis (Fig. 3) resolved all the S. z. hippolyta into a single clade that also contained S. cybele, as well as S. z. bremnerii.
Mean pairwise genetic distances for nuclear and mitochondrial sequences among the S. z. hippolyta clades M1 and M3 and S. cybele were consistent with the phylogenetic relationships just described. Mitochondrial p-distances (Table 3a, c) suggest that clades M1 and M3 of S. z. hippolyta differ from one another just as much as each does from S. cybele. However, nuclear p-distances between clades M1 and M3 are half as great as the distances between each clade and S. cybele (Table 3b, c).
Monophyly of other Speyeria subspecies
Few subspecies of any other Speyeria species were supported as monophyletic. However, few of these taxa were sufficiently well-sampled to allow a meaningful test (Table 1). Interestingly, despite a lack of broad sampling, all S. zerene subspecies except S. z. conchyliatus are polyphyletic in the COII analysis with individuals in clades M1, M3, and/or M4. S. z. conchyliatus is paraphyletic with respect to the inclusion of S. z. gunderi in a monophyletic polytomy (Fig. 2, left). The conchyliatus + gunderi group was unresolved by the nuclear data analysis.
Subspecies of some other Speyeria species were consistently paired into clades that may provide insight into their taxonomic affinities. For example, S. atlantis sorocko was never resolved with other S. atlantis, but they were supported as a sister taxon to S. hollandi in the mitochondrial, the nuclear and the full concatenated analyses (Figs. 2, 4).
Evidence of Wolbachia in the Speyeria lineage
Of 66 individuals screened for the Wolbachia 16S gene, we amplified two positive PCR products, one each from S. z. gunderi and S. z. picta (“Appendix 1”). Both were submitted for sequencing, and one provided clean sequence (contact authors for accession number). The top 100 best matches from NCBI BLAST searches of this sequence had 98–99 % identity (e-values = 0) with a strain of Wolbachia pipientis. All of the matches that were annotated were isolated from insect hosts.
Discussion
Western North American species of Speyeria are notable for their complex and often subtle morphological variation and for the difficulty they present for making accurate determinations of species and subspecies (Pyle 2002; Dunford 2009). Our analysis of patterns of mitochondrial and nuclear DNA variation does not provide a tidy resolution of this complexity. Patterns suggested by an analysis of the mitochondrial COII gene were rarely confirmed by an examination of nuclear genes. Nuclear genes that have proven to be useful markers in other Lepidoptera provided little phylogenetically informative variation, leaving relationships among many taxa unresolved. Different genes showed considerable variation in their ease of amplification, causing different analyses to contain different subsets of individuals. DNA from museum specimens proved difficult to extract or amplify, reducing the taxon sample for our target group, S. z. hippolyta. Nevertheless, we are able to draw some useful insights about S. z. hippolyta and the larger group to which it belongs.
Aside from S. cybele (clearly supported as monophyletic by our COII analysis), our analyses provide little support for the nominal species currently recognized for Speyeria, and even less support for the many subspecies designations. In many cases, the lack of pattern we have found should be regarded as quite tentative, because some taxa are represented by only a few individuals. However, even the species for which we have the largest sample, S. zerene, fails to emerge as a distinct group in any of our analyses. Interestingly, both the nuclear and mtDNA analyses suggest the possible existence of a previously unrecognized monophyletic group composed of S. atlantis sorocko and S. hollandi hollandi, including a putative hybrid between the two. But as a general rule, there is little molecular support for most of the nominal taxa in our sample.
These genetic findings stand in contrast to the subtle but distinctive morphological differences recognized by Speyeria experts and used to make consistent species and subspecies identifications. This contrast suggests that the evolutionary history of Speyeria in North America may be quite recent, allowing little opportunity for fixed molecular markers to diverge within lineages. Barriers to interbreeding in this group may be the consequence of morphological, behavioral or ecological traits that are expected to evolve more rapidly than neutral traits because they are driven by selection. In such cases we would expect neutral genetic variation to display a pattern consistent with incomplete lineage sorting, as observed here (Forister et al. 2008).
The existence of mtDNA patterns that are discordant with patterns of nuclear DNA raises questions about the origins of these discordances and their implications (Toews and Brelsford 2012). At least four mechanisms could contribute to discordances. First, as discussed above, they could be the result of incomplete lineage sorting. Secondly, they could be caused by introgression of mtDNA haplotypes into populations through hybridization. Because females are the heterogametic sex in Lepidoptera, it is expected, according to Haldane’s Rule, that females that result from interspecific hybridization will experience reduced viability relative to males. For this reason it is thought that Lepidoptera will be less prone to the introgression of maternally inherited genetic material when hybridization occurs (Sperling 2003). However, clear cases of mitochondrial introgression have been reported among Lepidoptera (Forister et al. 2008; Gompert et al. 2008; Zakharov et al. 2009). Given that some Speyeria localities have as many as eight sympatric species (Hammond 1974), and that some species, such as S. zerene, are considered relatively vagile (Hammond 1974), hybridization is a potentially important process in this group. Hammond and McCorkle (personal communication, Feb. 25, 2012) report that approximately 1/1,000 Speyeria individuals observed in the field appear to be hybrids on morphological grounds. Models of hybridization (Chan and Levin 2005) have shown that even occasional long-distance dispersal by a single migrant can lead to introgression. S. z. gloriosa is thought to be capable of migrating 80–160 km during its flight season (Hammond and McCorkle personal communication, Feb. 25, 2012).
A third possible source of discordance between mtDNA and nuclear variation patterns is Wolbachia infection. This bacterial symbiont is increasingly being recognized as posing a particular challenge to genetic studies of Lepidoptera and other arthropods (Nice et al. 2009). By conferring cytoplasmic incompatibility (Werren et al. 2008), Wolbachia infection can drive maternally inherited traits in the mitochondrial genome to spread through populations, causing patterns of mtDNA variation to depart from expectations (Hurst and Jiggins 2005; Galtier et al. 2009). Our data indicate that Wolbachia infection is present in at least some populations of S. zerene.
Heteroplasmy, the possession of multiple mitochondrial haplotypes by a single individual, represents a fourth potential source of mtDNA and nuclear DNA discordance. If PCR selectively amplifies only one of the possible haplotypes present in an individual, mtDNA will be a poor reflection of the true species tree. Nearly half of the bee species surveyed by Magnacca and Brown (2010) exhibited some degree of heteroplasmy for the COI barcoding gene. Additional data, perhaps obtained through pyrosequencing (White et al. 2005), are necessary to distinguish among these possible sources of discordance between mtDNA and nuclear DNA patterns.
As is true for the Karner blue butterfly (Gompert et al. 2006), our analysis suggests that COII does not accurately represent species and subspecies-level genetic relationships within Speyeria. Insofar as COII is closely linked to the classic barcode region of COI, Speyeria joins a growing list of taxa for which COI may not be a particularly useful “barcode” marker (Wahlberg et al. 2003a; Roe and Sperling 2007; Forister et al. 2008).
Our primary motivation for this study was to obtain clearer information about the phylogenetic status of the threatened Speyeria zerene hippolyta, in order to assess the appropriateness of its current classification as an ESU. Our results for this group were particularly perplexing. Bayesian analysis of the wingless gene resolves all of the S. z. hippolyta individuals and their morphologically-similar geographical neighbor S. z. bremnerii into a single clade with posterior probability = 1.0. The concatenated nuclear data, whose phylogenetic pattern reflects that of wingless, also resolves the five S. z. hippolyta included in this analysis into a single weakly-supported clade, N2 (posterior probability = 0.55). However, interpretation of the N2 clade is complicated by the fact that it also includes seven individuals of S. cybele. While S. z. hippolyta is a small-winged, sexually-monomorphic butterfly, S. cybele is one of the largest Speyeria species and is sexually dimorphic in the western U.S.A. (Hammond 1978). It is likely that S. cybele’s inclusion with S. z. hippolyta in the N2 clade was driven by their shared pattern for a single marker, wingless, which could be the result of convergence in this relatively invariant gene. No analyses of other individual genes, nuclear or mitochondrial, supported a S. cybele–S. z. hippolyta clade.
Results for the COII tree are quite different. Here, individuals of S. z. hippolyta are represented in two distinct clades whose average sequences differ by more than 3 %, more than each differs from some other nominal species. Furthermore, neither clade consists solely of S. z. hippolyta; each also includes five other subspecies of S. zerene and two other nominal Speyeria species. In addition, mean p-distances of the concatenated nuclear genes are similar within and among S. z. hippolyta individuals that are resolved in the mitochondrial clades M1 and M3 (Table 2). That these p-distances are relatively high is apparent from the long branches of the nuclear concatenated tree (Fig. 2, right). Combined, these data suggest that there is some genetic variability within and among S. z. hippolyta populations.
However, the pattern created by these data is not strong enough to override other evidence supporting S. z. hippolyta’s status as a distinct ESU. This group displays specific morphological, developmental, and ecological traits that McCorkle and Hammond (1988) described as adaptations to the salt-spray meadows and windswept headlands that characterize its coastal habitat. Unfortunately, we were unable to draw any inferences about the two other sensitive subspecies of S. zerene, S. z. behrensii and S. z. myrtleae. Our samples of these taxa were comprised only of museum specimens, and we were unable to amplify genes from any of them.
Though our results do not call into question’s S. z. hippolyta’s status as an ESU, they do have significant implications for current management practices for this group. Currently, individuals from the large, stable population at Mt. Hebo are being captively reared and released at Cascade Head and Rock Creek/Bray Point to augment the much smaller, declining populations there. Our study detected differences between the mitochondrial DNA haplotypes of the Mt. Hebo and Rock Creek populations, differences that in the future could either be erased by these augmentations or that might render the augmentations ineffective, if the genetic differences are great enough to provide barriers to interbreeding. Work in RVB’s laboratory is currently underway to determine what proportion of the Rock Creek population retains its distinctive mtDNA haplotype as opposed to having acquired the haplotype of the captively-reared individuals used for augmentation.
Our provocative finding that some populations of S. zerene are infected with Wolbachia raises additional concerns about population augmentations. While none of our S. z. hippolyta samples scored positively for Wolbachia, it is possible that we failed to detect Wolbachia in some infected individuals. Introducing Wolbachia-infected individuals into an uninfected population temporarily reduces its effective population size (until the infection is fixed or extinct), and thus could cause augmentation to have the opposite of its intended effect (Nice et al. 2009). Our screen was only preliminary and did not provide a comprehensive survey of Wolbachia infection. A more comprehensive screening of both extant and extinct populations of S. z. hippolyta is currently underway (Amy Truitt personal communication, Aug. 15 2011).
In conclusion, the recent ancestry that seems to characterize this group of butterflies creates challenges for delineation of ESUs. For several taxa in this group, knowledge of ESU boundaries has important implications for management actions. It is possible that ESU determination in Speyeria might be aided by the application of an integrative taxonomic approach (Crandall et al. 2000; Dayrat 2005; Roe and Sperling 2007; Padial and De la Riva 2010; Schlick-Steiner et al. 2010), combining molecular data with information about the morphological, ecological, and geographic variation of these taxa. However, in a group as evolutionarily dynamic as Speyeria appears to be, even the combination of these approaches is unlikely to produce a phylogeny characterized by reciprocally monophyletic taxa, particularly at the subspecies level (Roe and Sperling 2007). In such a situation, it may be best to err on the side of caution when making conservation decisions for the Oregon silverspot.
References
Beltrán M, Jiggins CD, Bull V, Linares M, Mallet J, McMillan WO, Bermingham E (2002) Phylogenetic discordance at the species boundary: comparative gene genealogies among rapidly radiating Heliconius butterflies. Mol Biol Evol 19(12):2176–2190
Brittnacher JG, Sims SR, Ayala FJ (1978) Genetic differentiation between species of the genus Speyeria (Lepidoptera: Nymphalidae). Evol 32:199–210
Brower AVZ (1994) Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc Natl Acad Sci 91:6491–6495
Brower AVZ, DeSalle R (1998) Patterns of mitochondrial versus nuclear DNA sequence divergence among nymphalid butterflies: the utility of wingless as a source of characters for phylogenetic inference. Insect Mol Biol 7:73–82
Chan KMA, Levin SA (2005) Leaky prezygotic isolation and porous genomes: rapid introgression of maternally inherited DNA. Evol 59:720–729
Crandall KA, Bininda-Emonds ORP, Mace GM, Wayne RK (2000) Considering evolutionary processes in conservation biology. Trends Ecol Evol 15(7):290–295
Crone EE, Pickering D, Schultz CB (2007) Can captive rearing promote recovery of endangered butterflies? An assessment in the face of uncertainty. Biol Conserv 139:103–112
Dayrat B (2005) Towards integrative taxonomy. Biol J Linnaean Soc 85:407–415
DeSalle R, Amato G (2004) The expansion of conservation genetics. Nat Rev Genet 5:702–712
dosPassos CF, Grey LP (1947) Systematic catalogue of Speyeria (Lepidoptera, Nymphalidae) with designation of types and fixations of type localities. Am Mus Novit 1370:1–30
Dunford JC (2007) The genus Speyeria and the Speyeria atlantis/Speyeria hesperis complex: species and subspecies accounts, systematics, and biogeography (Lepidoptera: Nymphalidae). Dissertation, University of Florida
Dunford JC (2009) Taxonomic overview of the greater fritillary genus Speyeria Scudder and the atlantis-hesperis species complexes, with species accounts, type images, and relevant literature. Insecta Mundi 0090:1–74
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797
Forister ML, Nice CC, Fordyce JA, Gompert Z, Shapiro AM (2008) Considering evolutionary processes in the use of single-locus genetic data for conservation, with examples from the Lepidoptera. J Insect Conserv 12:37–51
Galtier N, Nabholz B, Glemin S, Hurst GDD (2009) Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol Ecol 18:4541–4550
Gompert Z, Nice CC, Fordyce JA, Forister ML, Shapiro AM (2006) Identifying units for conservation using molecular systematics: the cautionary tale of the Karner blue butterfly. Mol Ecol 15:1759–1768
Gompert Z, Forister ML, Fordyce JA, Nice CC (2008) Widespread mito-nuclear discordance with evidence for introgressive hybridization and selective sweeps in Lycaeides. Mol Ecol 17(24):5231–5244
Hammond PC (1974) An ecological survey of the Nymphalid butterfly genus Speyeria. Dissertation, University of Nebraska
Hammond PC (1978) Geographic variation and speciation in the Nymphalid butterfly genus Speyeria. Dissertation, Virginia Polytechnic Institute and State University
Hammond PC (1981) The colonization of violets and Speyeria butterflies on the ash-pumice fields deposited by Cascadian volcanoes. J Res Lepidoptera 20(3):179–191
Hebert PDN, Cywinska A, Ball SL, deWard JR (2003) Biological identifications through DNA barcodes. Proc R Soc Lond B 270:313–321
Hovanitz W (1943) Geographic variation and racial structure of Argynnis callippe in California. Amer Nat 77:400–425
Huelsenbeck JP, Ronquist F (2001) MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Hurst GDD, Jiggins FM (2005) Problems with mitochondrial DNA as a marker in population, phylogeographic, and phylogenetic studies: the effects of inherited symbionts. Proc R Soc Lond B 272:1525–1534
Maddison WP, Maddison DR (2011) Mesquite: a modular system for evolutionary analysis. Version 2.75 http://mesquiteproject.org. Accessed April 13, 2011
Magnacca KN, Brown MJF (2010) Mitochondrial heteroplasmy and DNA barcoding in Hawaiian Hylaeus (Nesoprosopis) bees (Hymenoptera: Colletidae). BMC Evol Biol. doi:10.1186/1471-2148-10-174
McCorkle DV, Hammond PC (1988) Biology of Speyeria zerene hippolyta (Nymphalidae) in a marine-modified environment. J Lepidopterist’s Soc 42:184–195
Moritz C (1994) Defining ‘evolutionarily significant units’ for conservation. Trends Ecol Evol 9:373–375
Nice CC, Gompert Z, Forister ML, Fordyce JA (2009) An unseen foe in arthropod conservation efforts: the case of Wolbachia infections in the Karner blue butterfly. Biol Conserv 142:3137–3146
Padial JM, De la Riva I (2010) A response to recent proposals for integrative taxonomy. Biol J Linnaean Soc 101:747–756
Pelham JP (2008) A catalogue of the Butterflies of the United States and Canada with a complete bibliography of the descriptive and systematic literature. J Res Lepidoptera 40:1–65
Pyle RM (2002) The butterflies of Cascadia. Seattle Audubon Society, Seattle
Rambaut A, Drummond AJ (2007) Tracer v 1.4, available from http://beast.bio.ed.ac.uk/Tracer
Regier JC, Grant MC, Mitter C, Cook CP, Peigler RS, Rougerie R (2008) Phylogenetic relationships of wild silkmoths (Lepidoptera: Saturniidae) inferred from four protein-coding nuclear genes. Syst Entomol 33:219–228
Roe AD, Sperling FAH (2007) Population structure and species boundary delimitation of cryptic Dioryctria moths: an integrative approach. Mol Ecol 16:3617–3633
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Schlick-Steiner BC, Steiner FM, Seifert B, Stauffer C, Christian E, Crozier RH (2010) Integrative taxonomy: a multisource approach to exploring biodiversity. Ann Rev Entomol 55:421–438
Sperling FAH (2003) Butterfly molecular systematics: from species definitions to higher-level phylogenies. In: Boggs CL, Watt WB, Ehrlich PR (eds) Butterflies: ecology and evolution taking flight. University of Chicago Press, Chicago, pp 431–458
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Toews DPL, Brelsford A (2012) The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecology 21(16):3907–3930
U.S. Fish and Wildlife Service (2001) Oregon silverspot butterfly (Speyeria zerene hippolyta) revised recovery plan. U. S. Fish and Wildlife Service, Portland
Van Buskirk RW Jr (2000) Phylogeography, monitoring and conservation of Speyeria zerene hippolyta, the Oregon Silverspot Butterfly, Dissertation. University of California, Davis
Wahlberg N, Wheat CW (2008) Genomic outposts serve the phylogenomic pioneers: designing novel nuclear markers for genomic DNA extractions of Lepidoptera. Systematic Biol 57(2):231–242
Wahlberg N, Oliveira R, Scott JA (2003a) Phylogenetic relationships of Phyciodes butterfly species (Lepidoptera: Nymphalidae): complex mtDNA variation and species delimitations. Syst Entomol 28:257–273
Wahlberg N, Weingartner E, Nylin S (2003b) Towards a better understanding of the higher systematics of Nymphalidae (Lepidoptera: Papilionidae). Mol Phylogenetics Evol 28:473–484
Wahlberg N, Weingartner E, Warren AD, Nylin S (2009) Timing major conflict between mitochondrial and nuclear genes in species relationships of Polygonia butterflies (Nymphalidae: Nymphalini). BMC Evol Biol 9:92
Waples RS (1991) Pacific salmon, Onchorhynchus spp., and the definition of “species” under the Endangered Species Act. Marine Fisheries Rev 53(3):11–22
Watts PC, Thompson DJ, Allen KA, Kemp SJ (2007) How useful is DNA extracted from the legs of archived insects for microsatellite-based population genetic analyses? J. Insect Conserv 2:195–198
Werren JH, Windsor DM (2000) Wolbachia infection frequencies in insects: evidence of a global equilibrium? Proc R Soc Lond B 267:1277–1285
Werren JH, Baldo L, Clark ME (2008) Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol 6:741–751
White HE, Durston VJ, Sellar A, Fratter C, Harvey JF, Cross NCP (2005) Accurate detection and quantitation of heteroplasmic mitochondrial point mutations by pyrosequencing. Genetic Testing 9(3):190–199
Williams BL (2002) Conservation genetics, extinction, and taxonomic status: a case history of the regal fritillary. Conserv Biol 16(1):148–157
Xerces Society (2012) Red list of butterflies and moths. http://www.xerces.org/red-list-of-butterflies-and-moths/. Accessed 9 Jan 2012
Zakharov EV, Lobo NF, Nowak C, Hellmann JJ (2009) Introgression as a likely cause of mtDNA paraphyly in two allopatric skippers (Lepidoptera: Hesperiidae). Heredity 102:590–599
Acknowledgments
We benefited from Paul Hammond’s and David McCorkle’s many years of studying Speyeria in the field and laboratory, and the generosity with which they have shared their time, samples, knowledge, and insights. Other individuals and institutions providing materials or other assistance include Andrew Warren and the McGuire Center for Lepidoptera and Biodiversity; Chris Marshall, Dana Ross and the Oregon State University Arthropod Collection; Niklas Wahlberg; Gary Albright and the Tillamook County Pioneer Museum; Mike Patterson; Anne Warner of United States Fish and Wildlife Service; Debbie Pickering of The Nature Conservancy; and Mary Jo Anderson and David Shepherdson at the Oregon Zoo. We are grateful for the field assistance of Charlie Blackmar, Megan Siefert, Ryan Essman, Amanda Delzer, Steven Levitte, Becca Salesky, Terry Stratton, Marrissa Hirt, and Beka Feathers, and for the advice and assistance of Pamela Zobel-Thropp, Elise Maxwell and Wendy McLennan. Work at Lewis & Clark College was generously supported by Oregon Zoo’s Future for Wildlife Program, James Dunford, and Lewis & Clark College. Data collection and initial analysis by RVB was supported by the Center for Population Biology, University of California, Davis.
Author information
Authors and Affiliations
Corresponding author
Appendices
Rights and permissions
About this article
Cite this article
McHugh, A., Bierzychudek, P., Greever, C. et al. A molecular phylogenetic analysis of Speyeria and its implications for the management of the threatened Speyeria zerene hippolyta . J Insect Conserv 17, 1237–1253 (2013). https://doi.org/10.1007/s10841-013-9605-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10841-013-9605-5