
Abstract
VACTERL, the prototype for associated congenital anomalies, also has connections with functional issues such as pregnancy losses, prematurity, growth delays, perinatal difficulties, and parental subfertility. This segues into a broader association with similar connections even in the absence of malformations. DNA methylation disturbances in the ovum are a likely cause, with epigenetic links to individual components and to folate effects before conception, explaining diverse fetal and placental findings and providing a link to fetal origin hypothesis-related effects. The association encompasses the following: (1) Pre- and periconceptual effects, with frequent fertility issues and occasional imprinting disorders. (2) Early malformations. (3) Adverse pregnancy outcomes (APOs), as above. (4) Developmental destabilization that resolves soon after birth. This potentiates other causes of association findings, introducing multiple confounders. (5) Long-term fetal origins hypothesis-related risks. The other findings are exceptional when the same malformations have Mendelian origins, supporting a distinct pathogenesis. Expressions are facilitated by one-carbon metabolic issues, maternal and fetal stress, and decreased embryo size. This may be one of the commonest causes of adverse reproductive outcomes, but multifactorial findings, variable onsets and phenotypes, and interactions with multiple confounders make recognition difficult. This association supports VACTERL as a continuum that includes isolated malformations, extends the fetal origins hypothesis, explains adverse effects linked to maternal obesity, and suggests possible interventions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In 1972, Quan and Smith [1] described defects with “a nonrandom tendency... to associate together,” soon expanded to VACTERL (vertebral, ano-rectal, cardiac, tracheo-esophageal, renal, and limb). This was the prototype for associations, teratogenic derivatives shaped by developmental constraints [2]. While physical defects have been the primary focus [3], connections to prematurity, fetal growth restriction (FGR—third trimester weight and length deficiencies here), prenatal lethality, perinatal issues, and parental subfertility will also be shown, supporting an extended disorder. These findings are typically absent when the same malformations occur in Mendelian syndromes [4], indicating a distinct pathogenesis.
The key finding here is a variable association of both structural and non-structural issues. This goes beyond a simple expanded VACTERL association, since those anomalies only occur in a minority of cases, and the same factors show mutual correlations even without malformations (Table 1).
While gaps in our understanding of epigenetics make details uncertain, DNA methylation disturbances arising in the ovum are a likely cause, with links to individual components and folate-related issues that include fertility effects before conception. This explains diverse fetal and placental findings arising at different times, and rapid postnatal improvements consistent with ex utero epigenetic adaptations (“DNA methylation links” section, below). It also clarifies parental fertility links and justifies including later onset fetal origin hypothesis [5] effects.
Overall, this epigenetic association includes:
-
1.
Pre- and peri-conceptual effects, with fertility issues and occasional imprinting disorders.
-
2.
A variety of malformations arising early in development, especially VACTERL anomalies, both as isolated and as concordant findings.
-
3.
Adverse pregnancy outcomes (APOs), primarily miscarriages, stillbirths, prematurity, FGR, and perinatal difficulties, and possibly others as well.
-
4.
A general developmental destabilization that enhances other causes of malformations and APOs, introducing multiple confounders, and resolving soon after birth.
-
5.
Fetal origin hypothesis-related risks that can become manifest during adult life.
Delineation here starts with APO correlations without malformations. This is validated, and extended, by showing the same relationships using both VACTERL defects and SUA (single umbilical artery) as independent markers for the disorder with separate pathogenetic origins. A discussion of epigenetic links follows, which helps justify fetal origin hypothesis links. Finally, insights from twins and issues related to severity, frequency, and recurrences add further details.
This may be one of the most common factors affecting reproduction, but multifactorial findings with variable onsets, protean phenotypes, and multiple confounders make identification difficult. However, recognition clarifies a variety of issues, and suggests interventions, including some targeted to assisted reproductive technology (ART). Growth issues from 28 to 33 weeks may also involve a particular form of stress potentially responsive to folate (“Possible interventions” section, below).
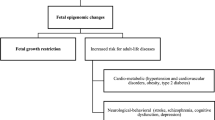
Interactions add complexity, and Fig. Fig. 1 represents a rough overview. Because different uses of the word association can be confusing, aside from quotes, I have reserved the term for the collective entity and used correlation for a statistical relationship.

EDA origins. Post-fertilization findings reflect susceptibilities to separate causative factors. Hypocellularity can be part of the association, or arise separately; in both cases, it enhances later EDA effects. Early and late growth deficiencies can be separate, and one-carbon issues can be non-continuous
Nonstructural correlations
Correlations between prematurity, FGR, prenatal lethality, perinatal issues, and parental subfertility in the absence of malformations may extend to other disorders, especially pre-eclampsia, which is omitted because of difficulties in excluding secondary complications.
As a start, growth delays with prematurity were originally recognized postnatally, e.g., 41% of 120 preterm infants had a birth weight and/or length 2 standard deviations below the mean for gestational age [6]. With the advent of accurate ultrasound biometry [7], this was extended [8], adding gestational age-related variations: Multiple parameters were delayed for very premature fetuses (24–29.9 weeks), but with moderate prematurity (30–36.9 weeks), much of the growth lag was later [9]. With prematurity, especially inducted, birth weights < 10th percentile were found in 30% of cases at 28 to 33 weeks, 15% at 22 to 24 weeks, and 19.6% at 34 to 36 weeks [10]. Differences here may be a clue to the timing of stress-related effects (“Lessons from twins” section, below). Similarly, fetal weight estimates at 32 weeks had a left skew and a lower median with prematurity compared to a normal distribution for term infants [11].
Customized standards improved accuracy, showing growth restriction in 13% of spontaneous, and 32% of medically indicated, late preterm births [12]. The high rate of medical indications suggests an increased vulnerability to perinatal difficulties. And here, as Basso et al. [13] noted, “the association between birth weight and mortality is among the strongest seen in epidemiology. While preterm delivery causes both small babies and high mortality, it does not explain this association... If, as some postulate, birth weight is not itself on the causal path to mortality, its relation with mortality would have to be explained by confounding factors that decrease birth weight and increase mortality.” Weinberg [14] similarly suggested that links between birth weight and health outcomes may not be causal. However, susceptibilities soon resolve, consistent with an epigenetic switch to accommodate a new physiology (“An epigenetic mechanism” section, below).
For stillbirths, Kapurubandara et al. [15] attributed 27% to congenital anomalies and 17% to growth restriction. For normally formed fetuses, FGR was the greatest risk factor [16]. A review found increased risks for perinatal death after one miscarriage, very preterm delivery after two or more, very preterm delivery and low birth weight after recurrent miscarriages, and preterm delivery with crown-rump length discrepancies [17]. Similarly, women with recurrent miscarriages had higher risks for preterm delivery, and for small for gestational age (SGA) infants with low Apgar scores [18].
For fertility, an increased time to pregnancy is a risk factor for preterm birth, low birth weight, placental disorders, and other issues [19]. A meta-analysis confirmed prematurity [20], and Zhu et al. [21] saw stillbirth rates of 0.3% for fertile, 0.4% for untreated subfertile, and 0.5% for treated subfertile couples and for spontaneous pregnancies with untreated subfertility, while Draper et al. [22] calculated a 3.3 adjusted odds ratio for perinatal mortality compared to normal fertility. Adjusting for age and parity, subfertile women had increased risks for low birth weight and preterm infants [23].
There are also links to imprinting disorders [24], with “increased prevalence of imprinting defects in patients with Angelman syndrome born to subfertile couples” suggesting a common cause for both [25]. Doornbos et al. [26] felt that increases in such disorders with assisted reproduction “can fully be explained by the increased fertility problems of the parents.” An imprinted gene polymorphism related to spontaneous abortions with assisted reproduction [27] supports other imprinting effects.
Associations with malformations
Malformation associations reflect teratogenic vulnerabilities and causes shaped by developmental constraints [2], e.g., the thalidomide embryopathy is also an association determined by both dosage and timing, with different findings with exposures on days 22 and 32, for example, and by susceptibility, with some animals insensitive to the prominent human prenatal effects [28].
In delineating a specific epigenetic association, APOs are mostly nonstructural and continuous, with definitional and ascertainment issues. However, malformations are distinct and less sensitive to other factors, making them useful markers.
VACTERL here refers to the associated acronymic idiopathic blastogenetic malformations (IBMs), primary structural anomalies arising during blastogenesis (from zygote formation until 28 days) [29]. As will be shown, the same APOs occur with both isolated and combined defects, supporting a continuum that includes single findings. Because major congenital anomalies are largely VACTERL defects, studies with birth defects as a general category are included here. However, there are some exceptions, e.g., isolated oral clefts were “not associated with impaired fetal growth or preterm delivery” [30].
“Idiopathic” excludes monozygous twinning and syndromic, chromosomal, and teratogenic causes. “Primary” eliminates SUA, which is typically a later atresia [31]. SUA can also occur both with and without other malformations, so that APO correlations with this anomaly presumably represent an independent confirmation malformation-related associations.
VACTERL and adverse outcomes
To start, sporadic malformations typically show decreased birth weights [32,33,34]. This applies specifically to VACTERL [35], anorectal [36], and gastrointestinal abnormalities, both isolated and combined [37], cardiac defects ranging from simple to complex [38, 39], TEF/EA (tracheo-esophageal fistula/esophageal atresia) [40], and radial findings alone [41], or with other anomalies [42]. Confirming differences between sporadic and genetic cases, newborns with isolated idiopathic preaxial polydactyly type I had lower birth weights than matched controls and familial cases [43].
Preterm deliveries (before 37 weeks of gestation) also increased with birth defects in general, and more with multiple than with isolated defects [44, 45]. A prematurity odds ratio for syndromic multiple findings was 0.9, but 1.5 for isolated anomalies [46]. Since syndromes are more often genetic, this again supports greater adversities with idiopathic anomalies than with genetic causes.
Specific defects with preterm increases include anorectal anomalies [47], TEF/EA either isolated or associated [40], longitudinal limb reductions [48], and congenital heart disease at varying frequencies according to type [49]. Oral clefts with other findings (but not when isolated) also showed high rates of both prematurity and SGA [30].
Contrawise, premature infants have higher risks for birth defects [34]. Dolan et al. [50] emphasized “overlapping outcomes” that Miquel-Verges et al. [45] extended to “a complex interaction between the development of birth defects, prematurity and intrauterine growth.”
For prenatal deaths, aneuploidy, the primary cause of early clinically unrecognized lethality, and about half of all later losses [51], is a confounder with all lethal phenotypes [52, 53]. However, exclusion still leaves considerable early losses with findings ranging from a few membranes, to an empty sac, with or without a cord stump, to a disorganized embryo, to multiple defects, to a single anomaly [54]. So, for 221 successfully karyotyped early missed abortions, 56 were euploid, 20 with growth disorganization, and 20 with single or multiple anomalies (the latter including 3 amniotic bands, which are disruptions, not malformations) [55].
As the number of previous spontaneous abortions increased, so did miscarriages, but aneuploidy decreased even with rising maternal ages [56]. With three or more consecutive losses, missed abortions on transcervical embryoscopy showed less aneuploidy than controls, but almost 80% of euploid embryos had either structural anomalies or growth disorganization [57].
With ongoing losses, malformation rates in the survivors progressively declined, but more slowly than aneupoldy decreased. Of 544-s trimester miscarriages, only 1.3% were aneuploid, while 71 had malformations, with 49 isolated [58]. Excluding 8 isolated club feet as deformations, clinodactyly and facial dysmorphism as later non-blastogenetic findings, and 11 SUA as secondary atresias, leaves 9.6% with anomalies, still high.
Serious birth defects rose from 2.5% with no prior pregnancy loss to 4.2% with three or more [59]. A 15-fold increased stillbirth risk with isolated ultrasound-detected anomalies more than doubled if growth restriction was also present [60]. Late second-trimester miscarriages with malformations also occurred about 3 weeks before those without [58]. For typically euploid sirenomelia, 47% of cases were liveborn, 71.2% premature, and 88.2% at under 2500 g, while 8% of informative cases were from multiple gestations, and 5% had diabetic mothers [61], consistent with involvement of the early hypocellularity that is a risk factor for VACTERL [4].
Opitz [62] stated that blastogenetic defects, particularly in associations, tended to be highly lethal, and had low recurrences. However, except for neural tube defects, blastogenetic anomalies, including VACTERL defects, had increased maternal histories of spontaneous abortions, leading Martínez-Frias and Frias to suggest that recurrences might not be rare [63].
Adverse perinatal effects can reflect malformations, low birth weight, prematurity, and other confounders, but may also represent a continuation of prenatal susceptibilities, so that “congenital anomalies in preterm birth are associated with a higher rate of pregnancy complications and are an independent risk factor for neonatal morbidity and perinatal mortality” [64].
Fertility effects are difficult to document, but parental subfertility was seen with VACTERL [35], and with anorectal defects with other major anomalies [47]. Similarly, singletons born of infertile couples had increasing congenital anomalies with rising times to pregnancy [21], which was also an independent risk for birth defects [65]. This also held for ART in a meta-analysis [66].
Single umbilical artery
While SUA is heterogeneous, a secondary atresia is the most common form [31], separating it from primary malformations, and providing a second type of association marker. The atresia probably reflects an early hypocellularity, as seen with conditions such as monozygotic twinning and maternal diabetes. This reduction, although not a direct cause for anomalies, is a predisposition to IBMs [4], and is a likely risk factor for other adverse pregnancy outcomes.
Correlations with early malformations are well established. Idiopathic SUA had a 20% incidence with TEF/EA plus two or more other VACTERL findings [67], as did 8% of VACTERL cases ascertained through radial anomalies [68]. Increases also occurred with isolated TEF/EA [69], and anorectal [70], renal, vertebral [71], and cardiac defects [72].
A meta-analysis found an increased incidence of SGA with SUA, both with or without preterm birth [73]. Isolated SUA rates rose with increases in growth restriction and prematurity in singletons [74] and twins [75]. With SUA without aneuploidy, malformations, low birth weight, and prematurity increased, and at least 36% of increased perinatal mortality was without malformations or low birth weight [76]. In hospital autopsies, SUA increased with malformations without aneuploidy, and with SGA fetuses [77].
Isolated SUA was also an independent risk factor for adverse labor and delivery outcomes [78, 79], perinatal mortality (where there were also parental histories of subfertility) [80], and for a poor outcome with congenital diaphragmatic hernia [81].
Contradictory results, even for SUA meta-analyses [73, 82, 83], are hardly surprising: Rates and findings vary with ascertainment, type and degree of investigation, and populations. Heifetz, in his landmark study [84], noted that “the incidence of SUA in autopsy series is about twice the incidence in prospective series. SUA has a much higher incidence among malformed, stillborn, or spontaneous abortuses than among apparently normal, liveborn, or induced abortuses.”
Still, overall SUA correlations with IBMs, FGR, prematurity, losses, perinatal issues, and parental fertility issues are consistent with a specific association. However, as with malformations, it most likely represents a marker for a secondary potentiation from an embryonic hypocellularity [4].
Epigenetic factors: background
In trying to tie together these diverse findings, epigenetics seems to be the most plausible common factor (below). Although other aspects may also be involved [85], the focus here is on DNA methylation, the best understood effect. This can also encompass spatial modifications [86] with in vivo interactions [87].
Somatic cell modifications are typically initiated and maintained by DNA methyltransferases [DNMTs] that repress transcription by adding CH3 to cytosine at a cytosine phosphorylated to guanidine (CpG) site [88]. Genome-wide effects are indicated by quantitation of repetitive DNA sequence methylation, especially long interspersed nuclear element-1 (LINE-1) and short Alu [89]. S-adenosylmethionine provides DNA methyl groups as transmethylation pathways intersect at methionine formation from homocysteine, connecting choline, methionine, methyltetrahydrofolate, and vitamins B-6 and B-12 [88].
Ethnic, ancestral, and population differences [90, 91], and maternal nutrition effects [92], support multifactorial contributions. “Genotype alone best explained ~25%... the best explanation for 75% of VMRs [variably methylated regions] was the interaction of genotype with different in utero environments, including maternal smoking, maternal depression, maternal BMI, infant birth weight, gestational age, and birth order” [93]. Even monozygotic twins have significant CpG site methylation profile differences, the largest component “attributed to the combined effects of nonshared intrauterine environment and stochastic factors” [94]. Alternatively, outliers with disrupted methylation at many CpG sites [95] show variable discontinuous effect as well.
DNA methylation links
An epigenetic pathogenesis is consistent with multiple placental and fetal effects [96], and is supported by connections between DNA methylation and individual components:
-
1.
Subfertility includes methylation-based imprinting disorders [24, 25], and an imprinted gene polymorphism is related to spontaneous abortions with assisted reproduction [27].
-
2.
For IBMs, normal developmental regulation connects fetal DNA methylation with congenital heart disease [97]. Differential methylation of a CpG locus in a cardiac morphogenesis gene was linked to perimembranous ventricular septal defects [98], and a DNMT 1 polymorphism affected risks for great artery transpositions [99]. Epigenetic factors are also related to kidney and urinary tract anomalies [100], and hyperhomocysteinemia, which affects DNA methylation, causes birth defects [101].
-
3.
With early pregnancy losses, DNMT 1 expression and global DNA methylation were reduced in chorionic villi, so “that insufficient embryonic maintenance methylation is associated with abnormal embryonic development in human early pregnancy loss” and “led to a decreased implantation rate of embryos, increased fetal absorption, and poor fetal and placental development” [102].
-
4.
For prematurity, two different aberrant DNA hypomethylation patterns suggest a heterogeneous pathophysiology [103]. With 6–19-week cervical DNA, increased methylation of prostaglandin E receptor 2 gene (PTGER2) showed longer gestations, while repetitive long interspersed nuclear element-1 Homo sapiens-specific (LINE 1-HS) were shorter. Maternal blood LINE-1 methylation at the end of the first, but not the second, trimester was correlated with prematurity [104, 105]. Third-trimester DNA methylation around 16.1 and 29.5 weeks decreased in 15 of 24 genes with spontaneous preterm delivery. Preterm FGR showed the lowest methylation, and preterm delivery mothers had lower dietary choline, a major source of methyl groups for DNA modification.
-
5.
DNA methylation links to fetal growth issues [106] include expressions of genes involved in energy homeostasis in placentas and cord blood in SGA newborns [107]. Chorionic villi DNMT3A protein was downregulated with early embryo growth arrest [108], and a promoter polymorphisms for the gene correlated with spontaneous abortion risk [109]. A correlation of low birth weight and cord blood DNA methylation disruption was more common with in vitro than in vivo conception, suggesting that “some individuals are more susceptible to environmentally mediated epigenetic alterations” [95], although this could also reflect a higher incidence of predisposed fetuses in the in vitro group.
-
6.
A rapid postnatal reduction in vulnerabilities is consistent with acute ex utero epigenetic shifts, e.g., DNA methylation establishes postnatal pancreatic β cell glucose-stimulated insulin secretion [110], and some highly prenatally expressed imprinted transcripts decrease after birth [111]. A similar effect occurs with imprinted disorders: Hypoglycemia in 30–50% of newborns with Beckwith-Wiedemann syndrome typically resolves within the first 3 days of life [112], while transient neonatal diabetes mellitus had a 1-day modal onset, and remission at 2 months [113].
Folate and methylation
A central role for folic acid, which is crucial for methylation, provides additional evidence for connections between epigenetic factors and association components.
For fertility, there are absolute physiological needs for endogenous folate in oocytes and before the two cell blastocyst stage [114]. Low folate and environmental effects on methylation around the time of conception are seen in animal models [92], and vitamin B12, which is also involved in the one-carbon pathway, can be influential as well [115].
Silvestris et al. [116] suggested pre-conception oxidative stress effects on DNA methylation involving the one-carbon cycle. Human follicular fluid folate suppresses inflammation and upregulates a high-density lipoprotein pathway source for steroid hormone synthesis [117]. Low folate also increases follicular fluid homocysteine, causing oocyte immaturity and poor early embryo quality, “while methylenetetrahydrofolate reductase (MTHFR) gene variants [are] associated with lower ovarian reserves, diminished response to follicular stimulation, and reduced live birth after in vitro fertilization… [I]mbalances in folate metabolism and related gene variants may impair female fecundity as well as compromise implantation and the chance of a live birth” [118].
High versus low maternal folate intake gave a 0.80 adjusted relative risk for spontaneous abortion [119]. Folate only before conception reduced growth delays, preterm births [120, 121], adverse perinatal outcomes [122], and links to fecundability [123]. Maternal periconceptional one-carbon metabolism links to human embryonic growth and development [124, 125], and a 62% lower fecundability among women born before 34 weeks themselves [126] support pre-fertilization effects. Similarly, a role for folate in autism prevention [127] may be related to preventing autism-related issues, such as prematurity [128], but there are also indications of direct connections to oocyte and early embryo methylation [129].
With ongoing developmental epigenetic methylation, later folate deficiencies can affect an already compromised fetus throughout pregnancy, and supplementation improved perinatal outcomes only when began pre-conception and continued beyond 12 weeks of pregnancy [123]. This combination of early and late effects might explain “mixed” findings with intervention studies [130].
Folate connections to the association are also supported by maternal obesity effects. Since serum folate levels are inversely related to pre-pregnancy body mass [131], association findings would be expected, and do occur. Despite late macrosomia with obesity-related metabolic disturbances, there is excess growth restriction early on [132], as well as increases in limb reductions; anorectal, renal, and cardiovascular anomalies [133,134,135]; fertility issues [136]; prematurity; early losses and stillbirth; and possibly perinatal difficulties [137].
Fetal origin links
The fetal origin hypothesis and its derivatives connect prenatal problems with postnatal disorders, including heart disease, hypertension, stroke, type 2 diabetes, and behavioral and mental issues. Environmentally caused growth delays and prematurity were first cited [5]; more recently, maternal stress, even without primary fetal issues, has been implicated [138]. Interactions are possible, and there may be other mechanisms as well, e.g., adenosine toxicities with oxidative stress [139].
But whatever the trigger, epigenetic responses are shown by extensive animal and human evidence “that prenatal stress could lead to lasting, broad and functionally organized signatures in DNA methylation which, in turn, could mediate exposure-phenotype associations” [140]. DNA methylation also affects typical late effect disorders such as cardiovascular disease and diabetes [141, 142], linking both long- and short-term effects.
As a result, epigenetic disruptions could increase fetal issues and adult onset methylation-related effects. Animal and human findings show “that dietary availability of methyl donors has an impact on the patterns of gene expression by affecting DNA methylation at regulatory regions,” with chromatin acting as a “nutrient sensor” [87]. With this, multiple newborn CpG methylations correlated with maternal plasma folate levels in pregnancy. Most affected genes lacked known connections to folate, but “some relate to birth defects other than neural tube defects, neurological functions or varied aspects of embryonic development” [143].
This is a plausible adaptation to intermittent food scarcities when reproduction is hard to sustain. Here, a lower maternal folate can cause infertility, APOs, and, perhaps incidentally, long-term epigenetic issues. If scarcities continue, folate remains low, complicating later pregnancy issues [115, 125, 126]. However, if food becomes more available, outcomes and survival will improve.
In support, a 1944–1945 Dutch famine “markedly reduced... conceptions resulting in birth” after about 2 months, with immediate recovery with adequate food [144], and altered DNA methylation patterns linked to growth and metabolism in surviving fetuses as adults [145]. Similarly, smaller infants and increased prematurity were seen with seasonal scarcities in The Gambia [146].
Possible links between short- and long-term DNA methylation effects suggest testable hypotheses about adult disorders:
-
1.
Connections to parental subfertility, with smaller sibships and longer inter-pregnancy intervals.
-
2.
An ongoing process with parental subfertility suggests increased adult risks to siblings of propositi.
Lessons from twins
Twins offer further insights into epigenetic effects.
First, hypocellularity, an apparent risk factor for SUA and associated APOs (above), can normalize: Despite an initial monozygotic division, late growth curves are the same for twins and singletons [147] until a 31–33-week twin deceleration consistent with crowding occurs [148].
Second, there is support for maternal influences on all members of a pregnancy: A discordance in about 20% of pairs that begins a bit earlier than the overall deceleration leads to an 18% or greater birth weight difference, with adverse effects regardless of zygosity that include increased perinatal mortality, individual morbidity, and composite perinatal morbidity for both twins [148].
Third, there are indications of specific times for fetal stress. Late association effects coincide with preparations for postnatal life that are largely achieved by 34 weeks or so [149], including what Ellison [150] calls a metabolic crisis as the placenta fails to keep pace with increasing energy demands, leading to the upregulation of fetal compensatory systems, including increased fetal corticosteroids. These stresses would accentuate epigenetic vulnerabilities. This may also apply to singletons, since non-induced SGA preterm births show a similar period of increasing issues with a greater frequency between 28 and 33 weeks of gestation compared to 22 to 24 weeks and 34 to 36 weeks [10].
Severity, frequency, and recurrences
Severity varies, with correlations between different findings. Rates and average numbers of major malformations rose as birth weights fell [33, 151], and idiopathic multiple defects had higher fetal death rates than isolated [152]. Stillbirths also occurred in 2.5% of isolated euploid TEF/EA but 10.1% with additional major malformations [40]. Similarly, prematurity rates are higher with multiple than with isolated birth defects [46], and increased congenital anomaly risks with multiple births rise with the early death of a co-twin [153]. Even without IBMs, growth delays were earlier for very premature fetuses than for moderate [9].
Variability makes frequencies difficult to ascertain, but some clues do exist. At a minimum, using major malformations for ascertainment, substantial correlations with adverse outcomes indicate that an appreciable percentage of recognized anomalies are likely to involve association effects. This also applies to typically unrecognized, but even more common, very early euploid losses [57], and to associated APO dyads, e.g., about a 15–20% incidence of FGR with prematurity [10, 12], and in 20% or more of stillbirths without defects [11, 13].
Specific factors can expose latencies. High versus low quintile maternal folate intake gave an adjusted relative risk of 0.80 for spontaneous abortions [119]. Similarly, a small embryo may be more sensitive to additional perturbations, and embryonic hypocellularity [4] probably contributes to a ≥ 25% discordance in 15% of monochorionic twins that represented “the optimal cutoff” for predicting stillbirth and neonatal mortality [154].
Overall, frequencies seem substantial, but specifics are uncertain. However, in the future, it may be possible to obtain statistical determinations through multifactorial [93] or discontinuous [95] methylation variations.
Severity and frequency also affect estimated recurrences indicated by ongoing fertility issues and positive family histories, while variable manifestations make ascertainment an issue. Pregnancy stresses can be crucial for expression, giving female biases [155], with links between preterm pregnancies and later maternal cardiovascular disease [156] and lower fecundability among women born early themselves [126].
Multifactorial contributions are likely, e.g., epigenetic modifications can be transmitted over generations [157,158,159], and folate metabolic polymorphisms are common (above).
Possible interventions
The proposed association suggests detectable high vulnerability subgroups involving both intrinsic and extrinsic factors that may benefit from monitoring and interventions.
Advances in analyzing DNA methylation patterns in single blastocysts [160] may make it possible to detect susceptible embryos before implantation, and placental biopsies may help detect modifiable aspects of ART with adverse effects [161].
While the efficacy of supplemental folate, both before and after fertilization, is well known, detection of at-risk groups may justify particularly aggressive approaches in specific cases. Timing-related effects may be important clues here, and the detection of abnormal growth from 28 to 33 weeks in both twins and singletons (“Lessons from twins” section, above) suggest trials of additional folate.
Conclusions
A distinct association of structural and non-structural findings includes
-
1.
Pre- and peri-conceptual, with frequent fertility issues and occasional imprinting disorders.
-
2.
IBMs, including both isolated and combined VACTERL defects, supporting a continuum that extends to single anomalies. A paucity of related findings when the same malformations have Mendelian origins shows a distinct pathogenesis.
-
3.
APOs such as prenatal lethality, prematurity, growth delays, and perinatal difficulties.
-
4.
Developmental destabilization that resolves soon after birth. This enhances certain teratogenic disturbances, and a variety of specific causes of APOs, introducing multiple confounders.
-
5.
Fetal origin hypothesis-related risks.
Epigenetic disturbances of DNA methylation in the ovum are a likely cause: Low maternal folate is a major risk before conception, and if deficits continue, later effects are enhanced, while embryonic hypocellularity and maternal and fetal stress potentiate findings. Embryos with methylation issues should be especially vulnerable, suggesting targeted interventions.
Frequencies and recurrences are uncertain, with variable interactions, non-specific and protean findings, timing differences, and multiple confounders obscuring recognition. Still, this may be one of the commonest contributions to adverse reproductive outcomes—at the very least, correlations involving malformations indicate a major role in the production of birth defects.
It is hardly novel to see the complex roles of folate metabolism pointing towards possible reproductive interventions. However, understanding the proposed association may facilitate the targeting of high-risk groups with tailored therapies.
Change history
02 June 2018
The original version of this article unfortunately contained a mistake. The last 3 words of the article title were omitted. With this, the original article was corrected and the correct article title is now presented in here.
References
Quan L, Smith DW. The VATER association: vertebral defects, anal atresia, tracheoesophageal fistula with esophageal atresia, radial dysplasia. Birth Defects Orig Art Ser. 1972;8(1):75–8.
Lubinsky M. The VACTERL association as a disturbance of cell fate determination. Am J Med Genet A. 2015;167(1):2582–8.
Solomon BD. Vacterl/Vater association. Orphanet J Rare Dis. 2011;6(1):56. https://doi.org/10.1186/1750-1172-6-56.
Lubinsky M. Embryonic hypocellularity, blastogenetic malformations, and fetal growth restriction. Am J Med Genet A. 2017;173(1):151–6.
Almond D, Currie J. Killing me softly: the fetal origins hypothesis. J Econ Perspect. 2011;25(3):153–72.
Heinonen K, Matilainen R, Koski H, Launiala K. Intrauterine growth retardation (IUGR) in pre-term infants. J Perinat Med. 1985;13(4):171–81.
Hadlock FP, Harrist RB, Sharman RS, Deter RL, Park SK. Estimation of fetal weight with the use of head, body and femur measurements—a prospective study. Am J Obstet Gynecol. 1985;151:333–7.
Zhang J, Merialdi M, Platt LD, Kramer MS. Defining normal and abnormal fetal growth: promises and challenges. Am J Obstet Gynecol. 2010;202(6):522–8.
Doubilet PM, Benson CB, Wilkins-Haug L, Ringer S. Fetuses subsequently born premature are smaller than gestational age-matched fetuses not born premature. J Ultrasound Med. 2003;22(4):359–63.
Zeitlin J, Ancel PY, Saurel-Cubizolles MJ, Papiernik E. The relationship between intrauterine growth restriction and preterm delivery: an empirical approach using data from a European case-control study. Br J Obstet Gynecol. 2000;107(6):750–8.
Gardosi JO. Prematurity and fetal growth restriction. Early Hum Dev. 2005;81(1):43–9.
Carreno CA, Costantine MM, Holland MG, Ramin SM, Saade GR, Blackwell SC. Approximately one-third of medically indicated late preterm births are complicated by fetal growth restriction. Am J Obstet Gynecol. 2011;204(3):263. e1-e4
Basso O, Wilcox AJ, Weinberg CR. Birth weight and mortality: causality or confounding? Am J Epidemiol. 2006;164(4):303–11.
Weinberg CR. Invited commentary: troubling trends in birth weight. Am J Epidemiol. 2015;183(1):24–5.
Kapurubandara S, Melov SJ, Shalou ER, Mukerji M, Yim S, Rao U, et al. A perinatal review of singleton stillbirths in an Australian metropolitan tertiary centre. PLoS One. 2017;12(2):e0171829.
Gardosi J, Madurasinghe V, Williams M, Malik A, Francis A. Maternal and fetal risk factors for stillbirth: population based study. Br Med J. 2013;346(2):f108.
Van Oppenraaij RH, Jauniaux E, Christiansen OB, Horcajadas JA, Farquharson RG, Exalto N. Predicting adverse obstetric outcome after early pregnancy events and complications: a review. Hum Reprod Update. 2009;15(4):409–21.
Fawzy M, Saravelos S, Li TC, Metwally M. Do women with recurrent miscarriage constitute a high-risk obstetric population? Hum Fertility. 2016;19(1):9–15.
Wise LA, Mikkelsen EM, Sørensen HT, Rothman KJ, Hahn KA, Riis AH, et al. Prospective study of time to pregnancy and adverse birth outcomes. Fertil Sterility. 2015;103(4):1065–73.
Messerlian C, Maclagan L, Basso O. Infertility and the risk of adverse pregnancy outcomes: a systematic review and meta-analysis. Hum Reprod. 2013;28(1):125–37.
Zhu JL, Basso O, Obel C, Bille C, Olsen J. Infertility, infertility treatment, and congenital malformations: Danish national birth cohort. Br Med J. 2006;333(7370):679–81.
Draper ES, Kurinczuk JJ, Abrams KR, Clarke M. Assessment of separate contributions to perinatal mortality of infertility history and treatment: a case–control analysis. Lancet. 1999;353:1746–49.
Thomson F, Shanbhag S, Templeton A, Bhattacharya S. Obstetric outcome in women with subfertility. Br J Obstet Gynecol. 2005;112(5):632–7.
Seggers J, de Walle HE, Bergman JE, Groen H, Hadders-Algra M, Bos ME, et al. Congenital anomalies in offspring of subfertile couples: a registry-based study in the northern Netherlands. Fertil Steril. 2015;103(4):1001–10.
Ludwig M, Katalinic A, Gross S, Sutcliffe A, Varon R, Horsthemke B. Increased prevalence of imprinting defects in patients with Angelman syndrome born to subfertile couples. J Med Genet. 2005;42(4):289–91.
Doornbos ME, Maas SM, McDonnell J, Vermeiden JP, Hennekam RC. Infertility, assisted reproduction technologies and imprinting disturbances: a Dutch study. Hum Reprod. 2007;22(9):2476–80.
Liu Y, Tang Y, Ye D, Ma W, Feng S, Li X, et al. Impact of abnormal DNA methylation of imprinted loci on human spontaneous abortion. Reprod Sci. 2017;25(1):131–9. https://doi.org/10.1177/1933719117704906.
Kim JH, Scialli AR. Thalidomide: the tragedy of birth defects and the effective treatment of disease. Toxicol Sci. 2011;122(1):1–6.
Opitz JM, Zanni G, Reynolds JF, Gilbert-Barness E. Defects of blastogenesis. Am J Med Genet A. 2002;115(4):269–86.
Stoll C, Alembik Y, Dott B, Roth MP. Associated malformations in cases with oral clefts. Cleft Palate Craniofac J. 2000;37(1):41–7.
Benirschke J, Kaufmann P. Anatomy and pathology of the umbilical cord and major fetal vessels. In: Benirschke J, Kaufmann P, editors. Pathology of the human placenta. NY: Springer; 2000. p. 355–98.
Khoury MJ. James LM, Erickson JD. On the measurement and interpretation of birth defect associations in epidemiologic studies. Am J Med Genet 1990; 37:229–36.
Mili F, Edmonds LD, Khoury MJ, McClearn AB. Prevalence of birth defects among low-birth-weight infants. A population study. Am J Dis Child. 1991;145:1313–8.
Rasmussen SA, Moore CA, Paulozzi LJ, Rhodenhiser EP. Risk for birth defects among premature infants: a population-based study. J Pediatr. 2001;138(5):668–73.
Czeizel A, Ludányi I. An aetiological study of the VACTERL-association. Eur J Pediatr. 1985;144(4):331–7.
Wijers CH, Rooij IA, Marcelis CL, Brunner HG, Blaauw I, Roeleveld N. Genetic and nongenetic etiology of nonsyndromic anorectal malformations: a systematic review. Birth Defects Res C: Embryo Today. 2014;102:382–400.
Tárnok A, Méhes K. Gastrointestinal malformations, associated congenital abnormalities, and intrauterine growth. J Pediatr Gastroenterol Nutr. 2002;34:406–9.
Malik S, Cleves MA, Zhao W, Correa A, Hobbs CA. Association between congenital heart defects and small for gestational age. Pediatr. 2007;119:e976–82.
Wallenstein MB, Harper LM, Odibo AO, Roehl KA, Longman RE, Macones GA, et al. Fetal congenital heart disease and intrauterine growth restriction: a retrospective cohort study. J Matern Fetal Neonatal Med. 2012;25:662–5.
Depaepe A, Dolk H, Lechat MF. The epidemiology of tracheo-oesophageal fistula and oesophageal atresia in Europe. EUROCAT Working Group. Arch Dis Childh. 1993;68:743–8.
Czeizel AE, Vitéz M, Kodaj I, Lenz W. A family study on isolated congenital radial and tibial deficiencies in Hungary, 1975–1984. Clin Genet. 1993;44:32–6.
Evans JA, Vitez M, Czeizel A. Congenital abnormalities associated with limb deficiency defects: a population study based on cases from the Hungarian Congenital Malformation Registry (1975–1984). Am J Med Genet A. 1994;49A:52–66.
Materna-Kiryluk A, Jamsheer A, Wisniewska K, Wieckowska B, Limon J, Borszewska-Kornacka M, et al. Epidemiology of isolated preaxial polydactyly type I: data from the Polish Registry of Congenital Malformations (PRCM). BMC Pediatr. 2013;13:26.
Honein MA, Kirby RS, Meyer RE, Xing J, Skerrette NI, Yuskiv N, et al. The association between major birth defects and preterm birth. Matern Child Health J. 2009;13:164–75.
Miquel-Verges F, Mosley BS, Block AS, Hobbs CA. A Spectrum project: preterm birth and small-for-gestational age among infants with birth defects. J Perinatol. 2015;35:198–203.
Egbe A, Uppu S, Lee S, Stroustrup A, Ho D, Srivastava S. Congenital malformations in the newborn population: a population study and analysis of the effect of sex and prematurity. Pediatr Neonatol. 2015;56:25–30.
Wijers CH, van Rooij IA, Rassouli R, Wijnen MH, Broens PM, Sloots CE, et al. Parental subfertility, fertility treatment, and the risk of congenital anorectal malformations. Epidemiology. 2015;26:169–76.
Källén B. Epidemiology of Human Congenital Malformations. Chapter 25. Limb reduction defects. Springer Verlag 2014: pp. 123–127.
Laas E, Lelong N, Thieulin AC, Houyel L, Bonnet D, Ancel PY, et al. Preterm birth and congenital heart defects: a population-based study. Pediatr. 2012;130:e829–37.
Dolan SM, Callaghan WM, Rasmussen SA. Birth defects and preterm birth: overlapping outcomes with a shared strategy for research and prevention. Birth Defects Res A. 2009;85:874–8.
van den Berg MM, van Maarle MC, van Wely M, Goddijn M. Genetics of early miscarriage. Biochim Biophys Acta. 2012;1822:1951–9.
Byrne J, Warburton D, Kline J, Blanc W, Stein Z. Morphology of early fetal deaths and their chromosomal characteristics. Teratology. 1985;32:297–315.
Minelli E, Buchi C, Granata P, Meroni E, Righi R, Portentoso P, et al. Cytogenetic findings in echographically defined blighted ovum abortions. Ann Genet. 1993;36:107–10.
Hardy K, Hardy PJ. 1st trimester miscarriage: four decades of study. Transl Pediatr. 2015;4:189–200.
Philipp T, Philipp K, Reiner A, Beer F, Kalousek DK. Embryoscopic and cytogenetic analysis of 233 missed abortions: factors involved in the pathogenesis of developmental defects of early failed pregnancies. Hum Reprod. 2003;18:1724–32.
Ogasawara M, Aoki K, Okada S, Suzumori K. Embryonic karyotype of abortuses in relation to the number of previous miscarriages. Fertil Steril. 2000;73:300–4.
Feichtinger M, Walner E, Hartman B, Reiner A, Phillip T. Transcervical embryoscopic and cytogenetic findings reveal distinctive differences in primary and secondary recurrent pregnancy loss. Fertil Steril. 2017;107:144–9.
Joó JG, Beke A, Berkes E, Papp Z, Rigó J Jr, Papp C. Fetal pathology in second-trimester miscarriages. Fetal Diagn Ther. 2009;25:186–91.
Khoury MJ, Erickson JD. Recurrent pregnancy loss as an indicator for increased risk of birth defects: a population-based case–control study. Paediatr Perinat Epidemiol. 1993;7:404–16.
Frey HA, Odibo AO, Dicke JM, Shanks AL, Macones GA, Cahill AG. Stillbirth risk among fetuses with ultrasound-detected isolated congenital anomalies. Obstet Gynecol. 2014;124:91–8.
Orioli IM, Amar E, Arteaga-Vazquez J, Bakker MK, Bianca S, Botto LD, et al. Sirenomelia: an epidemiologic study in a large dataset from the International Clearinghouse of Birth Defects Surveillance and Research, and literature review. Am J Med Genet C. 2011;157:358–73.
Opitz JM. Blastogenesis and the “primary field” in human development. Birth Defects Orig Artic Ser. 1993;29(1):3–37.
Martínez-Frías ML, Frias JL. Are blastogenetic anomalies sporadic? Am J Med Genet A. 1997;68:381–5.
Linhart Y, Bashiri A, Maymon E, Shoham-Vardi I, Furman B, Vardi H, et al. Congenital anomalies are an independent risk factor for neonatal morbidity and perinatal mortality in preterm birth. Eur J Obstet Gynecol Reprod Bio. 2000;90:43–9.
Mburia-Mwalili A, Yang W. Interpregnancy interval and birth defects. Birth Defects Res A. 2015;103:904–12.
Hansen M, Kurinczuk JJ, Milne E, de Klerk N, Bower C. Assisted reproductive technology and birth defects: a systematic review and meta-analysis. Hum Reprod Update. 2013;19:330–53.
de Jong EM, Felix JF, Deurloo JA, van Dooren MF, Aronson DC, Torfs CP, et al. Non-VACTERL-type anomalies are frequent in patients with esophageal atresia/tracheo-esophageal fistula and full or partial VACTERL association. Birth Defects Res A. 2008;82:92–7.
Carli D, Garagnani L, Lando M, Fairplay T, Bernasconi S, Landi A, et al. VACTERL (vertebral defects, anal atresia, tracheoesophageal fistula with esophageal atresia, cardiac defects, renal and limb anomalies) association: disease spectrum in 25 patients ascertained for their upper limb involvement. J Pediatr. 2014;164:458–62.
Torfs CP, Curry CJ, Bateson TF. Population-based study of tracheoesophageal fistula and esophageal atresia. Teratology. 1995;52:220–32.
Zwink N. Risk factors for congenital anorectal malformations. Medical dissertation abstract. Medizinische Fakultät Heidelberg. 2012; http://www.ub.uni-heidelberg.de/archiv/14882
Martínez-Frías ML, Bermejo E, Rodríguez-Pinilla E, Prieto D. Does single umbilical artery (SUA) predict any type of congenital defect? Clinical–epidemiological analysis of a large consecutive series of malformed infants. Am J Med Genet A. 2007;146:15–25.
Araujo Júnior E, Palma-Dias R, Martins WP, Reidy K, da Silva Costa F. Congenital heart disease and adverse perinatal outcome in fetuses with confirmed isolated single functioning umbilical artery. J Obstet Gynaecol. 2015;35:85–7.
Kim HJ, Kim JH, Chay DB, Park JH, Kim MA. Association of isolated single umbilical artery with perinatal outcomes: systemic review and meta-analysis. Obstet Gynecol Sci. 2017;60:266–73.
Mailath-Pokorny M, Worda K, Schmid M, Polterauer S, Bettelheim D. Isolated single umbilical artery: evaluating the risk of adverse pregnancy outcome. Eur J Obstet Gynecol Reprod Biol. 2015;184:80–3.
Stout MJ, Odibo AO, Longman R, Shanks AL, Cahill AG. The incidence of isolated single umbilical artery in twins and adverse pregnancy outcomes. Prenat Diagn. 2013;33:269–72.
Lilja M. Infants with single umbilical artery studied in a national registry. 2: survival and malformations in infants with single umbilical artery. Paediatr Perinat Epidemiol. 1992;6:416–22.
Prucka S, Clemens M, Craven C, McPherson E. Single umbilical artery: what does it mean for the fetus? A case-control analysis of pathologically ascertained cases. Genet Med. 2004;6:54–7.
Ashwal E, Melamed N, Hiersch L, Edel S, Bardin R, Wiznitzer A, et al. The impact of isolated single umbilical artery on labor and delivery outcome. Prenat Diagn. 2014;34:581–5.
Naveiro-Fuentes M, Carrillo-Badillo MP, Malde-Conde J, Gallo-Vallejo JL, Puertas-Prieto A. Perinatal outcomes in singleton pregnancies with a single umbilical artery. J Matern Fetal Neonatal Med. 2016;29:1562–5.
Burshtein S, Levy A, Holcberg G, Zlotnik A, Sheiner E. Is single umbilical artery an independent risk factor for perinatal mortality? Arch Gynecol Obstet. 2011;283:191–4.
Bianco K, Feldstein V, Norton M, Farrell JA, Keller R. OP04. 08: the prognostic significance of a single umbilical artery (SUA) in fetuses with prenatally detected congenital diaphragmatic hernia (CDH). Ultrasound Obstet Gynecol. 2011;38(S1):68.
Voskamp BJ, Fleurke-Rozema H, Oude-Rengerink K, Snijders RJM, Bilardo CM, Mol BWJ, et al. Relationship of isolated single umbilical artery to fetal growth, aneuploidy and perinatal mortality: systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2013;42:622–8.
Xu Y, Ren L, Zhai S, Luo X, Hong T, Liu R, et al. Association between isolated single umbilical artery and perinatal outcomes: a meta-analysis. Med Sci Monit. 2016;22:1451–9.
Heifetz SA. Single umbilical artery. A statistical analysis of 237 autopsy cases and review of the literature. Perspect Pediatr Pathol. 1984;8:345–78.
Yiu TT, Li W. Pediatric cancer epigenome and the influence of folate. Epigenom. 2015;7:961–73.
Mourad R, Cuvier O. Predicting the spatial organization of chromosomes using epigenetic data. Genome Biol. 2015;16:182.
Navarro E, Funtikova AN, Fíto M, Schröder H. Prenatal nutrition and the risk of adult obesity: long-term effects of nutrition on epigenetic mechanisms regulating gene expression. J Nutr Biochem. 2017;39:1–4.
Niculescu MD, Zeisel SH. Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and choline. J Nutr. 2002;132:2333S–5S.
Yang AS, Estécio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:38e–38.
Lam LL, Emberly E, Fraser HB, Neumann SM, Chen E, Miller GE, et al. Factors underlying variable DNA methylation in a human community cohort. Proc Nat Acad Sci. 2012;109(Suppl 2):17253–60.
Mozhui K, Smith AK, Tylavsky FA. Ancestry dependent DNA methylation and influence of maternal nutrition. PLoS One. 2015;10(3):e0118466.
Dominguez-Salas P, Moore SE, Baker MS, Bergen AW, Cox SE, Dyer RA, et al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat Commun. 2014;5:3746.
Teh AL, Pan H, Chen L, Ong ML, Dogra S, Wong J, et al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res. 2014;24:1064–74.
Gordon L, Joo JE, Powell JE, Ollikainen M, Novakovic B, Li X, et al. Neonatal DNA methylation profile in human twins is specified by a complex interplay between intrauterine environmental and genetic factors, subject to tissue-specific influence. Genome Res. 2012;22:1395–406.
Ghosh J, Mainigi M, Coutifaris C, Sapienza C. Outlier DNA methylation levels as an indicator of environmental exposure and risk of undesirable birth outcome. Hum Mol Genet. 2016;25:123–9.
Macaulay EC, Bloomfield FH. Unravelling the link between the placental epigenome and pregnancy outcomes. Biol Reprod. 2016;94:81–2.
Serra-Juhé C, Cuscó I, Homs A, Flores R, Torán N, Pérez-Jurado LA. DNA methylation abnormalities in congenital heart disease. Epigenetics. 2015;10:167–77.
Wijnands KP, Chen J, Liang L, Verbiest MM, Lin X, Helbing WA, et al. Genome-wide methylation analysis identifies novel CpG loci for perimembranous ventricular septal defects in human. Epigenomics. 2017;9:241–51.
Lei L, Lin H, Zhong S, Zhang Z, Chen J, Yu X, et al. DNA methyltransferase 1 rs16999593 genetic polymorphism decreases risk in patients with transposition of great arteries. Gene. 2017;615:50–6.
Nicolaou N, Renkema KY, Bongers EM, Giles RH, NVm K. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol. 2015;11:720–31.
Iacobazzi V, Infantino V, Castegna A, Andria G. Hyperhomocysteinemia: related genetic diseases and congenital defects, abnormal DNA methylation and newborn screening issues. Mol Genet Metab. 2014;113:27–33.
Yin LJ, Zhang Y, Lv PP, He WH, Wu YT, Liu AX, et al. Insufficient maintenance DNA methylation is associated with abnormal embryonic development. BMC Med. 2012;10:26. https://doi.org/10.1186/1741-7015-10-26.
Bhavnani SK, Dang B, Kilaru V, Caro M, Visweswaran S, Saade G, Smith AK, Menon R. Methylation differences reveal heterogeneity in preterm pathophysiology: results from bipartite network analyses. J Perinat Med 2017;0. doi: https://doi.org/10.1515/jpm-2017-0126
Burris HH, Baccarelli AA, Motta V, Byun HM, Just AC, Mercado-Garcia A, et al. Association between length of gestation and cervical DNA methylation of PTGER2 and LINE 1-HS. Epigenetics. 2014;9:1083–91.
Burris HH, Rifas-Shiman SL, Baccarelli A, Tarantini L, Boeke CE, Kleinman K, et al. Associations of long interspersed nuclear element-1 DNA methylation with preterm birth in a prospective cohort study. J Develop Orig Health Dis. 2012;3:173–81.
Chen X, Bai G, Scholl TO. Spontaneous preterm delivery, dietary choline intake and DNA hypomethylation of tumor related genes in pregnant women. FASEB J. 2016;30(Suppl 1):912.2.
Díaz M, García C, Sebastiani G, de Zegher F, López-Bermejo A, Ibáñez L. Placental and cord blood methylation of genes involved in energy homeostasis: association with fetal growth and neonatal body composition. Diabetes. 2017;66:779–84.
Gu H, Gao J, Guo W, Zhou Y, Kong Q. The expression of DNA methyltransferases3A is specifically downregulated in chorionic villi of early embryo growth arrest cases. Mol Med Rep. 2017;16:591–6.
Liu Y, Zheng H, Guo P, Feng S, Zhou X, Ye D, et al. DNA methyltransferase 3A promoter polymorphism is associated with the risk of human spontaneous abortion after assisted reproduction techniques and natural conception. J Assist Reprod Genet. 2017;34:245–52.
Dhawan S, Tschen SI, Zeng C, Guo T, Hebrok M, Matveyenko A, et al. DNA methylation directs functional maturation of pancreatic β cells. J Clin Invest. 2015;125:2851–60.
Iglesias-Platas I, Martin-Trujillo A, Petazzi P, Guillaumet-Adkins A, Esteller M, Monk D. Altered expression of the imprinted transcription factor PLAGL1 deregulates a network of genes in the human IUGR placenta. Hum Mol Genet. 2014;23:6275–85.
DeBaun MR, King AA, White N. Hypoglycemia in Beckwith-Wiedemann syndrome. Semin Perinatol. 2000;24:164–71.
Docherty LE, Kabwama S, Lehmann A, Hawke E, Harrison L, Flanagan SE, et al. Clinical presentation of 6q24 transient neonatal diabetes mellitus (6q24 TNDM) and genotype–phenotype correlation in an international cohort of patients. Diabetologia. 2013;56:758–62.
Meredith M, MacNeil AH, Trasler JM, Baltz JM. Growing mouse oocytes transiently activate folate transport via folate receptors as they approach full size. Biol Reprod. 2016; biolreprod-115. doi: https://doi.org/10.1095/biolreprod.115.137687.
Steegers-Theunissen RP, Twigt J, Pestinger V, Sinclair KD. The periconceptional period, reproduction and long-term health of offspring: the importance of one-carbon metabolism. Hum Reprod Update. 2013;19:640–55.
Silvestris E, Cohen M, Menezo Y. Oxidative stress (OS) and DNA methylation errors in reproduction a place for a support of the one carbon cycle (1-C Cycle) before conception. Womens Health Gynecol. 2016; 2(4).
Twigt JM, Bezstarosti K, Demmers J, Lindemans J, Laven JS, Steegers-Theunissen RP. Preconception folic acid use influences the follicle fluid proteome. Eur J Clin Investig. 2015;45:833–41.
Laanpere M, Altmäe S, Stavreus-Evers A, Nilsson TK, Yngve A, Salumets A. Folate-mediated one-carbon metabolism and its effect on female fertility and pregnancy viability. Nutr Rev. 2010;68:99–113.
Gaskins AJ, Rich-Edwards JW, Hauser R, Williams PL, Gillman MW, Ginsburg ES, et al. Maternal prepregnancy folate intake and risk of spontaneous abortion and stillbirth. Obstet Gynecol. 2014;124:23–31.
Hodgetts VA, Morris RK, Francis A, Gardosi J, Ismail KM. Effectiveness of folic acid supplementation in pregnancy on reducing the risk of small-for-gestational age neonates: a population study, systematic review, and meta-analysis. Br J Obstet Gynecol. 2015;122:478–90.
Zheng JS, Guan Y, Zhao Y, Zhao W, Tang X, Chen H, et al. Pre-conceptional intake of folic acid supplements is inversely associated with risk of preterm birth and small-for-gestational-age birth: a prospective cohort study. Br J Nutr. 2016;115:509–16.
De-Regil LM, Fernández-Gaxiola AC, Dowswell T, Peña-Rosas JP. Effects and safety of periconceptional folate supplementation for preventing birth defects. Cochrane Libr 2010.
Cueto HT, Riis AH, Hatch EE, Wise LA, Rothman KJ, Sørensen HT, et al. Folic acid supplementation and fecundability: a Danish prospective cohort study. Eur J Clin Nutr. 2016;70:66–71.
Parisi F, Rousian M, Koning AH, Willemsen SP, Cetin I, Steegers EA, et al. Periconceptional maternal biomarkers of one-carbon metabolism and embryonic growth trajectories: the Rotterdam Periconceptional Cohort (Predict Study). Fertil Steril. 2017;107:691–698.e1.
Parisi F, Rousian M, Koning AH, Willemsen SP, Cetin I, Steegers-Theunissen RP. Periconceptional maternal one-carbon biomarkers are associated with embryonic development according to the Carnegie stages. Hum Reprod 2017; doi: https://doi.org/10.1093/humrep/dew349.
Wildenschild C, Riis AH, Ehrenstein V, Hatch EE, Wise LA, Rothman KJ, et al. A prospective cohort study of a woman’s own gestational age and her fecundability. Hum Reprod. 2015;30:947–56.
Surén P, Roth C, Bresnahan M, Haugen M, Hornig M, Hirtz D, et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. J Am Med Assn. 2013;309:570–7.
Erdei C, Dammann O. The perfect storm: preterm birth, neurodevelopmental mechanisms, and autism causation. Perspect Biol Med. 2014;57:470–81.
Ménézo Y, Mares P, Cohen M, Brack M, Viville S, Autism EK. Imprinting and epigenetic disorders: a metabolic syndrome linked to anomalies in homocysteine recycling starting in early life? J Assist Reprod Genet. 2011;28:1143–5.
Abu-Saad K, Fraser D. Maternal nutrition and birth outcomes. Epidemiol Rev. 2010;32:5–25.
Bjørke-Monsen AL, Ulvik A, Nilsen RM, Midttun Ø, Roth C, Magnus P, et al. Impact of pre-pregnancy BMI on B vitamin and inflammatory status in early pregnancy: an observational cohort study. Nutrients. 2016;8:776.
Radulescu L, Munteanu O, Popa F, Cirstoiu M. The implications and consequences of maternal obesity on fetal intrauterine growth restriction. J Med Life. 2013;6:292–8.
Stothard KJ, Tennant PW, Bell R, Rankin J. Maternal overweight and obesity and the risk of congenital anomalies: a systematic review and meta-analysis. J Am Med Assn. 2009;301:636–50.
Zwink N, Jenetzky E, Brenner H. Parental risk factors and anorectal malformations: systematic review and meta-analysis. Orphanet J Rare Dis. 2011;6:25.
Macumber I, Schwartz S, Leca N. Maternal obesity is associated with congenital anomalies of the kidney and urinary tract in offspring. Pediatr Nephrol. 2016;17:1–8.
Khairy M, Rajkhowa M. Effect of obesity on assisted reproductive treatment outcomes and its management: a literature review. Obstet Gynaecol. 2017;19:47–54.
Poston L, Caleyachetty R, Cnattingius S, Corvalán C, Uauy R, Herring S, et al. Preconceptional and maternal obesity: epidemiology and health consequences. Lancet Diabetes Endocrinol. 2016;4:1025–36.
Mulder EJ, De Medina PR, Huizink AC, Van den Bergh BR, Buitelaar JK, Visser GH. Prenatal maternal stress: effects on pregnancy and the (unborn) child. Early Hum Dev. 2002;70:3–14.
Rivkees SA, Wendler CC. Long-term consequences of disrupting adenosine signaling during embryonic development. Mol Aspects Med. 2017;55:110–7. https://doi.org/10.1016/j.mam.2017.02.001.
Cao-Lei L, De Rooij SR, King S, Matthews SG, Metz GA, Roseboom TJ, et al. Prenatal stress and epigenetics. Neurosci Biobehav Rev. 2017;7634(16):30726–6. https://doi.org/10.1016/jneubiorev201705.016.
Hedman ÅK, Mendelson MM, Marioni RE, Gustafsson S, Joehanes R, Irvin MR, Zhi D, Sandling JK, Yao C, Liu C, Liang L. Epigenetic patterns in blood associated with lipid traits predict incident coronary heart disease events and are enriched for results from genome-wide association studies. Circ Cardiovasc Genet. 2017; 10(1). pii: e001487. doi: https://doi.org/10.1161/CIRCGENETICS.116.001487.
Volkov P, Bacos K, Ofori JK, Esguerra JL, Eliasson L, Rönn T, et al. Whole-genome bisulfite sequencing of human pancreatic islets reveals novel differentially methylated regions in type 2 diabetes pathogenesis. Diabetes. 2017;66:1074–85.
Joubert BR, Herman T, Felix JF, Bohlin J, Ligthart S, Beckett E, et al. Maternal plasma folate impacts differential DNA methylation in an epigenome-wide meta-analysis of newborns. Nat Commun. 2016;7:10577.
Stein Z, Susser M. Fertility, Fecundity, famine: food rations in the Dutch famine 1944/5 have a causal relation to fertility, and probably to fecundity. Hum Biol. 1975;47:131–54.
Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 2014;5:5592. https://doi.org/10.1038/ncomms6592.
Moore SE. Early life nutritional programming of health and disease in the Gambia. J Dev Orig Health Dis. 2016;7:123–31.
Gielen M, Lindsey PJ, Derom C, Loos RJ, Souren NY, Paulussen AD, et al. Twin-specific intrauterine “growth” charts based on cross-sectional birthweight data. Twin Res Hum Genet. 2008;11:224–35.
Breathnach FM, Malone FD. Fetal growth disorders in twin gestations. Sem Perinatol. 2012;36(3):75–181.
Ancel PY, Goffinet F, Kuhn P, Langer B, Matis J, Hernandorena X, et al. Survival and morbidity of preterm children born at 22 through 34 weeks’ gestation in France in 2011: results of the EPIPAGE-2 cohort study. JAMA Pediatr. 2015;169:230–8.
Ellison PT. Evolutionary perspectives on the fetal origins hypothesis. Am J Hum Biol. 2005;17:113–8.
Khoury MJ, Erickson JD, Cordero JF, McCarthy BJ. Congenital malformations and intrauterine growth retardation: a population study. Pediatr. 1988;82:83–90.
Calzolari E, Barisic I, Loane M, Morris J, Wellesley D, Dolk H, et al. Epidemiology of multiple congenital anomalies in Europe: a EUROCAT population-based registry study. Birth Defects Res A. 2014;100:270–6.
Pharoah PO, Glinianaia SV, Rankin J. Congenital anomalies in multiple births after early loss of a conceptus. Hum Reprod. 2008;24:726–31.
D’Antonio F, Khalil A, Dias T, Thilaganathan B. the Southwest Thames Obstetric Research Collaborative (STORK). Weight discordance and perinatal mortality in twins: analysis of the Southwest Thames Obstetric Research Collaborative (STORK) multiple pregnancy cohort. Ultrasound Obstet Gynecol. 2013;41:643–8.
Cheong JN, Wlodek ME, Moritz KM, Cuffe JS. Programming of maternal and offspring disease: impact of growth restriction, fetal sex and transmission across generations. J Physiol. 2016;594:4727–40.
Tanz LJ, Stuart JJ, Williams PL, Rimm EB, Missmer SA, Rexrode KM, et al. Preterm delivery and maternal cardiovascular disease in young and middle-aged adult women. Circulation. 2017;135:578–89.
Ly L, Chan D, Aarabi M, Landry M, Behan NA, MacFarlane AJ, et al. Intergenerational impact of paternal lifetime exposures to both folic acid deficiency and supplementation on reproductive outcomes and imprinted gene methylation. Mol Hum Reprod. 2017;23:461–77.
Sales VM, Ferguson-Smith AC, Patti ME. Epigenetic mechanisms of transmission of metabolic disease across generations. Cell Metab. 2017;25:559–71.
Serpeloni F, Radtke K, De Assis SG, Henning F, Nätt D, Elbert T. Grandmaternal stress during pregnancy and DNA methylation of the third generation: an epigenome-wide association study. Transl Psychiatry. 2017;7(8):e1202. https://doi.org/10.1038/tp.2017.153.
Huntriss J, Woodfine K, Huddleston JE, Murrell A, Picton HM. Analysis of DNA methylation patterns in single blastocysts by pyrosequencing®. Methods Mol Biol. 2015;1315:259–70.
Ghosh J, Coutifaris C, Sapienza C, Mainigi M. Global DNA methylation levels are altered by modifiable clinical manipulations in assisted reproductive technologies. Clin Epigenetics. 2017;9:14.
Author information
Authors and Affiliations
Corresponding author
Additional information
The original version of this article was revised: a modification has been made to the article title.
Rights and permissions
About this article

Cite this article
Lubinsky, M. An epigenetic association of malformations, adverse reproductive outcomes, and fetal origins hypothesis related effects. J Assist Reprod Genet 35, 953–964 (2018). https://doi.org/10.1007/s10815-018-1197-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-018-1197-2