
Abstract
Integration of molecular genetic techniques and geometric morphometrics represent a valuable tool in the resolution of taxonomic uncertainty and the identification of significant units for conservation. We combined mitochondrial DNA cytochrome c oxidase subunit II gene sequence data and geometric morphometric analysis to examine taxonomic status and identify units for conservation in four species of the hypogean beetle Duvalius (Coleoptera, Trechinae) using mainly museum specimens collected in central Italy. Previous taxonomic studies based on morphological traits described several subspecies often inhabiting geographically distinct caves. Phylogenetic analysis identified two well supported monophyletic lineages and a number of different clades with relatively small genetic differences, suggesting a short divergence time in line with known geological history of the study area. Geometric morphometrics, on the other hand, recovered a high level of distinctiveness among specimens. Both genetic and morphometric analyses did not entirely corroborate former taxonomic nomenclature, suggesting possible rearrangements and the definition of evolutionary significant units. Beetles of the genus Duvalius are protected by regional laws and the majority of taxa considered in this study inhabit caves located outside protected areas. Our study advocates the importance of devoting protection efforts to networks of cave ecosystems rather than single locations or species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Taxonomic distinction and assessment of units for conservation rely on different diagnostic concepts including ecological and reproductive isolation, common ancestor, evolutionary history, genotypic and morphological clustering and differential adaptation with enhanced fitness components (de Queiroz 2007; Petit and Excoffier 2009; Hausdorf 2011). Morphological traits often undergo strong selection due to environmental pressures or reproductive mechanisms and may not entirely reflect the evolutionary history of species. Nevertheless, taxonomy has been traditionally based on morphological characters. In the last decades, molecular markers provided an additional set of characters to recognize species and address phylogenetic relationships (Vogler and Monaghan 2007; Sperling and Roe 2009). Critics of taxonomy based either on molecular or morphological features were raised and integrative approaches were eventually promoted for objective resolution of questions (Schlick-Steiner et al. 2010; Padial et al. 2010). However, the use of both genetic and morphological characters may lead to contrasting results, so that careful examination of evolutionary processes explaining such divergence is often necessary (see Schlick-Steiner et al. 2010 for a review).
Discrepancy between genetic and morphological data is relatively common in cave organisms. A number of studies have described the occurrence of low levels of genetic divergence among relatively young but morphologically distinct species (e.g. Leys et al. 2003; Juan et al. 2010 and references therein). In hypogean environments, in fact, physical traits may depend on sets of genes influenced by micro- and macro-environmental factors. This may allow for morphological plasticity to shape adaptive characters in a relatively short time (Allegrucci et al. 1992; Caccone and Sbordoni 2001). On the other hand, strong selection for traits linked to hypogean life may create instances of morphological convergence among taxa showing high genetic divergence (Faille et al. 2010; Juan and Emerson 2010). In both cases it may be difficult to recognize the evolutionary histories of allopatric populations living in different caves. However, populations showing distinctive traits encompass important contributions to biodiversity and should be considered as significant units for conservation (Fraser and Bernatchez 2001; Moritz 2002).
In this study, we applied an integrative approach on the hypogean Coleopteran genus Duvalius. In this group adaptation to hypogean life is expected to significantly affect both genetics and morphology of species and populations. Members of this genus are known from Mediterranean area to northwestern China. They live mainly in caves and in the superficial hypogean compartment (MSS, Milieu Souterrain Superficiel) (Giachino and Vailati 2010), where micro-environmental factors are expected to produce strong selective pressure. As a result, taxonomists have identified, on the basis of morphological characters, a plethora of species and subspecies often characterized by very limited distributions. The description of new species and their phylogenetic relationships have been so far based upon subjective evaluation of morphological characters and male copulatory apparatus (Jeannel 1928). Since many of these morphological characters show minor differences, the taxonomy of this group has been subject to different interpretations, especially at the sub-specific level. Studies on this genus based on molecular analysis have not been conducted yet, mainly because of difficulties in finding live specimens. Most Duvalius species are in fact extremely localized and are known from secluded, hard to reach caves.
We combined geometric morphometrics of external characters and molecular analysis of mitochondrial DNA cytochrome c oxidase subunit II (COII) gene to investigate the current taxonomy of a number of key species of the genus Duvalius in central Italy and assess the presence of significant units for conservation. We used museum specimens to minimize logistical and conservation problems (e.g. Crandall et al. 2009). Results from our study may be useful to evaluate whether current status of a subset of caves included in protected areas and sites of community importance under the European Commission Habitat Directive is effective in preserving intra- and interspecific biodiversity or alternative measures should be considered for the protection of cave organism diversity.
Methods
Study group
The genus Duvalius includes more than 300 species (Moravec et al. 2003) and shows morphological adaptations to subterranean habitats, including elongation of appendages, absence of wings, depigmentation, eye degeneration and development of specific sensory organs (Vandel 1964). In Italy, Duvalius is represented by approximately 80 species distributed from the Alps to Sardinia and Sicily (Magrini 1997, 1998). The Tuscan-Aemilian area comprises 11 species and 24 subspecies, most of which are protected under regional laws (Tuscany Regional Law n. 56/2000 and Emilia-Romagna Regional Law n. 15/2006) due to their endemic or limited distribution. According to Magrini (1997), Tuscan species are divided into six groups. Our study focused on the species-richest andreinii group, which shows a relatively high taxonomic uncertainty. This group includes four species with several subspecies: Duvalius andreinii (Gestro, 1907), D. minozzii (Dodero, 1917), D. jureceki (Dodero, 1917) and D. apuanus (Dodero, 1917). Duvalius andreinii, D. jureceki and D. apuanus are endemic to Tuscany, while D. minozzii is distributed in Tuscany and Emilia-Romagna. Some of these taxa were originally assigned to other genera and subgenera. The majority of the subspecies now assigned to Duvalius minozzii were considered distinct species, and three of the four subspecies now assigned to Duvalius apuanus were originally described as belonging to D. jureceki (Magrini 1997).
Sampling
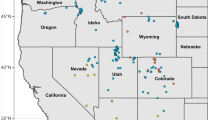
Specimens were collected in 20 caves and one superficial hypogean site in Tuscany and Emilia-Romagna, Italy (Fig. 1; Table 1 and Online Resource 1 Table S1) and stored dried at the Natural History Museum of the University of Florence and in private collections. A total of 108 specimens were used for morphometric examination. Of these, 102 samples were collected from 1942 to 1990 using mostly pitfall traps. Six samples were collected in 2008 and 2009 and preserved in 96 % ethanol. We used only males in order to avoid biases due to sexual dimorphism. Molecular analyses were performed on the six recently collected individuals and on other 20 samples, representative of all subspecies and sampling sites. Specimens from Liguria, northwestern Italy, belonging to Duvalius doriai doriai (Fairmaire, 1859) and D. canevai (Gestro, 1885) and Trechus quadristriatus (Schrank, 1781), also belonging to the subfamily Trechinae, were included as outgroups. Phylogenetic trees were rooted using T. quadristriatus, which is part of a clade different from Duvalius (Faille et al. 2010).
DNA extraction, amplification and sequencing
DNA was extracted from whole individuals after abdomen excision. Recently collected specimens were incubated overnight in 600 μl of lysis buffer (100 mM Tris–HCl, 5 mM EDTA, 100 mM NaCl, 0.5 % SDS, pH 8.0) with 10 μl of proteinase K (20 mg/ml) at 37 °C. DNA was then obtained using standard phenol–chloroform methods (Sambrook and Russell 2001). Museum specimens were processed using protocols similar to, but not as stringent as methods used for ancient DNA (Wandeler et al. 2007). Cross-contamination among archival specimens and between new and archival specimens was reduced by carrying out DNA extraction and amplification in separated rooms. Instruments, reagents and solutions were sterilized and dedicated to museum samples. Archival samples can be subject to different levels of DNA degradation depending on age of specimens and storage conditions (Zimmermann et al. 2008) and solutions used in pitfall traps can damage DNA (Stoeckle et al. 2010). Since silica-based methods have proven most effective in DNA extraction from museum specimens (Hajibabaei et al. 2005), we used the NucleoSpin Tissue XS kit (Machery-Nagel, Düren, Germany) following the manufacturer’s protocol. Two independent extractions were performed for each specimen along with a separate reaction using lysis buffer as negative control. All DNA extractions were stored in TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 7.6) at −80 °C.
The mitochondrial cytochrome c oxidase subunit II (COII) gene of six recently collected specimens was amplified and sequenced using the light-strand primer TrCOIIL3056 (5′-TATGGCAGAATAGTGTAATG-3′) and the heavy-strand primer TrCOIIH3927 (5′-TTATTGGGGCTATTTGTGGAA-3′) designed specifically for this project on the leucine tRNA and ATP synthase 8 genes, respectively. Primer numbers refer to the 3′ base of the published Trachypachus holmbergi Mannerheim, 1853 mitochondrial genome sequence (Sheffield et al. 2008). Polymerase chain reaction (PCR) amplification was conducted in a total volume of 25 μl with 15–100 ηg of total DNA, 1× PCR buffer, 1.5 mM MgCl2, 100 μM of each dNTP, 0.5 μM of each primer and 1 unit of Taq DNA polymerase (Invitrogen). Thermal profiles consisted of an initial denaturation step of 5 min at 94 °C, followed by 35 cycles of 30 s at 94 °C, 30 s at 48 °C and 90 s at 72 °C, with a final extension step of 10 min at 72 °C. PCR products (911 bp) were cycle-sequenced using BigDye Terminator v3.1 chemistry (Applied Biosystems) according to the manufacturer’s protocol. Cycle sequencing reactions were resolved on an Applied Biosystems 3100 DNA analyzer and raw sequence chromatographs from both strands were edited and aligned using CodonCode Aligner 3.0.1 (CodonCode Corporation). The resulting consensus sequence consisted of a total of 684 nucleotides corresponding to the complete Duvalius COII gene sequence (Genbank accession numbers: JX486748-JX486751).
Mitochondrial gene sequences of archival samples are generally difficult to obtain because of DNA sharing. Short amplicons can be therefore produced that cover the entire sequence of interest (Van Houdt et al. 2010). Partial COII sequences of archival specimens of Duvalius were obtained using a set of species-specific internal primers designed to amplify three short overlapping fragments (Table 2). Polymerase chain reactions were done in a 25 μl reaction volume containing 3 μl of extracted DNA, 1× Restorase buffer, 200 μM of each dNTP, 0.5 μM of each primer and 1.25 units of Restorase DNA Polymerase (Sigma-Aldrich), a blend of high quality Taq DNA polymerase and a DNA repair enzyme which has proved effective in the amplification of damaged DNA (Hajibabaei et al. 2005). PCR mix was incubated for 15 min at 37 °C and then for 5 min at 72 °C. Primers were then added to the mix prior to amplification. PCR cycles consisted of an initial denaturation step of 2 min at 94 °C, followed by 40 cycles of 30 s at 94 °C, 30 s at 50 °C and 60 s at 72 °C, with a final extension step of 5 min at 72 °C. Two independent PCR amplifications were performed for each DNA extraction. Both strands of PCR products were sequenced and assembled to generate a 504 bp partial COII sequence for each museum specimens (Genbank accession numbers: JX486736-JX486747). A partial COII sequence was also obtained for the outgroup species T. quadristriatus (Genbank accession number: JX486752).
Genetic diversity and phylogenetic analysis
Mitochondrial DNA diversity was estimated by alignment of the partial 504 bp COII sequence for both recent and museum specimens. No evidence of pseudogenes was detected. Light and heavy strands always provided a perfect match and consensus sequences were checked for insertions, deletions and stop codons that would result in a non-functional protein. Sequences were checked for deviation from neutrality using the Tajima’s D implemented in ARLEQUIN 3.5 (Excoffier and Lischer 2010). Best fit of molecular evolution model to our data was assessed using JMODELTEST (Posada 2008) under the Bayesian Information Criterion. Likelihood values were calculated for 88 models using a maximum likelihood optimization of tree topology implemented in Phyml (Guindon and Gascuel 2003). The model of sequence evolution that best fit our data was the Hasegawa, Kishino and Yano (HKY) model (Hasegawa et al. 1985), which assumes a time-reversible process, a non-uniform distribution of nucleotides and different rates for transitions and transversions.
Overall mean maximum likelihood (ML) distances among specimens were calculated using MEGA 5 (Tamura et al. 2011). Phylogenetic relationships were inferred by Bayesian inference using Metropolis-coupled Markov chain Monte Carlo implemented in MRBAYES 3.1.2 (Ronquist and Huelsenbeck 2003). Approximation of the posterior probabilities of trees was performed by two independent runs starting with default prior values, initial random trees and three heated and one cold Markov chains that ran for 106 generations sampled every 1,000 generations. Stationarity of the analysis was determined by examining the standard deviation of split frequencies between the two simultaneous runs and the potential scale reduction factor (Ronquist and Deans 2010). The first 25 % of trees were discarded as burn-in so that trees were used for analysis only after the chain became stable. The remaining trees were used to construct a 50 %-majority rule consensus tree. Consensus trees with branch lengths and posterior clade probabilities were edited using TREEVIEW (Page 1996).
Geometric morphometrics
Morphometric analyses were conducted on external characters. Inspection of male copulatory apparatus, a character used for identification of several Coleoptera species, was not considered because in the andreinii group the aedeagus morphology is not very informative and the copulatory lamella can show high levels of deformation due to weak sclerotization (Magrini 1997). Specimens were placed perpendicular to a Nikon D2XS camera with a 60 mm F 2.8 lens and paired with a mm scale bar. Images were taken of the dorsal side of the right antennae, head, pronotum and elytrae. For each antenna, we identified 43 landmarks (Fig. 2). For the head, pronotum and elytrae, landmarks were located on homologous and well identifiable anatomical structures (Bookstein 1997). We identified sliding semi-landmarks as points not located on clear homologous structure, and allowed to slide along the outline (Bookstein 1997). For the dorsal side of the head, pronotum and elytrae we identified a total of 22, 32 and 34 landmarks and semilandmarks, respectively (Fig. 2). Digital data were processed using TPSDIG 2.16 and definition of sliders was conducted using TPSUTIL 1.46.
We used MORPHOJ 1.02j (Klingenberg 2011) to remove the effect of asymmetry by creating medium symmetric dispositions with respect to the sagittal axis. Generalized Procrustes Analysis (GPA) was applied separately to the landmarks of the antenna, head, pronotum and elytrae in order to remove non-shape variation in location, scale and orientation and to superimpose the objects in a common coordinate system (Bookstein 1997). Partial warps were calculated using GPA shape residuals. Relative warps (PCs) were obtained by principal component analysis of partial warps and visualized by thin-plate spline (TPS) deformation grids, which allows for comparison of shape differences. GPA, calculation of partial and relative warps and TPS visualization were conducted using TPSRELW 1.49. We also considered centroid size for each character (Bookstein 1991) in order to evaluate the importance of overall dimensions. TPS program series can be downloaded at “http://life.bio.sunysb.edu/morph/”.
We applied a full cross validation discriminant analysis to assign each specimen to sampling sites on the basis of morphological traits. Discriminant analysis is biased when number of predictors is much higher than cases. Thus, we included only the four centroid sizes and the first 10 PCs for each character together showing a cumulative variance higher than 90 %. We did not consider populations with two samples since full cross validation analysis may give unreliable results for groups of only two individuals (Hair et al. 2009). Although full cross validation discriminant analysis is considered a conservative approach, this method is not suited to recover the overall variation because ordination along discriminant functions does not represent a large part of the shape variation among specimens (Mitteroecker and Bookstein 2011). We therefore performed a Partial Least Squares Discriminant Analysis (PLSDA) to identify specific PCs and centroid sizes that could account for differences among populations using the mixOmics R package. PLSDA components are built to find a proper compromise between describing the set of explanatory variables (PCs and centroid size) and predicting the response variables (sampling site membership). Unlike discriminant analysis, PLSDA is not affected by high numbers of variable and allows inclusion of all PCs and centroid sizes in the analysis.
Results
Mitochondrial DNA sequence diversity
A total of 14 mtDNA COII haplotypes, characterized by 24 polymorphic sites (4.76 % of sequence length) were found for 26 Duvalius specimens. The highest level of diversity was found at the third codon position (58.3 % of total variation). A relatively high transition-to-transversion (Ti/Tv) ratio was observed, a substitution pattern characteristic of mitochondrial, not nuclear genes. The average Ti/Tv ratio was 3.4, a high frequency of transitions suggesting a pattern of nucleotide substitution not yet saturated. As expected in coleopteran mtDNA, particularly for the suborder Adephaga (Simon et al. 1994; Sheffield et al. 2008), we recorded a high A + T content and a very low C and G content in the third codon position (Table 3). The pattern of amino acid substitution showed a low level of variation (3.6 %).
No mtDNA sequence diversity was found among individuals sampled in the same locality. Maximum likelihood (ML) mean distance among all specimens considered in this study was low (0.017 ± 0.005 SE). A relatively higher intraspecific mean ML distance was recorded for D. apuanus and D. minozzii, which included the highest number of subspecies analyzed. Interspecific pairwise ML mean distances were low between D. apuanus and D. jureceki and between D. minozzii and D. andreinii, while higher values were recorded for all other comparisons (Table 4). No genetic difference was found at the subspecies level between D. apuanus lanzai from cave 7 and D. a. apuanus collected in cave 1, 2 and 3, and between D. minozzii aspettatii from cave 15 and D. m. magrinii from cave 17 and 18.
Phylogenetic analyses
Stationarity of the Bayesian analysis was confirmed by two convergence diagnostic parameters, the value of the standard deviation of split frequencies between two simultaneous runs (0.0076) and the potential scale reduction factor, which approached a value of 1. The topology of the Bayesian tree revealed two well supported monophyletic lineages. The first lineage included all subspecies of D. apuanus and D. jureceki, while D. andreinii and all subspecies of D. minozzii were part of a second lineage (Fig. 3). Within the first lineage, three clades could be distinguished. The first and second clades were supported with posterior probability values of 65 and 78 % respectively, while the third one was strongly supported with 97 % posterior probability. The first clade was represented by samples of D. apuanus apuanus from the Apuan Alps and Pizzorne mountains, and one sample of D. a. lanzai from the Apennines. The second clade included samples of D. a. apuanus from the Apuan Alps, and D. a. intermedius and D. a. rasettii from the southern Apuan Alps and Lucca mountains, respectively. The third clade comprised the two subspecies of D. jureceki from the Apennines. The second lineage showed two distinct monophyletic clades. The first one included specimens of D. andreinii from the northwestern Apennines and the second clade described two D. minozzi subspecies from the Apennines and an Apennine cave close to the Emilia-Romagna plains. D. minozzii was paraphyletic with respect to D. andreinii, and D. minozzi bernii was basal to the other taxa.
Geometric morphometrics analysis
We obtained 82, 40, 60 and 64 relative warps from the analyses of the antennae, head, pronotum and elytrae, respectively. The full cross validation discriminant analysis correctly attributed 60.6 % of specimens to their sampling site against a mean probability of correct assignment by chance of 5.3 % (1/19; 21 sampling sites minus two sites with less than three specimens). In particular, specimens from localities 2, 7 and 14 were perfectly assigned to their sampling site and a further 63 specimens from 11 localities were assigned to their sampling locations with percentages ranging from 50 to 85.7 % (Table 1; Fig. 4). Percentage of correct assignment increased significantly when assignments to named subspecies (50–100 %) and species (75–92.3 %) were used as grouping variables (Table 1).
This result was supported by PLSDA. Specimens from all 21 sampling sites showed a good level of clustering when the first four components were combined. The first PLSDA component was highly correlated to the centroids of all morphological characters and to the PC3 of antennae. The second component was mostly correlated to the PC1 of the head. Both components 1 and 2 were correlated to the PC2 of elytrae and PC1 of pronotum (Fig. 5). Specimens of D. apuanus showed a high level of variability in components 1 and 2, while D. jureceki showed less variation. Specimens of D. minozzii were highly distinct by reduced size and less elongated antennae, while D. andreinii had intermediate component 1 and 2 values between D. apuanus and D. minozzii. D. apuanus e D. jureceki were characterized by larger size and by more elongated antennae. Such characteristics were particularly evident in D. apuanus lanzai (no.7 in Fig. 5), which revealed high divergence in both components 1 and 2, while D. apuanus apuanus from sites 1 and 2 had an opposite trend. Elytrae PC2 and pronotum PC1 were correlated with both component 1 and 2 and were linked to the position of the anterior discal elytral setae and elongation of pronotum. In particular, D. minozzii and D. jureceki were fully differentiated by both PLSDA components, indicating that D. jureceki had setae in a more distal position and a larger shape in the rear part of the pronotum than D. minozzii (Fig. 5). The third component of PLSDA was mainly correlated to the pronotum PC3 (Online Resource 1 Fig. S1), representing a change in width and a different form of its basal part. D. minozzii bernii (no. 19 in Online Resource 1 Fig. S1) showed a large pronotum with a narrow basal part, while specimens of D. apuanus apuanus from sites 2 and 6 had a longer and narrower pronotum with a larger basal section. Specimens of D. apuanus and D. andreinii showed a high and low level of variability, respectively. Most populations of D. jureceki, D. minozzii and D. apuanus were well differentiated by component 3. Both components 3 and 4 were correlated to the PC3 of elytrae. The PC2 (Fig. 5) and PC3 (Online Resource 1 Fig. S1) of the elytrae represented different positions of anterior discal elytral setae with respect to lateral setae and to the anterior margin of the elytrae.
Discussion
In this study, we used an integrated approach to define levels of divergence among cave-dwelling coleopterans of the genus Duvalius from central Italy and recovered a number of discrepancies between genetic and geometric morphometrics data and former taxonomic assessments. We worked mainly on museum and private natural history collections which proved to be crucial resource when studying rare and hard to collect specimens (Wandeler et al. 2007; Crandall et al. 2009). Phylogenetic analysis of mitochondrial DNA COII gene recovered two monophyletic lineages and a number of different clades. Genetic differences among clusters were weak, suggesting a relatively short divergence time. This could mirror the geological events that led to the formation of the Apuan Alps and the Tuscan-Aemilian Apennines. The Apuan Alps were formed between the Middle and Upper Miocene with karstic phenomena dating back approximately 2 My ago, while the Apennine area had an even more recent development during the Plio-Pleistocene (Piccini 2002; Bartolini 2003). Geometric morphometrics, on the other hand, recovered a high level of distinctiveness among specimens collected in different locations. Both genetic and morphometric results did not entirely corroborate the current taxonomic nomenclature but suggested the definition of evolutionary significant units as a result of rapid microevolutionary events which often occur in hypogean environments (Caccone and Sbordoni 2001).
Phylogenetic analysis defined a first, well supported monophyletic lineage including specimens of D. apuanus and D. jureceki, and a second monophyletic clade comprising samples of D. andreini and D. minozzii. Mean ML distance between D. apuanus and D. jureceki was relatively low and similar to divergence values recorded among the most closely related species of Trechinae (e.g. Faille et al. 2010). Similarities between D. apuanus and D. jureceki were defined by former taxonomists on a morphological basis (Magrini 1997) and some subspecies currently assigned to D. apuanus were originally described as subspecies of D. jureceki. However, the strong monophyly of D. jureceki suggests that this taxon should be considered as a separate species. Moreover, all known specimens of D. jureceki are characterized by the lack of the second discal elytral seta, occurring in all other Italian Duvalius. On the other hand, different clades of D. apuanus had low topology support and showed minor intraspecific genetic distances. In the second monophyletic lineage, D. minozzii was paraphyletic and shared a common ancestor with D. andreinii, which could be therefore classified as conspecific to D. minozzii. Moreover, D. minozzi bernii showed strong genetic divergence, higher than values recorded between D. apuanus and D. jureceki. Genetic distinction and peripheral geographic location of D. minozzi bernii suggested a possible assignment to a separate species. Our genetic analysis was limited to a relatively small sample size due to inherent difficulties in finding live specimens of rare and protected Duvalius taxa and to known issues associated to DNA extraction and amplification from archival samples. However, we provided evidence for low mtDNA sequence variation among caves (individuals from the same cave always showed no haplotype variation) and between caves. Although a larger sample size would be desirable, these results suggest that for some hypogean taxa even a small number of individuals may be fairly representative of the haplotypic diversity of single cave populations. Low number of samples has been used in other studies of the subfamily Trechinae due to the rarity of these hypogean species (e.g. Caccone and Sbordoni 2001; Faille et al. 2010).
Morphometric analysis showed a strong pattern of differentiation among specimens from different sampling sites and corroborated results of mtDNA analysis but in a few cases, which made morphometric data relatively similar to former taxonomy. In particular, D. apuanus showed the highest variability in morphological traits among sampling locations. Despite genetic data revealed no significant differences between D. apuanus lanzai and the other named subspecies of the same clade, this population was clearly morphologically differentiated. This result also mirrored the separation of D. apuanus lanzai on the left side of the Serchio river (Fig. 1). Samples of D. a. rasettii, D. a. intermedius, D. minozzii aspettatii and D. m. magrinii were also differentiated by morphometric analysis although no clear divergence was recovered by genetic data. Discrepancy between mtDNA and morphological data may be due to hypogean selective pressure on nuclear gene complexes affecting adaptive physical characters rather than energetic pathways. Several studies have, in fact, described low levels of mtDNA genetic divergence in morphologically distinct cave taxa (see Juan et al. 2010 and references therein). On the other hand, D. andreinii revealed intermediate morphological characteristics between D. apuanus and D. minozzii and showed the lowest morphological distinction with 25 % erroneous assignment. This pattern was supported by phylogenetic analysis and questioned the former definition of D. andreinii as a separate species. Conformity between morphometric, genetic and geographic data were also found for D. minozzi berni.

Map of sampling locations in Tuscany and Emilia-Romagna, central Italy. Numbers show locations of 20 caves and one superficial hypogean sampling site (circled number). Symbols represent four Duvalius species: D. apuanus (bullets), D. jureceki (black triangles), D. minozzii (open triangles), D. andreinii (circles). Sampling locations of Duvalius subspecies are reported in Table 1

Schematic representation of fixed landmarks (bullets) and sliding semi-landmarks (circles) used for geometric morphometric analyses of antennae (a), head (b), pronotum (c) and elytrae (d) of Duvalius



First and second components of Partial Least Squares Discriminant Analysis (PLSDA) of Duvalius morphological characters. Numbers within circles represent collection sites (see Fig. 1). Standard deviation of component values are reported for each sampling location. Double arrows indicate the positive and negative contributions of PCs and body size to PLSDA components. Variations in character shapes are shown in thinplate spline deformation grides. Antennae variation in elongation; Pronotum relative warp representing front to rear compression of the pronotum; Elytrae position of frontal setae relative to lateral setae; Head relative warp representing overall elongation of the head
Integration of genetic and morphometric analysis partially substantiated former classification of taxa of the andreinii group. Although reproductive isolation is difficult to assess in strictly vicariant populations, our results may allow for taxonomic revision in light of an unified species concept (de Queiroz 2007). Subspecific taxa may be sustained when they represent allopatric, phenotypically distinct groups with diagnosable genetic characters, some of which lacking reciprocal monophyly (Braby et al. 2012). The definition of such operational taxonomic units, representatives of intraspecific biological diversity, as an objective approach for prioritizing protection at the intraspecific level, may be of great importance to the conservation of cave organisms (Ryder 1986; Moritz 2002). Genetic evidence of evolutionary significant units is often based on either non coding DNA sequences or genes involved in general metabolic pathways that may not be subject to rapid selection under local micro-habitat pressure, so that ecological, morphological and demographic traits are also considered as crucial for biodiversity conservation (Crandall et al. 2000; Fraser and Bernatchez 2001). From this perspective, morphometrics retains an important role in the study of speciation and biodiversity and can evolve rapidly in ecosystems with rather strong selective pressures (e.g. Caccone and Sbordoni 2001 and references therein). Subterranean species of Coleoptera, particularly in Trechinae and Cholevidae, can show similar morphological and physiological modifications which can be interpreted as convergent adaptations to specific hypogean environment (Caccone and Sbordoni 2001; Faille et al. 2010). Several characters that distinguished specimens from different sampling locations, like the overall elongation of body structures (e.g. antennae), are among the main features used to diagnose different adaptations to subterranean life (Culver et al. 1990). Our study provided an example of species particularly adapted to cave life with respect to some morphological characters rather than other traits. For instance, D. jureceki had long antennae and head but short pronotum, D. minozzi presented elongated head but short antennae while D. apuanus encompassed most of the variation found for the other species. A combination of such morphological features distinguished most specimens from different sampling sites, suggesting micro-adaptation to single cave habitats and/or strong drift effect caused by isolation.
Integration of genetic and morphometric data support D. apuanus, D. jureceki and D. minozzii as distinct species, while D. andreinii may be considered as conspecific to D. minozzii. From a conservation perspective, the northwestern Apuan Alps include three genetically similar but morphologically and geographically distinct populations of D. a. apuanus (caves 1–3), which should be considered as separate units for conservation. Similarly, D. a. lanzai, although genetically similar to the D. a. apuanus populations from the northwestern Apuan Alps, represents an important unit for conservation on both morphological and geographical grounds. D. apuanus intermedius from cave 8 and D. a. rasettii represent two distinct taxa, but no clear distinction advocates assignment of the two D. a. rasettii populations northwest of the Lucca mountains (caves 9, 10) to separate units for conservation. A similar conclusion can be drawn for D. jureceki jureceki from cave 11 and the two populations of D. j. maginianus (caves 12, 13), respectively. Individuals of D. minozzii bernii from cave 19 represent a clear unit for conservation on genetic, morphological and geographical basis. Finally, the geographically and morphologically distinct population of D. andreinii from cave 20 in the northwestern Apennine and D. minozzii minozzii from cave 14 both represent separate units worth conservation efforts. On the other hand, no clear morphological and/or genetic divergence support distinct conservation of specific D. minozzii populations from caves 15 to 18.
The rarity and limited distribution of species and the very particular morphological and physiological adaptations that allow successful colonization of the hypogean environment, make cave beetles populations particularly susceptible to habitat changes (Culver 1970; Slaney and Weinstein 1997). Caves, as islands, are in fact isolated environments lacking a rescue effect (Reboleira et al. 2011). Major threats to cave ecosystems include destruction of karsts by quarrying, alteration of groundwater flow, pollution, removal of food sources, disturbance during exploratory visits and excessive sampling of specimens (Reboleira et al. 2011). Comprehensive knowledge on the distribution and level of isolation of hypogean Carabidae is therefore important for devising effective protection plans. Large areas of the northern Italian Apennines are protected by law. However, only eight out of 21 sites considered in this study are currently part of protected regional or national reserves. Populations from cave number 3, 7 and 14, assigned to D. apuanus apuanus, D. a. lanzai and D. minozzii, respectively, are all located outside of protected areas but show clear individualistic traits thus deserving conservation efforts. In Tuscany and Emilia-Romagna, protection enforcement for many insect groups, including the genus Duvalius is sometimes hindered by difficulties in providing comprehensive, updated information on sites harbouring distinct taxa or significant units for conservation. Our study provides an example on how the integration of molecular and morphometric analyses can help improving the knowledge on specific patterns of insect biodiversity and advocates the importance of devoting protection efforts to networks of cave ecosystems (Sharratt et al. 2000).
References
Allegrucci G, Caccone A, Cesaroni D, Sbordoni V (1992) Evolutionary divergence in Dolichopoda cave crickets: a comparison of single copy DNA hybridization data with allozymes and morphometric distances. J Evol Biol 5:121–148
Bartolini C (2003) When did the Northern Apennine become a mountain chain? Quat Int 101–102:75–80
Bookstein FL (1991) Morphometric tools for landmark data: geometry and biology. Cambridge University Press, New York
Bookstein FL (1997) Landmark methods for forms without landmarks: localizing group differences in outline shape. Med Image Anal 1:225–243
Braby MF, Eastwood R, Murray N (2012) The subspecies concept in butterflies: has its application in taxonomy and conservation biology outlived its usefulness? Biol J Linn Soc Lond 106:699–716
Caccone A, Sbordoni V (2001) Molecular biogeography of cave life: a study using mitochondrial DNA from Bathysciine beetles. Evolution 55:122–130
Crandall KA, Bininda-Emonds ORP, Mace GM, Wayne RK (2000) Considering evolutionary processes in conservation biology. Trends Ecol Evol 15:290–295
Crandall KA, Robison HW, Buhay JE (2009) Avoidance of extinction through nonexistence: the use of museum specimens and molecular genetics to determine the taxonomic status of an endangered freshwater crayfish. Conserv Genet 10:177–189
Culver DC (1970) Analysis of simple cave communities: niche separation and species packing. Ecology 51:949–958
Culver DC, Kane TC, Fong DW, Jones R, Taylor MA, Sauereisen SC (1990) Morphology of cave organisms—is it adaptative? Mem Biospeol 17:13–26
De Queiroz K (2007) Species concept and species delimitation. Syst Biol 56:879–886
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Faille A, Ribera I, Deharveng L, Bourdeau C, Garnery L, Quéinnec E, Deuve T (2010) A molecular phylogeny shows the single origin of the Pyrenean subterranean Trechini ground beetles (Coleoptera: Carabidae). Mol Phylogenet Evol 54:97–106
Fraser DJ, Bernatchez L (2001) Adaptive evolutionary conservation: towards a unified concept for defining conservation units. Mol Ecol 10:2741–2752
Giachino PM, Vailati D (2010) The subterranean environment. Hypogean life, concepts and collecting techniques. L’ambiente sotterraneo. Vita ipogea, concetti e tecniche di raccolta. WBA Handbooks 3, Verona
Guindon S, Gascuel O (2003) A simple, fast and accurate algorithm to estimate large phylogenies by maximum-likelihood. Syst Biol 52:696–704
Hair JF Jr, Black WC, Babin BJ, Anderson RE (2009) Multivariate data analysis. Prentice Hall, Upper Saddle River
Hajibabaei M, deWaard JR, Ivanova NV, Ratnasingham S, Dooh RT, Kirk SL, Mackie PM, Hebert PDN (2005) Critical factors for assembling a high volume of DNA barcodes. Phil Trans R Soc B 360:1959–1967
Hasegawa M, Kishino H, Yano T (1985) Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174
Hausdorf B (2011) Progress toward a general species concept. Evolution 65:923–931
Jeannel R (1928) Monographie des Trechinae. Morphologie comparée et distribution d’un groupe de Coléoptères.Troisième Livraison: les Trechini cavernicoles. L’Abeille 35:1–808
Juan C, Emerson BC (2010) Evolution underground: shedding light on the diversification of subterranean insects. J Biol 9:17
Juan C, Guzik MT, Jaume D, Cooper SJB (2010) Evolution in caves: Darwin’s “wrecks of ancient life” in the molecular era. Mol Ecol 19:3865–3880
Klingenberg CP (2011) MorphoJ: an integrated software package for geometric morphometrics. Mol Ecol Resour 11:353–357
Leys R, Watts CHS, Cooper SJB, Humphreys WF (2003) Evolution of subterranean diving beetles (Coleoptera: Dytiscidae: Hydroporini, Bidessini) in the arid zone of Australia. Evolution 57:2819–2834
Magrini P (1997) Les Duvalius et leur complexe evolutif. Espèces Françaises, Italiennes & Parasites. Troisième partie (Les espèces Italiennes). C R Lab Entomol Faune Hypogée Endogée Nice 2:202–292
Magrini P (1998) Les Duvalius et leur complexe evolutif. Atlas biogéograhique. Deuxième partie (Les espèces Italiennes). C R Lab Entomol Faune Hypogée Endogée Nice 3:55–150
Mitteroecker P, Bookstein FL (2011) Linear discrimination, ordination, and the visualization of selection gradients in modern morphometrics. Evol Biol 38:100–114
Moravec P, Uéno S-I, Belousov IA (2003) Tribe Trechini Bonelli, 1810. In: Löbl I, Smetana A (eds) Catalogue of Palaearctic Coleoptera.1. Archostemata-Myxophaga-Adephaga. Apollo Books, Stenstrup, pp 288–346
Moritz C (2002) Strategies to protect biological diversity and the evolutionary processes that sustain it. Syst Biol 51:238–254
Padial JM, Miralles A, De la Riva I, Vences M (2010) The integrative future of taxonomy. Front Zool 7:16
Page RDM (1996) TREEVIEW: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 12:357–358
Petit RJ, Excoffier L (2009) Gene flow and species delimitation. Trends Ecol Evol 24:386–393
Piccini L (2002) The Plio-Pleistocene evolution of karst in Tuscany. In: Le voragini catastrofiche. Un Nuovo problema per la Toscana. Edizioni Regione Toscana, Firenze, pp 99–109
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256
Reboleira ASPS, Borges PAV, Gonçalves F, Serrano ARM, Oromí P (2011) The subterranean fauna of a biodiversity hotspot region—Portugal: an overview and its conservation. Int J Speleol 40:23–37
Ronquist F, Deans AR (2010) Bayesian phylogenetics and its influence on insect systematics. Annu Rev Entomol 55:189–206
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Ryder OA (1986) Species conservation and systematics: the dilemma of subspecies. Trends Ecol Evol 1:9–10
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Schlick-Steiner BC, Steiner FM, Seifert B, Stauffer C, Christian E, Crozier RH (2010) Integrative taxonomy: a multisource approach to exploring biodiversity. Annu Rev Entomol 55:421–438
Sharratt NJ, Picker MD, Samways MJ (2000) The invertebrate fauna of the sandstone caves of the Cape Peninsula (South Africa): patterns of endemism and conservation priorities. Biodivers Conserv 9:107–143
Sheffield NC, Song H, Cameron SL, Whiting MF (2008) A comparative analysis of mitochondrial genomes in Coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol Biol Evol 25:2499–2509
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P (1994) Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am 87:651–701
Slaney DP, Weinstein P (1997) Conservation of cave fauna: more than just bats. Mem Natl Mus Vic 56:591–596
Sperling F, Roe A (2009) Molecular dimensions of insect taxonomy. In: Footitt RG, Adler PH (eds) Insect biodiversity: science and society. Wiley-Blackwell, Malden, pp 397–415
Stoeckle BC, Dworschak K, Gossner MM, Kuehn R (2010) Influence of arthropod sampling solutions on insect genotyping reliability. Entomol Exp Appl 135:217–223
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Van Houdt JKJ, Breman FC, Virgilio M, De Meyer M (2010) Recovering full DNA barcodes from natural history collections of Tephritid fruitflies (Tephritidae, Diptera) using mini barcodes. Mol Ecol Resour 10:459–465
Vandel A (1964) Biospéologie.La Biologie des Animaux Cavernicoles. Gauthier-Villars, Paris
Vogler AP, Monaghan MT (2007) Recent advances in DNA taxonomy. J Zool Syst Evol Res 45:1–10
Wandeler P, Hoeck PEA, Keller LF (2007) Back to the future: museum specimens in population genetics. Trends Ecol Evol 22:634–642
Zimmermann J, Hajibabaei M, Blackburn DC, Hanken J, Cantin E, Posfai J, Evans TC (2008) DNA damage in preserved specimens and tissue samples: a molecular assessment. Front Zool 5:18
Acknowledgments
We thank Marco Bastianini, Giovanni Bertagni, Corrado Busi, Augusto Degiovanni, Andrea Petrioli and Stefano Taiti for granting access to private Duvalius collections and help in collecting wild specimens and Chiara Natali for her assistance during laboratory work. Leonardo Piccini provided information on the geological history of the study area. We also thank Saulo Bambi for taking images used for morphometric analyses and Saverio Vicario for constructive comments on phylogenetic analyses. We also thank two anonymous referees for their constructive suggestions.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Zinetti, F., Dapporto, L., Vanni, S. et al. Application of molecular genetics and geometric morphometrics to taxonomy and conservation of cave beetles in central Italy. J Insect Conserv 17, 921–932 (2013). https://doi.org/10.1007/s10841-013-9573-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10841-013-9573-9