
Abstract
Trophectoderm biopsy is increasingly performed for pre-implantation genetic testing of aneuploidies and considered a safe procedure on short-term clinical outcome, without strong assessment of long-term consequences. Poor biological information on human trophectoderm is available due to ethical restrictions. Therefore, most studies have been conducted in vitro (choriocarcinoma cell lines, embryonic and pluripotent stem cells) and on murine models that nevertheless poorly reflect the human counterpart. Polarization, compaction, and blastomere differentiation (e.g., the basis to ascertain trophectoderm origin) are poorly known in humans. In addition, the trophectoderm function is poorly known from a biological point of view, although a panoply of questionable and controversial microarray studies suggest that important genes overexpressed in trophectoderm are involved in pluripotency, metabolism, cell cycle, endocrine function, and implantation. The intercellular communication system between the trophectoderm cells and the inner cell mass, modulated by cell junctions and filopodia in the murine model, is obscure in humans. For the purpose of this paper, data mainly on primary cells from human and murine embryos has been reviewed. This review suggests that the trophectoderm origin and functions have been insufficiently ascertained in humans so far. Therefore, trophectoderm biopsy should be considered an experimental procedure to be undertaken only under approved rigorous experimental protocols in academic contexts.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The trophectoderm (TE) is the external cell mass of the blastocyst that develops into the placenta and the other extraembryonic membranes. For implantation to occur, the embryo needs to acquire a well-differentiated functional TE, whose aim is to interact with the endometrium, start implantation, and provide a proper placental development. Due to ethical restrictions, research on the human embryo is scant and conducted on very few embryos [1,2,3] sometimes obtained from in vitro matured oocytes [4, 5] which do not necessarily reflect the physiological counterpart. Studies on TE cells have been preferentially undertaken on animal embryos and in vitro models (choriocarcinoma cell lines, embryonic and pluripotent stem cells) as they are more available and morphologically similar to the human counterpart. Nevertheless, these models display marked differences in gene expression, epigenetic changes, chromatin remodeling, transcriptional activity, genome activation, timing of polarization and compaction, blastocyst formation, implantation, and placental development compared with humans [6,7,8,9].
TE biopsy coupled with pre-implantation genetic testing for aneuploidy (PGT-A) is increasingly used to select euploid blastocysts and is intended to increase in vitro fertilization (IVF) success rates. The rationale for moving from blastomere to TE biopsy is based on evidence that the blastomere biopsy is detrimental to the embryo [10]. However, the safety of TE biopsy is still controversial and its long-term sequelae are presently unknown [11, 12]. Furthermore, PGT-A has failed to demonstrate any clear advantage over the sequential transfer of untested embryos [11, 13], and its clinical value is controversial based on a number of unanswered questions including the real association of embryo aneuploidy with IVF failure and the reliability of a small number of focally biopsied TE cells in reflecting the genetic/chromosomal status of the inner cell mass (ICM) [rev. in [14]. Manipulation of a delicate cell layer like the human TE should be based on sufficient knowledge of its embryological origin and function due to the possible serious impact of early embryo manipulation on the subsequent embryo-fetal development. This review aims to demonstrate that such knowledge is not available to date because biological data on human embryo compaction, polarization, TE function, cell junctions, intercellular communication, and the biological sequelae of TE biopsy is poorly ascertained from a biological perspective.
Polarization
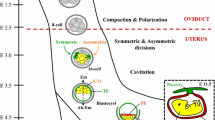
In the murine model, polarization occurs in committed blastomeres within 1–3 h from compaction [15]. At the 8-cell stage, a polar blastomere side with dense microvilli and few cell junctions is detected opposite a basolateral cell side with fewer microvilli and rich in cell junctions [16]. Upon asymmetric blastomere division, the polar side is inherited by only one of the cell couplet and two different daughter cells are generated which occupy either the outer or the inner part of the embryo [17]. Together with surface polarization, cytoplasmic polarization also occurs, characterized by the spatial reorganization of the actin cytoskeleton, endocytic organelles, and the nucleus leading to a preferential endocytic activity localized at the apical rather than the basolateral cell side [17]. Polarization thus relies on the action of the cytoskeleton [18] and it is strictly dependent on Ca2+ and cell-cell contacts [15, 18,19,20]. At the 8-cell stage, an apical domain is established at the polar side of blastomeres playing a pivotal role in TE differentiation [rev. in 21]. Briefly, the apical domain is mainly composed of the actin-binding protein Ezrin and the complex between partitioning defective proteins Par3-Par6 and atypical protein kinase C (aPKC) [22], a common mechanism required for apico-basal cell polarity in different epithelial cell types [23]. The apical domain becomes progressively localized at the polar side of blastomeres while the adhesion molecule E-cadherin, concentrated in the adherens junctions (zonula aderentes), localizes at the basolateral cell side together with Par-1, junction adhesion molecules (JAM), and Na+/K+ ATPase. Starting at compaction, E-cadherin and catenins relocate to the contact cell points at the basolateral cell side [24]. Post-translational events activate both E-cadherin, which binds homotopically on the external cell surface, and catenins which bind E-cadherin to the actin cytoskeleton [25]. The presence or absence of an apical domain dictates the diverging fate of blastomeres leading to either TE or ICM differentiation [21]. These programs are mainly regulated by the Hippo/Yes-associated protein (YAP) pathway in synergy with Notch signaling [22, 26,27,28] and angiomotin (Amot) [29]. AMOT is a scaffold protein whose main function is to tether the Hippo/YAP signaling. In polar cells, AMOT is sequestered at the apical domain. This interferes with the activation of the Hippo/YAP signaling cascade and YAP phosphorylation. The unphosphorylated YAP is thus free to migrate into the nucleus and activate the TE cells transcription program mainly mediated by Cdx2 [29]. Polarized distribution of mitochondria and regulatory proteins is also observed in murine blastomeres [30, 31].
In humans, the timing of polarization is controversial. Some have found signs of polarization at the 2-cell stage before genome activation [32] while others have observed no polarization until the 8-cell stage, similarly to the murine model [33, 34]. The blastomeres at the 2-cell stage display signs of both surface and cytoplasmic polarization. An apical region free of cell junctions, rich in microvilli, and displaying active endocytotic membrane transport has been observed, together with a basolateral region with abundant cell junctions, loss of microvilli, and no sign of active endocytotic membrane transport [32]. No study has been found on the apical domain and Par proteins in human embryos. Conversely, E-cadherin and JAM have been described in human compacted blastomeres [35,36,37,38]. In normally compacting embryos, E-cadherin relocates from the blastomere cytoplasm to the cell-cell contact points leading to adherens junction assembly. Conversely, in abnormally compacted fragmented embryos, relocation fails to occur, E-cadherin is described as “erratic” or “absent,” and polarity fails to be stabilized [35]. To the best of our knowledge, YAP and AMOT have not been described in human blastomeres. At the blastocyst stage, it has been found that, differently from the murine model in which the transcription coactivator YAP is excluded from the nuclei of ICM cells [39], in human embryos YAP is localized in the nuclei of both ICM (“diffuse pattern”) and TE (“more heterogeneous pattern”). YAP is considered important for the maintenance of pluripotency in human TE [40], differently from the murine model where it induces TE differentiation [39]. In the human embryo, AMOTL2 is localized in the TE lateral adherens junctions; its knockout does not affect differentiation while blocking blastocyst hatching from the zona pellucida [41]. Similarly to the murine model, a polarized distribution of mitochondria and regulatory proteins has been observed in human blastomeres [31, 42, 43].
Compaction
In mice, compaction starts at the 8-cell stage and relies on two diverging models. The first is based on the function of filopodia, E-cadherin-rich plasma membrane protrusions of 11 μm long also including α- and β-catenins (binding E-cadherin to the cytoskeleton), F-actin, and myosin-X [44]. The protrusions can be observed as from the 8-cell stage and disappear at the 16–32-cell stage. The blastomeres extend filopodia cyclically and retract them from the neighboring cells only before dividing. Slightly fewer than 60% of cells form filopodia; of these, about 70% divide symmetrically, while the others divide asymmetrically. Upon elongation of filopodia, the blastomeres acquire a flattened shape, while they revert back to a round shape upon filopodia retraction or laser ablation. E-cadherin, catenins, or myosin knockout decreases the number of filopodia without abolishing them, suggesting that compensatory mechanisms may intervene. On the contrary, the ablation of F-actin completely inhibits filopodia formation and compaction [44]. According to the second model for compaction, E-cadherin clears actomyosin from the blastomere contact points. This decreases the contractility forces at the cell-cell contact points, thus facilitating the cell-autonomous contraction of the contractile shell at the cell-medium interface [45]. E-cadherin knockout mice fail compaction, TE formation, pre-implantation development, and implantation [46, 47].
In humans, the knowledge of compaction is mostly based on a panoply of observational and time-lapse studies [48,49,50,51,52,53,54,55,56,57,58,59,60,61,62] while molecular mechanisms remain obscure. Compaction initiates traditionally at the 10–18-cell stage [63], but more recent time-lapse studies have shown an earlier start at or just after the 8-cell stage similarly to the murine model [64]. Most embryos initiating compaction earlier than the 8-cell stage develop into poor quality blastocysts with high incidence of cytokinetic failure and multinucleated blastomeres [53]. At compaction, human blastomeres display a flattened shape, with reduced clefts between each other. At the morula stage, most embryos are compacted although not homogenously: 75% blastomeres are compacted and display an increased density of microvilli over the free surface, while uncompacted blastomeres display a lower microvilli density. In the contact areas of partially disaggregated blastomeres, small abundant vesicles of unknown significance are also observed [33]. Single blastomeres isolated from embryos at the 4-cell stage undergo normal compaction and cavitation and differentiate into both the ICM and the TE generating small blastocysts [65]. In human embryos, filopodia have not been described. However, long “microvilli” extending over adjacent blastomeres have been observed in baboon embryos by scanning electron microscopy [66].
The establishment of cell junctions
In the murine model, adherens junctions mature in blastomeres at the compaction stage, playing a pivotal role in the polarization and differentiation processes. They are maintained equally in both the TE and the ICM [67]. Once the TE and ICM cell fates have diverted, the embryo starts to be filled by fluid. This process requires the establishment of tight junctions and desmosomes which are critical for the differentiation of the TE lineage and the formation of the blastocyst [68,69,70]. Mature tight junctions are observed at the compaction stage and maintained later only in the TE cell layer just apically to the adherens junctions. Tight junctions mature by progressive synthesis and assembly of constitutive proteins, including the transmembrane proteins occludin, claudins, and JAM and the cytoplasmic proteins ZO-1, ZO-2, and ZO-3, together with cingulin (which contributes to binding the tight junctions to the cytoskeleton) and Rab13 (involved in intracellular transport processes). The maturation process of tight junctions involves E-cadherin adhesion whose inhibition however does not affect cell adhesion or polarization probably by residual maternal transcripts [46, 69, 70]. Once tight junctions are matured, paracellular permeability is blocked and the morula progresses to blastocyst by fluid accumulation and blastocoel formation [71]. Actin rings stabilize adherens and tight junction components [72], while increased hydraulic pressure leads to the maturation of functional tight junctions mediated by vinculin, a membrane protein involved in the linkage of integrin adhesion molecules to the actin cytoskeleton [73]. Later on, at the 32-cell stage, desmosome (macula aderentes) formation starts contributing to the maintenance of the epithelium integrity during blastocyst expansion. Similarly to the tight junctions, the desmosomes are localized only in the TE cell layer and are composed of plakoglobin, desmoplakin, and the membrane glycoprotein desmocollin 2 (DSC2) [rev. in 67]. Gap junction (nexus) assembly initiates at the 8-cell stage in mouse embryos and involves post-translationally regulated proteins. Gap junctions are composed of connexins, a family of integral membrane proteins classified according to their molecular mass [74]. The role of gap junctions in the normal development of mouse embryos is controversial [75,76,77,78]. However, their roles in cell communication mediated by the passage of low-molecular-weight molecules or ions and in post-implantation events are established [75, 79].
In the human embryo, the assembly of cell junctions starts at the 2-cell stage, i.e., before the genome activation which is apparently not required for the formation of adherens and gap junctions, while it is essential for tight junction assembly and embryo viability [32]. In cleavage-stage human embryos, E-cadherin is distributed in blastomere cytoplasm; later on, it relocates to the cell-cell contact points in normally compacting embryos, while in abnormally compacted fragmented embryos, relocation fails to occur and E-cadherin is described as “erratic” or “absent.” At the blastocyst stage, normal TE cells are surrounded by a strong band of cadherin fluorescence (“belt” pattern) which is not observed in disorganized TE [35]. Mature tight junctions are detected first in the cavitating human morula and located apically in TE cells [80]. Coxsackievirus and adenovirus receptor (CAR), a member of the junctional adhesion molecule family of adhesion receptor, is necessary for the adherens and tight junction assembly/biogenesis during pre-implantation development being therefore critical to blastocyst formation. In humans, CAR has been found in all stages of pre-implantation development, located in the nucleus in earlier stages, then co-localized with tight junction proteins (occludin and ZO-1) starting from the compaction stage, finally relocated back from the membrane to nucleus in hatching blastocysts [81]. Gap junctions have been first detected in apposing cell membranes at the 4-cell stage and found increasingly organized and functional as development proceeds [82]. Gap junctions are not involved in intercellular communication until the morula stage, while in blastocyst, they contribute to the communication between TE and ICM cells where they are localized with different patterns [83, 84]. In human blastocyst, connexins 31 and 43 are the main co-expressed isoforms in early and late pre-implantation stages of development, while connexin 45 is less translated until the 4-cell stage and connexins 32 and 26 are observed occasionally in the TE of late blastocysts [84]. Connexin 40, which is expressed by the extravillous trophoblast in the early human placenta, was not found to be expressed in TE from which placental trophoblast develops [85]. Interestingly, expression levels of gap junction proteins are variable in morphologically normal embryos which often show either as extensive, disorganized overexpression or as reduced expression suggesting that a normal embryo morphology may not be a reliable indicator of future viability [84]. Desmosome-like structures have been observed in human embryos at the 2-cell stage of development [86]. Controversially, others found desmosome-like structures starting from the 6-cell stage [34], while desmosome mRNA and protein have been detected only at the blastocyst stage [36, 83]. Together with tight junctions, TE desmosomes contribute to intercellular sealing and tissue integrity, critical for vectorial transport and blastocoel cavity formation. In the implantation process, endometrial epithelial cells share desmosomes with TE cells before displacing laterally to allow TE cells to form a penetration cone [87]. Desmosomes fail to anchor to TE cells in abortive human blastocysts unable to hatch [88].
Failure to form proper cell junctions negatively impacts embryo viability without any detectable morphological compromise [38, 89]. Also, in vitro culture and abnormal metabolic activity have been found to impair the assembly of cell junctions and embryo development without any evident morphological compromise [37, 38, 90]. Human embryos cultured in vitro show a relatively inefficient membrane assembly of junction proteins compared with the murine model [38, 90].
TE differentiation and functions in the human embryo
Similarly to the murine model, also in humans, a complex network of transcription factors intervenes in ICM and TE lineage specification, including OCT-4 and NANOG1 (markers of ICM) and CDX2 and GATA3 (markers of TE); however, significant differences in human have been found compared with mice [2, 3, 91, 92]. Most information on TE lineage specification derives from a human embryo in vitro culture [93,94,95,96]. In humans, no stemness marker specifically identifies cells allocated to ICM or TE [3]. The nuclear expression of NANOG (some not all cells) and SOX2 appears restricted to the ICM only after lineage segregation which occurs at the late blastocyst stage [4]. In human blastocysts, OCT-4 and its stemness isoform are expressed in both the TE and the ICM [1, 2, 5, 97,98,99,100] in accordance with studies on animal models [101,102,103]. However, unlike in primates, in which NANOG expression precedes that of OCT-4 [104], OCT-4 has been found to co-localize with NANOG in human blastocysts [105]. Interestingly, in humans, disaggregated TE cells from pre-hatched blastocysts are able to express NANOG upon re-positioning in the center of the embryo becoming unable to revert back to TE [100]. This is in accordance with studies on the murine model, in which TE cells are able to relocate to the inner position and acquire ICM characteristics by downregulating CDX2 expression finally contributing to the ICM [106]. Human TE cells also regain Cyclin E1 and NANOG under embryonic stem cell culture conditions [107]. Interestingly, CAR has been found to express in both TE and ICM, thus suggesting a role in maintaining pluripotency in TE cells [81].
All the above data, although limited, suggest that in pre-hatched blastocysts of humans and other species, TE cell developmental direction may not yet be definite while it commits to differentiation only in hatched blastocysts concomitant with the expression of CDX2 [106, 108]. Also, TE cells retain stemness genes common to embryonic stem cells and ICM. Available data also indicates that the animal model poorly reflects TE lineage specification.
The study of human TE functions has been attempted by transcriptome analysis based on low numbers of immunosurgically or mechanically dissected embryos. The main studies are summarized in Table 1 [1,2,3,4,5] and suggest that (1) a complex and still unclear network of transcription factors intervenes in polarization and TE lineage specification; (2) many genes are overexpressed in TE cells compared with day 3 blastomeres; (3) stemness genes are expressed in both ICM and TE, but none is able to identify the totipotent cells during human pre-implantation development; (4) TE cells overexpress many genes linked to protein biosynthesis, regulation of cell growth, and differentiation including apoptosis, DNA damage repair, DNA methylation, cytoskeleton, cell junction biogenesis, blastocoel formation, placental function, and trophoblast invasion; (5) TE cells show overexpression of genes linked to glycolysis, sterol biosynthesis, and androgen, lipid, and estrogen metabolism suggesting important metabolic and endocrine functions; (6) the number of genes that are expressed exclusively in immunosurgically isolated ICM or TE is rather low; and (7) many TE-specific genes have no known function. Other studies show differences between the murine model and humans; in particular, transcription of FGFRs is rarely observed in humans, and FGFR2 protein is not detected in human blastocysts, while being strongly expressed in mice blastocysts [109].
Interestingly, in murine blastocysts derived by intracytoplasmic sperm injection (ICSI) versus IVF, the expression of genes was found to differ including those regulating structural and organ-specific processes, several metabolic pathways, and signaling and transport mechanisms [110, 111]. In addition, several studies have shown that genes involved in the implantation process are overexpressed in human TE allowing for distinction among viable implanting blastocysts and those which fail to implant [112,113,114,115]. TE biopsy implicates an extended embryo culture to blastocyst without any clear clinical advantage over day 3 culture [116]. However, it has been shown that human embryos are highly sensitive to the environment under in vitro culture conditions which can alter the gene expression pattern [117,118,119]. Interestingly, it has been shown that cryopreservation, quite mandatory in PGT-A due to the time necessary to get the results of comprehensive chromosome screening, affects the normal pattern of gene expression in human pre-implantation embryos [120, 121]. Importantly, in the murine model, TE produces approximately 80% of the energy generated in the embryo which is mainly consumed by the Na(+), K(+)ATPase and is responsible for 90% of amino acid turnover compared with the ICM [122].
Embryonic intercellular communication
In the murine model, long filopodia transverse the blastocoel and maintain the communication between mural abembryonic TE cells and ICM, probably by the exchange of free molecules [123]. Also, TE cells connect to each other by thin filopodia [124]. In addition, filopodia traversing the blastocoelic cavity display receptors for growth factors including the fibroblast growth factor (FGFR). FGFR2 is known to be localized on mural TE, and its ligand (FGF4) is released by ICM to maintain TE cell mitotic activity [125]. The establishment of a communication system between ICM and TE was supposed long time ago in which ICM induces a high rate of proliferation in the polar TE which migrates to the proximal and distal mural regions [126,127,128]. Also, TE processes modulate the totipotency of ICM cells [129].
In humans, very poor information about the communication system was found. TE cells are linked by dense arrays of gap junctions between them, while ICM cells are linked by small, punctate gap junctions, as stated above [82, 83]. In human embryos, filopodia have not been described.
TE biopsy: biological effects
In both human and animal models, the biological effects of TE biopsy are limited to the observation of simplistic parameters like blastocoel re-expansion, hatching from the zona pellucida, and embryo survival which are apparently reassuring [130,131,132,133,134,135,136,137,138]. However, these parameters may not be fully informative regarding the biological status of the embryo, as is the case of “assisted” hatching which does not necessarily reflect the capacity of the embryo to hatch spontaneously if left untouched and can artificially rescue embryos otherwise not developing [80, 139]. Several studies of both the animal and human models have suggested impairment in developmental capacity and a lower percentage of live birth rates after TE biopsy [11, 12, 128, 132, 138, 140,141,142]. Importantly, in both the primate and the human models, increasing the number of TE cells biopsied from both high and low morphological grade blastocysts impacts negatively on proteins involved in implantation like hCG and VEGF and decreases both the implantation potential and the live birth rate [143,144,145,146,147,148]. The only randomized study specifically designed to ascertain the biological safety of TE biopsy in humans has been performed by removing only up to five cells [10], a number with no demonstrable impact on implantation potential and live birth rate [146] but nevertheless considered insufficient to determine embryo ploidy [149]. Therefore, the study setting is not applicable to routine cycles where the exact number of cells removed is technically difficult to ascertain due to the uncertain visualization of the cell nuclei and therefore may well exceed five (Fig. 1).

Schematic description of some unanswered questions on both the safety of the TE biopsy and the clinical value of PGT-A. (a) Assisted hatching: potential rescue of abnormal embryos. (b) Uncertain number of biopsied TE cells. (c) Higher number of biopsied TE cell: impaired HCG and VEGF secretion and increased risk of implantation failure. (d) Tight junction disruption and developmental compromise. (e) Focally biopsied TE cells do not necessarily reflect the ploidy of the ICM. (f) The developmental fate of TE cells is uncertain at the time of biopsy. (g) Impaired ICM cell load following TE cell extraction. (h) Cell communication system disruption. ICM, inner cell mass; TE, trophectoderm; ZP, zona pellucida; HCG, human chorionic gonadotropin; VEGF, vascular endothelial growth factor; AH, assisted hatching; TJ, tight junction; PGT-A, pre-implantation genetic testing for aneuploidy
Discussion
Polarization, compaction, and TE differentiation are poorly known in humans. Timing of polarization is controversial and the knowledge of surface and cytoplasmic polarity is limited to observational studies while the molecular mechanisms of polarization are presently unknown. Components of the Hippo/YAP signaling cascade have been sporadically studied in human embryos, demonstrating localization and functions differently from the murine model. A panoply of observational and time-lapse studies have described the shape changes in blastomeres at compaction and attempted inconclusively a correlation with IVF outcome, while the molecular mechanisms of compaction remain to be elucidated in humans. The origin of mechanical forces leading to the flattening of blastomeres is presently unknown in humans compared with the murine model, where convincing models have been proposed suggesting that sufficient tensile forces by either filopodia or the contractile shell at the cell-medium interface are sufficient to modify the cell shape. However, filopodia have not been detected in the human embryo, although similar structures have been found in non-human primates.
In humans, subtle modifications and assembly of cell junction proteins may cause severe impairment of embryo development mostly linked to metabolic factors, without any morphological detectable compromise. The disruption of the “belt” pattern of TE cadherin distribution is associated with a disorganized TE, with reasonable consequences on embryo viability and implantation. Tight junction assembly and maintenance in the TE cell layer impact paracellular sealing and blastocoel formation. Gap junctions are localized in both TE and ICM, and although their role in pre-implantation embryo development is presently uncertain, they take part in the TE and ICM communication system and play a pivotal role in post-implantation development. Together with tight junctions, TE desmosomes contribute to intercellular sealing and tissue integrity, critical for vectorial transport and blastocoel cavity formation. As well as gap junctions, desmosomes play a pivotal role in the implantation process. Surprisingly, we were unable to find data on more subtle consequences of TE biopsy on cell junctions in the human embryo which, similarly to the epithelial models, would not be revealed by simplistic morphological observations.
Complex data on lineage specification and TE cell differentiation have been reported in detail elsewhere. For the purpose of this review, I focalized on the stem cell nature of TE at the time of blastocyst biopsy. When TE is biopsied, its developmental fate is not yet definite. Reversal of TE destiny is easily obtainable both in vivo and in vitro and TE can acquire ICM characteristics. Also, in humans, TE and ICM are not strictly distinguishable on the basis of their molecular pattern. TE cells maintain a stem cell state, whose significance is presently unknown but certainly not negligible. It cannot be excluded that TE is involved in the maintenance of ICM cell load. Therefore, the subtraction of an even limited number of TE cells by biopsy may bear consequences of unknown significance.
The exploration of human TE functions has been attempted by microarray technology on very few dissected embryos, generating a panoply of studies aimed to define a “TE signature.” Although many genes overexpressed in TE have no known function, TE cells seem to possess multiple important functions, including metabolic and endocrine functions, common cell cycle mechanisms, cell junction biogenesis, blastocoel formation, placental function, and trophoblast invasion. The main functions of human TE are metabolic, endocrine and involved in the implantation mechanism. It may be speculated that the disruption of the TE cell layer by biopsy may perturb the physiological TE functions by modulation of the implicated genes. Interestingly, in murine blastocysts derived by ICSI versus IVF, the expression of many genes differ including those regulating structural and organ-specific processes, several metabolic pathways, and signaling and transport mechanisms. It can be speculated that, in a context of no clear clinical advantage of PGT-A compared with standard IVF cycles, the use of ICSI in PGT-A may perturb the embryo functions by altering gene expression. Also, TE biopsy implicates an extended embryo culture and cryopreservation, mandatory in preferred PGT-A protocols due to the time necessary to get the results of comprehensive chromosome screening, which may affect the normal pattern of gene expression in pre-implantation embryos.
Intercellular communication is virtually unknown in the human embryo. In the murine model, however, an active communication system has been ascertained between TE and ICM cells, mediated by cell junctions and filopodia. Importantly, ICM seems to maintain the TE cell load by active release of growth factors and polar TE cell migration to proximal and mural regions. It may be postulated that the subtraction of mural TE cells by biopsy may induce a greater ICM activity which can finally result in depauperation of the ICM cell load. Conversely, TE cells modulate the pluripotency of ICM cells, which may also be changed by TE biopsy. I was unable to find data on the sequelae of the disruption of the cell junction communication system in humans. Importantly, in the murine model, in accordance with the strong metabolic activity of TE, 80% of the embryo energy is generated by the TE and destined to the formation of blastocoel. Again, it may be inferred that the decrease of the TE cells caused by the biopsy, although limited, may have a negative impact on the overall embryonic energy burden and subsequent events including implantation. To the best of our knowledge, in humans, none of these topics has been addressed so far.
PGT-A is currently performed by TE biopsy as a routine procedure in many private IVF centers, despite the fact that its efficacy has not been demonstrated [11, 13]. The reason for this has to be seen from several points of view, including an economical one. Data on the short-term safety of TE biopsies seems reassuring; nevertheless, no strong evidence has been provided on the long-term consequences [150]. In vitro studies on the impact of TE biopsy and PGT-A on embryo viability are observational, sometimes using simplistic uninformative “safety” parameters like hatching from the zona pellucida and blastocyst short-term morphological recovery. However, PGT-A clinical outcome (biochemical/clinical loss rate, delivery rates, etc.) is the object of misinterpretation due to a number of factors including embryo selection biases independent from ploidy.
The safety of TE biopsy needs a more careful evaluation which may take advantage from the basic knowledge acquired from the animal models and the human embryo long-term culture systems. These models, although different from the human model, may nevertheless offer a study system useful to address some unanswered questions still open. Possible alternatives to invasive diagnosis of the human embryo are currently under study and may prove effective in selecting euploid embryos. For example, the analysis of cell-free DNA in spent culture media sounds encouraging [151, 152].
In the light of the data provided by this review, which demonstrates insufficient knowledge of the origin and function of TE, no attempt should be made to disrupt this delicate and unknown cell layer, outside strictly approved experimental protocols in academic contexts.
Data availability
Not applicable
References
Adjaye J, Huntriss J, Herwig R, BenKahla A, Brink TC, Wierling C, et al. Primary differentiation in the human blastocyst: comparative molecular portraits of inner cell mass and trophectoderm cells. Stem Cells. 2005;23:1514–25.
Assou S, Boumela I, Haouzi D, Monzo C, Dechaud H, Kadoch IJ, et al. Transcriptome analysis during human trophectoderm specification suggests new roles of metabolic and epigenetic genes. PLoS One. 2012;7:e39306.
Bai Q, Assou S, Haouzi D, Ramirez JM, Monzo C, Becker F, et al. Dissecting the first transcriptional divergence during human embryonic development. Stem Cell Rev Rep. 2012;8:150–62.
Cauffman G, De Rycke M, Sermon K, et al. Markers that define stemness in ESC are unable to identify the totipotent cells in human preimplantation embryos. Hum Reprod. 2009;24:63–70.
Cauffman G, Van de Velde H, Liebaers I, et al. Oct-4 mRNA and protein expression during human preimplantation development. Mol Hum Reprod. 2005;11:173–81.
Niakan KK, Han J, Pedersen RA, Simon C, Pera RAR. Human pre-implantation embryo development. Development. 2012;139:829–41.
Niakan KK, Eggan K. Analysis of human embryos from zygote to blastocyst reveals distinct gene expression patterns relative to the mouse. Dev Biol. 2013;375:54–64.
Fogarty NME, McCarthy A, Snijders KE, Powell BE, Kubikova N, Blakeley P, et al. Genome editing reveals a role for OCT4 in human embryogenesis. Nature. 2017;550:67–73.
Rossant J, Tam PPL. New insights into early human development: lessons for stem cell derivation and differentiation. Cell Stem Cell. 2017;20:18–28.
Scott RT Jr, Upham KM, Forman EJ, Zhao T, Treff NR. Cleavage-stage biopsy significantly impairs human embryonic implantation potential while blastocyst biopsy does not: a randomized and paired clinical trial. Fertil Steril. 2013;100:624–30.
Munné S, Kaplan B, Frattarelli JL, Child T, Nakhuda G, Shamma FN, et al. Preimplantation genetic testing for aneuploidy versus morphology as selection criteria for single frozen-thawed embryo transfer in good-prognosis patients: a multicenter randomized clinical trial. Fertil Steril. 2019;112:1071–9.
Pagliardini L, Viganò P, Alteri A, Corti L, Somigliana E, Papaleo E. Shooting STAR: reinterpreting the data from the ‘Single Embryo TrAnsfeR of Euploid Embryo’ randomized clinical trial. Reprod BioMed Online. 2020;40(4):475–8.
Dahdouh EM, Balayla J, García-Velasco JA. Impact of blastocyst biopsy and comprehensive chromosome screening technology on preimplantation genetic screening: a systematic review of randomized controlled trials. Reprod BioMed Online. 2015;30:281–9.
Gleicher N, Orvieto R. Is the hypothesis of preimplantation genetic screening (PGS) still supportable? A review. J Ovarian Res. 2017;10:21.
Humięcka M, Szpila M, Kłoś P, Maleszewski M, Szczepańska K. Mouse blastomeres acquire ability to divide asymmetrically before compaction. PLoS One. 2017;12:e0175032.
Ziomek CA, Johnson MH. Cell surface interaction induces polarization of mouse 8-cell blastomeres at compaction. Cell. 1980;21:935–42.
Fleming TP, Pickering SJ. Maturation and polarisation of the endocytotic system in outside blastomeres during mouse preimplantation development. J Embryol Exp Morphol. 1985;89:175–208.
Johnson MH, Maro B. A dissection of the mechanisms generating and stabilizing polarity in mouse 8- and 16-cell blastomeres: the role of cytoskeletal elements. J Embryol Exp Morphol. 1985;90:311–34.
Shirayoshi Y, Okada TS, Takeichi M. The calcium-dependent cell-cell adhesion system regulates inner cell mass formation and cell surface polarization in early mouse development. Cell. 1983;35:631–8.
Kimber SJ, Surani MA, Barton SC. Interactions of blastomeres suggest changes in cell surface adhesiveness during the formation of inner cell mass and trophectoderm in the preimplantation mouse embryo. J Embryol Exp Morphol. 1982;70:133–52.
Zhu M, Zernicka-Goetz M. Building an apical domain in the early mouse embryo: lessons, challenges and perspectives. Curr Opin Cell Biol. 2020;62:144–9.
Hirate Y, Hirahara S, Inoue K, Kiyonari H, Niwa H, Sasaki H. Par-aPKC-dependent and -independent mechanisms cooperatively control cell polarity, Hippo signaling, and cell positioning in 16-cell stage mouse embryos. Develop Growth Differ. 2015;57:544–56.
Doe CQ. Cell polarity: the PARty expands. Nat Cell Biol. 2001;3:E7–9.
Pey R, Vial C, Schatten G, Hafner M. Increase in intracellular Ca2+ and relocation of E-cadherin during experimental decompaction of mouse embryos. Proc Natl Acad Sci U S A. 1998;95:12977–82.
Aberle H, Schwartz H, Kemler R. Cadherin-catenin complex: protein interactions and their implications for cadherin function. J Cell Biochem. 1996;61:514–23.
Rayon T, Menchero S, Nieto A, Xenopoulos P, Crespo M, Cockburn K, et al. Notch and hippo converge on Cdx2 to specify the trophectoderm lineage in the mouse blastocyst. Dev Cell. 2014;30:410–22.
Watanabe Y, Miyasaka KY, Kubo A, Kida YS, Nakagawa O, Hirate Y, et al. Notch and Hippo signaling converge on Strawberry Notch 1 (Sbno1) to synergistically activate Cdx2 during specification of the trophectoderm. Sci Rep. 2017;7:46135.
Anani S, Bhat S, Honma-Yamanaka N, Krawchuk D, Yamanaka Y. Initiation of Hippo signaling is linked to polarity rather than to cell position in the pre-implantation mouse embryo. Development. 2014;141:2813–24.
Leung CY, Zernicka-Goetz M. Angiomotin prevents pluripotent lineage differentiation in mouse embryos via Hippo pathway-dependent and –independent mechanisms. Nat Commun. 2013;4:2251.
Kameyama Y, Ohnishi H, Shimoi G, Hashizume R, Ito M, Smith LC. Asymmetrical allocation of mitochondrial DNA to blastomeres during the first two cleavages in mouse embryos. Reprod Fertil Dev. 2010;22:1247–53.
Antczak M, Van Blerkom J. Oocyte influences on early development: the regulatory proteins leptin and STAT3 are polarized in mouse and human oocytes and differentially distributed within the cells of the preimplantation stage embryo. Mol Hum Reprod. 1997;3:1067–86.
Tesarík J. Involvement of oocyte-coded message in cell differentiation control of early human embryos. Development. 1989;105:317–22.
Nikas G, Ao A, Winston RM, et al. Compaction and surface polarity in the human embryo in vitro. Biol Reprod. 1996;55:32–7.
Dale B, Tosti E, Iaccarino M. Is the plasma membrane of the human oocyte reorganised following fertilisation and early cleavage? Zygote. 1995;3:31–6.
Alikani M. Epithelial cadherin distribution in abnormal human pre-implantation embryos. Hum Reprod. 2005;20:3369–75.
Ghassemifar MR, Eckert JJ, Houghton FD, Picton HM, Leese HJ, Fleming TP. Gene expression regulating epithelial intercellular junction biogenesis during human blastocyst development in vitro. Mol Hum Reprod. 2003;9:245–52.
Eckert JJ, Houghton FD, Hawkhead JA, Balen AH, Leese HJ, Picton HM, et al. Human embryos developing in vitro are susceptible to impaired epithelial junction biogenesis correlating with abnormal metabolic activity. Hum Reprod. 2007;22:2214–24.
Campbell S, Swann HR, Seif MW, Kimber SJ, Aplin JD. Cell adhesion molecules on the oocyte and preimplantation human embryo. Hum Reprod. 1995;10:1571–8.
Nishioka N, Inoue K, Adachi K, Kiyonari H, Ota M, Ralston A, et al. The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Dev Cell. 2009;16:398–410.
Qin H, Hejna M, Liu Y, Percharde M, Wossidlo M, Blouin L, et al. YAP induces human naive pluripotency. Cell Rep. 2016;14:2301–12.
Hildebrand S, Hultin S, Subramani A, Petropoulos S, Zhang Y, Cao X, et al. The E-cadherin/AmotL2 complex organizes actin filaments required for epithelial hexagonal packing and blastocyst hatching. Sci Rep. 2017;7:9540.
Van Blerkom J, Davis P, Alexander S. Differential mitochondrial distribution in human pronuclear embryos leads to disproportionate inheritance between blastomeres: relationship to microtubular organization, ATP content and competence. Hum Reprod. 2000;15(12):2621–33.
Antczak M, Van Blerkom J. Temporal and spatial aspects of fragmentation in early human embryos: possible effects on developmental competence and association with the differential elimination of regulatory proteins from polarized domains. Hum Reprod. 1999;14:429–47.
Fierro-González JC, White MD, Silva JC, Plachta N. Cadherin-dependent filopodia control preimplantation embryo compaction. Nat Cell Biol. 2013;15:1424–33.
Maître JL, Niwayama R, Turlier H, Nédélec F, Hiiragi T. Pulsatile cell-autonomous contractility drives compaction in the mouse embryo. Nat Cell Biol. 2015;17:849–55.
Larue L, Ohsugi M, Hirchenhain J, Kemler R. E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc Natl Acad Sci U S A. 1994;91:8263–7.
Riethmacher D, Brinkmann V, Birchmeier C. A targeted mutation in the mouse E-cadherin gene results in defective preimplantation development. Proc Natl Acad Sci U S A. 1995;92:855–9.
Ebner T, Moser M, Shebl O, Sommergruber M, Gaiswinkler U, Tews G. Morphological analysis at compacting stage is a valuable prognostic tool for ICSI patients. Reprod BioMed Online. 2009;18:61–6.
Landry DW, Zucker HA, Sauer MV, Reznik M, Wiebe L. Hypocellularity and absence of compaction as criteria for embryonic death. Regen Med. 2006;1:367–71.
Ivec M, Kovacic B, Vlaisavljevic V. Prediction of human blastocyst development from morulas with delayed and/or incomplete compaction. Fertil Steril. 2011;96:1473–8.
Skiadas CC, Jackson KV, Racowsky C. Early compaction on day 3 may be associated with increased implantation potential. Fertil Steril. 2006;86:1386–91.
Le Cruguel S, Ferre-L’Hotellier V, Moriniere C, et al. Early compaction at day 3 may be a useful additional criterion for embryo transfer. J Assist Reprod Genet. 2013;30:683–90.
Iwata K, Mio Y. Observation of human embryonic behavior in vitro by high-resolution time-lapse cinematography. Reprod Med Biol. 2016;15:145–54.
Campbell A, Fishel S, Bowman N, Duffy S, Sedler M, Hickman CFL. Modelling a risk classification of aneuploidy in human embryos using non-invasive morphokinetics. Reprod BioMed Online. 2013;26:477–85.
Desai N, Abdelhafez F, Bedaiwy MA, et al. Clinical pregnancy and live births after transfer of embryos vitrified on day 3. Reprod BioMed Online. 2010;20:808–13.
Fernandez Gallardo E, Spiessens, D’Hooghe T, et al. Effect of day 3 embryo morphometrics and morphokinetics on survival and implantation after slow freezing-thawing and after vitrification-warming: a retrospective cohort study. Reprod Biol Endocrinol. 2017;15:79.
Berger DS, Abdelhafez F, Russell H, et al. Severe teratozoospermia and its influence on pronuclear morphology, embryonic cleavage and compaction. Reprod Biol Endocrinol. 2011;9:37.
Desai N, Gill P, Tadros NN, Goldberg JM, Sabanegh E, Falcone T. Azoospermia and embryo morphokinetics: testicular sperm-derived embryos exhibit delays in early cell cycle events and increased arrest prior to compaction. J Assist Reprod Genet. 2018;35:1339–48.
Van Langendonckt A, Demylle D, Wyns C, et al. Comparison of G1.2/G2.2 and Sydney IVF cleavage/blastocyst sequential media for the culture of human embryos: a prospective, randomized, comparative study. Fertil Steril. 2001;76:1023–31.
Paternot G, Spiessens M, Verstreken D, van Bauwel J, Debrock S, D’Hooghe T, et al. Is there a link between blastomere contact surfaces of day 3 embryos and live birth rate? Reprod Biol Endocrinol. 2012;10:78.
Ebner T, Balaban B, Moser M, Shebl O, Urman B, Ata B, et al. Automatic user-independent zona pellucida imaging at the oocyte stage allows for the prediction of preimplantation development. Fertil Steril. 2010;94:913–20.
Fawzy M, AbdelRahman MY, Zidan MH, et al. Humid versus dry incubator: a prospective, randomized, controlled trial. Fertil Steril. 2017;108:277–83.
Edwards RG, Purdy JM, Steptoe PC, Walters DE. The growth of human preimplantation embryos in vitro. Am J Obstet Gynecol. 1981;141:408–16.
Iwata K, Yumoto K, Sugishima M, Mizoguchi C, Kai Y, Iba Y, et al. Analysis of compaction initiation in human embryos by using time-lapse cinematography. J Assist Reprod Genet. 2014;31:421–6.
Van de Velde H, Cauffman G, Tournaye H, et al. The four blastomeres of a 4-cell stage human embryo are able to develop individually into blastocysts with inner cell mass and trophectoderm. Hum Reprod. 2008;23:1742–7.
Fléchon JE, Panigel M, Kraemer DC, et al. Surface ultrastructure of preimplantation baboon embryos. Anat Embryol (Berl). 1976;149:289–95.
Fleming TP, Sheth B, Fesenko I. Cell adhesion in the preimplantation mammalian embryo and its role in trophectoderm differentiation and blastocyst morphogenesis. Front Biosci. 2001;6:D1000–7.
Ducibella T, Albertini DF, Anderson E, Biggers JD. The preimplantation mammalian embryo: characterization of intercellular junctions and their appearance during development. Dev Biol. 1975;45:231–50.
Fleming TP, Javed Q, Collins J, Hay M. Biogenesis of structural intercellular junctions during cleavage in the mouse embryo. J Cell Sci Suppl. 1993;17:119–25.
Wang H, Ding T, Brown N, Yamamoto Y, Prince LS, Reese J, et al. Zonula occludens-1 (ZO-1) is involved in morula to blastocyst transformation in the mouse. Dev Biol. 2008;318:112–25.
Kim J, Gye MC, Kim MK. Role of occludin, a tight junction protein, in blastocoel formation, and in the paracellular permeability and differentiation of trophectoderm in preimplantation mouse embryos. Mol Cells. 2004;17:248–54.
Zenker J, White MD, Gasnier M, Alvarez YD, Lim HYG, Bissiere S, et al. Expanding actin rings zipper the mouse embryo for blastocyst formation. Cell. 2018;173:776–91.
Chan CJ, Costanzo M, Ruiz-Herrero T, Mönke G, Petrie RJ, Bergert M, et al. Hydraulic control of mammalian embryo size and cell fate. Nature. 2019;571:112–6.
Beyer EC, Paul DL, Goodenough DA. Connexin43: a protein from rat heart homologous to a gap junction protein from liver. J Cell Biol. 1987;105:2621–9.
Kidder GM, Winterhager E. Intercellular communication in preimplantation development: the role of gap junctions. Front Biosci. 2001;6:D731–6.
De Sousa PA, Juneja SC, Caveney S, et al. Normal development of preimplantation mouse embryos deficient in gap junctional coupling. J Cell Sci. 1997;110:1751–8.
Lee S, Gilula NB, Warner AE. Gap junctional communication and compaction during preimplantation stages of mouse development. Cell. 1987;51:851–60.
Becker DL, Davies CS. Role of gap junctions in the development of the preimplantation mouse embryo. Microsc Res Tech. 1995;31:364–74.
Houghton FD. Role of gap junctions during early embryo development. Reproduction. 2005;129:129–35.
Gualtieri R, Santella L, Dale B. Tight junctions and cavitation in the human pre-embryo. Mol Reprod Dev. 1992;32:81–7.
Krivega M, Geens M, Van de Velde H. CAR expression in human embryos and hESC illustrates its role in pluripotency and tight junctions. Reproduction. 2014;148:531–44.
Dale B, Gualtieri R, Talevi R, Tosti E, Santella L, Elder K. Intercellular communication in the early human embryo. Mol Reprod Dev. 1991;29:22–8.
Hardy K, Warner A, Winston RM, et al. Expression of intercellular junctions during preimplantation development of the human embryo. Mol Hum Reprod. 1996;2:621–32.
Bloor DJ, Wilson Y, Kibschull M, Traub O, Leese HJ, Winterhager E, et al. Expression of connexins in human preimplantation embryos in vitro. Reprod Biol Endocrinol. 2004;2:25.
Holding C, Bolton V, Monk M. Detection of human novel developmental genes in cDNA derived from replicate individual preimplantation embryos. Mol Hum Reprod. 2000;6:801–9.
Pereda J, Coppo M. Ultrastructure of a two-cell human embryo. Anat Embryol (Berl). 1987;177:91–6.
Bentin-Ley U, Horn T, Sjogren A, et al. Ultrastructure of human blastocyst-endometrial interactions in vitro. J Reprod Fertil. 2000;120:337–50.
Sathananthan H, Menezes J, Gunasheela S. Mechanics of human blastocyst hatching in vitro. Reprod BioMed Online. 2003;7:228–34.
Bloor DJ, Metcalfe AD, Rutherford, et al. Expression of cell adhesion molecules during human preimplantation embryo development. Mol Hum Reprod. 2002;8:237–45.
Houghton FD, Hawkhead JA, Humpherson PG, et al. Non-invasive amino acid turnover predicts human embryo developmental capacity. Hum Reprod. 2002;17:999–1005.
Blakeley P, Fogarty NM, del Valle I, et al. Defining the three cell lineages of the human blastocyst by single-cell RNA-seq. Development. 2015;142:3151–65.
Yan L, Yang M, Guo H, Yang L, Wu J, Li R, et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol. 2013;20:1131–9.
Hertig AT, Rock J, Adams EC. A description of 34 human ova within the first 17 days of development. Am J Anat. 1956;98:435–93.
Shahbazi MN, Jedrusik A, Vuoristo S, Recher G, Hupalowska A, Bolton V, et al. Self-organization of the human embryo in the absence of maternal tissues. Nat Cell Biol. 2016;18:700–8.
Deglincerti A, Croft GF, Pietila LN, Zernicka-Goetz M, Siggia ED, Brivanlou AH. Self-organization of the in vitro attached human embryo. Nature. 2016;533:251–4.
Morris SA. Human embryos cultured in vitro to 14 days. Open Biol. 2017;7:170003.
Cauffman G, Liebaers I, Van Steirteghem A, et al. POU5F1 isoforms show different expression patterns in human embryonic stem cells and preimplantation embryos. Stem Cells. 2006;24:2685–91.
Hansis C, Tang YX, Grifo JA, Krey LC. Analysis of Oct-4 expression and ploidy in individual human blastomeres. Mol Hum Reprod. 2001;7:155–61.
Hansis C, Grifo JA, Krey LC. Oct-4 expression in inner cell mass and trophectoderm of human blastocysts. Mol Hum Reprod. 2000;6:999–1004.
De Paepe C, Cauffman G, Verloes A, et al. Human trophectoderm cells are not yet committed. Hum Reprod. 2013;28:740–9.
Mitalipov SM, Kuo HC, Hennebold JD, Wolf DP. Oct-4 expression in pluripotent cells of the rhesus monkey. Biol Reprod. 2003;69:1785–92.
Kirchhof N, Carnwath JW, Lemme E, Anastassiadis K, Schöler H, Niemann H. Expression pattern of Oct-4 in preimplantation embryos of different species. Biol Reprod. 2000;63:1698–705.
Szczepańska K, Stańczuk L, Maleszewski M. Oct4 protein remains in trophectoderm until late stages of mouse blastocyst development. Reprod Biol. 2011;11:145–56.
Harvey AJ, Armant DR, Bavister BD, Nichols SM, Brenner CA. Inner cell mass localization of NANOG precedes OCT3/4 in rhesus monkey blastocysts. Stem Cells Dev. 2009;18:1451–8.
Hambiliki F, Ström S, Zhang P, Stavreus-Evers A. Co-localization of NANOG and OCT4 in human pre-implantation embryos and in human embryonic stem cells. J Assist Reprod Genet. 2012;29:1021–8.
Toyooka Y, Oka S, Fujimori T. Early preimplantation cells expressing Cdx2 exhibit plasticity of specification to TE and ICM lineages through positional changes. Dev Biol. 2016;411:50–60.
Krivega MV, Geens M, Heindryckx B, Santos-Ribeiro S, Tournaye H, van de Velde H. Cyclin E1 plays a key role in balancing between totipotency and differentiation in human embryonic cells. Mol Hum Reprod. 2015;21:942–56.
Sritanaudomchai H, Sparman M, Tachibana M, Clepper L, Woodward J, Gokhale S, et al. CDX2 in the formation of the trophectoderm lineage in primate embryos. Dev Biol. 2009;335:179–87.
Kunath T, Yamanaka Y, Detmar J, MacPhee D, Caniggia I, Rossant J, et al. Developmental differences in the expression of FGF receptors between human and mouse embryos. Placenta. 2014;35:1079–88.
Bridges PJ, Jeoung M, Kim H, Kim JH, Lee DR, Ko CM, et al. Methodology matters: IVF versus ICSI and embryonic gene expression. Reprod BioMed Online. 2011;23:234–44.
Kohda T. Effects of embryonic manipulation and epigenetics. J Hum Genet. 2013;58:416–20.
Aghajanova L, Shen S, Rojas AM, Fisher SJ, Irwin JC, Giudice LC. Comparative transcriptome analysis of human trophectoderm and embryonic stem cell-derived trophoblasts reveal key participants in early implantation. Biol Reprod. 2012;86:1–21.
Jones GM, Cram DS, Song B, Kokkali G, Pantos K, Trounson AO. Novel strategy with potential to identify developmentally competent IVF blastocysts. Hum Reprod. 2008;23:1748–59.
Ntostis P, Kokkali G, Iles D, Huntriss J, Tzetis M, Picton H, et al. Can trophectoderm RNA analysis predict human blastocyst competency? Syst Biol Reprod Med. 2019;65:312–25.
Kirkegaard K, Villesen P, Jensen JM, Hindkjær JJ, Kølvraa S, Ingerslev HJ, et al. Distinct differences in global gene expression profiles in non-implanted blastocysts and blastocysts resulting in live birth. Gene. 2015;571:212–20.
Martins WP, Nastri CO, Rienzi L, van der Poel SZ, Gracia C, Racowsky C. Blastocyst vs cleavage-stage embryo transfer: systematic review and meta-analysis of reproductive outcomes. Ultrasound Obstet Gynecol. 2017;49:583–91.
Lonergan P, Fair T, Corcoran D, Evans ACO. Effect of culture environment on gene expression and developmental characteristics in IVF-derived embryos. Theriogenology. 2006;65:137–52.
Kleijkers SH, Eijssen LM, Coonen E, et al. Differences in gene expression profiles between human preimplantation embryos cultured in two different IVF culture media. Hum Reprod. 2015;30:2303–11.
McEwen KR, Leitch HG, Amouroux R, et al. The impact of culture on epigenetic properties of pluripotent stem cells and pre-implantation embryos. Biochem Soc Trans. 2013;41:711–9.
Tachataki M, Winston RML, Taylor DM. Quantitative RT-PCR reveals tuberous sclerosis gene, TSC2, mRNA degradation following cryopreservation in the human preimplantation embryo. Mol Hum Reprod. 2003;9:593–601.
Shaw L, Sneddon SF, Brison DR, Kimber SJ. Comparison of gene expression in fresh and frozen-thawed human preimplantation embryos. Reproduction. 2012;144:569–82.
Houghton FD. Energy metabolism of the inner cell mass and trophectoderm of the mouse blastocyst. Differentiation. 2006;74:11–8.
Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH. Nanotubular highways for intercellular organelle transport. Science. 2004;303:1007–10.
Salas-Vidal E, Lomelí H. Imaging filopodia dynamics in the mouse blastocyst. Dev Biol. 2004;265:75–89.
Chai N, Patel Y, Jacobson K, McMahon J, McMahon A, Rappolee DA. FGF is an essential regulator of the fifth cell division in preimplantation mouse embryos. Dev Biol. 1998;198:105–15.
Copp AJ. Interaction between inner cell mass and trophectoderm of the mouse blastocyst. I. A study of cellular proliferation. J Embryol Exp Morphol. 1978;48:109–25.
Copp AJ. Interaction between inner cell mass and trophectoderm of the mouse blastocyst. II. The fate of the polar trophectoderm. J Embryol Exp Morphol. 1979;51:109–20.
Cruz YP, Pedersen RA. Cell fate in the polar trophectoderm of mouse blastocysts as studied by microinjection of cell lineage tracers. Dev Biol. 1985;112:73–83.
Fleming TP, Warren PD, Chisholm JC, et al. Trophectodermal processes regulate the expression of totipotency within the inner cell mass of the mouse expanding blastocyst. J Embryol Exp Morphol. 1984;84:63–90.
Rowson LEA, Moor R. Development of sheep conceptus during the first fourteen days. J Anat. 1966;100:777–85.
Carson SA, Gentry WL, Smith AL, Buster JE. Trophectoderm microbiopsy in murine blastocysts: comparison of four methods. J Assist Reprod Genet. 1993;10:427–33.
Burwinkel TH, Kim HN, Buster JE, Minhas BS, Carson SA. Embryo survival after pronuclear microinjection and trophectoderm biopsy. Am J Obstet Gynecol. 1994;170:1199–203.
Sheardown SA, Findlay I, Turner A, Greaves D, Bolton VN, Mitchell M, et al. Preimplantation diagnosis of a human beta-globin transgene in biopsied trophectoderm cells and blastomeres of the mouse embryo. Hum Reprod. 1992;7:1297–303.
Muggleton-Harris AL, Glazier AM, Pickering SJ. Biopsy of the human blastocyst and polymerase chain reaction (PCR) amplification of the beta-globin gene and a dinucleotide repeat motif from 2-6 trophectoderm cells. Hum Reprod. 1993;8:2197–205.
McArthur SJ, Leigh D, Marshall JT, et al. Pregnancies and live births after trophectoderm biopsy and preimplantation genetic testing of human blastocysts. Fertil Steril. 2005;84:1628–36.
Muggleton-Harris AL, Findlay I. In-vitro studies on ‘spare’ human preimplantation embryos in culture. Hum Reprod. 1991;6:85–92.
Dokras A, Sargent IL, Ross C, Gardner RL, Barlow DH. Trophectoderm biopsy in human blastocysts. Hum Reprod. 1990;5:821–5.
Cimadomo D, Capalbo A, Levi-Setti PE, Soscia D, Orlando G, Albani E, et al. Associations of blastocyst features, trophectoderm biopsy and other laboratory practice with post-warming behavior and implantation. Hum Reprod. 2018;33:1992–2001.
Fong CY, Bongso A, Sathananthan H, Ho J, Ng SC. Ultrastructural observations of enzymatically treated human blastocysts: zona-free blastocyst transfer and rescue of blastocysts with hatching difficulties. Hum Reprod. 2001;16:540–6.
Monk M, Muggleton-Harris AL, Rawlings E, Whittingham DG. Pre-implantation diagnosis of HPRT-deficient male and carrier female mouse embryos by trophectoderm biopsy. Hum Reprod. 1988;3:377–81.
Gardner RL, Edwards RG. Control of the sex ratio at fullterm in the rabbit by transferring sexed blastocysts. Nature. 1968;218:346–9.
Nijs M, Van Steirteghem A. Developmental potential of biopsied mouse blastocysts. J Exp Zool. 1990;256:232–6.
Gentry WL, Critser ES. Growth of mouse pups derived from biopsied blastocysts. Obstet Gynecol. 1995;85:1003–6.
Dokras A, Sargent IL, Gardner RL, Barlow DH. Human trophectoderm biopsy and secretion of chorionic gonadotrophin. Hum Reprod. 1991;6:1453–9.
Summers PM, Campbell JM, Miller MW. Normal in-vivo development of marmoset monkey embryos after trophectoderm biopsy. Hum Reprod. 1988;3:389–93.
Zhang S, Luo K, Cheng D, Tan Y, Lu C, He H, et al. Number of biopsied trophectoderm cells is likely to affect the implantation potential of blastocysts with poor trophectoderm quality. Fertil Steril. 2016;105:1222–7.
Molbay M, Kipmen-Korgun D, Korkmaz G, Ozekinci M, Turkay Korgun E. Human trophoblast progenitor cells express and release angiogenic factors. Int J Mol Cell Med. 2018;7:203–11.
Neal SA, Franasiak JM, Forman EJ, Werner MD, Morin SJ, Tao X, et al. High relative deoxyribonucleic acid content of trophectoderm biopsy adversely affects pregnancy outcomes. Fertil Steril. 2017;107:731–6.
Gleicher N, Metzger J, Croft G, Kushnir VA, Albertini DF, Barad DH. A single trophectoderm biopsy at blastocyst stage is mathematically unable to determine embryo ploidy accurately enough for clinical use. Reprod Biol Endocrinol. 2017;15:33.
Berger JJ. Primum non nocere: are we closer to saying that the trophectoderm biopsy does no harm? Fertil Steril. 2019;112:35–6.
Vera-Rodriguez M, Diez-Juan A, Jimenez-Almazan J, Martinez S, Navarro R, Peinado V, et al. Origin and composition of cell-free DNA in spent medium from human embryo culture during preimplantation development. Hum Reprod. 2018;33:745–56.
Huang L, Bogale B, Tang Y, Lu S, Xie XS, Racowsky C. Noninvasive preimplantation genetic testing for aneuploidy in spent medium may be more reliable than trophectoderm biopsy. Proc Natl Acad Sci U S A. 2019;116:14105–12.
Code availability
Not applicable
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Not applicable
Consent to participate
Not applicable
Consent for publication
Not applicable
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tocci, A. The unknown human trophectoderm: implication for biopsy at the blastocyst stage. J Assist Reprod Genet 37, 2699–2711 (2020). https://doi.org/10.1007/s10815-020-01925-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-020-01925-0