
Abstract
Lysophosphatidylethanolamine acyltransferase (LPEAT) plays an important role in acyl remodeling of phospholipid via the Lands’ cycle, and consequently alters fatty acid compositions in triacylglycerol (TAG). In the present study, MiLPEAT was cloned from an arachidonic acid (ArA)-rich green microalga Myrmecia incisa. Its full-length cDNA was 1,303 bp containing a 981-bp open reading frame that encoded a 326-amino acid protein. Comparing the cDNA to its corresponding cloned DNA sequence showed that MiLPEAT possessed 6 introns. Bioinformatics analysis of LPEAT indicated that a phosphate acyltransferase domain, PlsC, consisting of 4 typical motifs, NH(x)4D, GCxYVxR, FPEGT, and PVxPVx, was present at the C-terminus of MiLPEAT while two (at least one) transmembrane domains at the N-terminus. The cDNA corresponding to C-terminal 224-residues was thereby subcloned into the vectors pET-28a and pMAL-c2X independently for production of recombinant MiLPEAT (rMiLPEAT). The purified soluble rMiLPEAT fused with maltose-binding protein was used for enzyme assay, and thin-layer chromatography profiles of the catalytic products demonstrated that rMiLPEAT could acylate lysophosphatidylethanolamine to phosphatidylethanolamine, thus functionally identifying MiLPEAT. Anti-MiLPEAT polyclonal antibody was generated against the purified rMiLPEAT fused with 6×His tag. Immuno-electron microscopic results with this polyclonal antibody illustrated that MiLPEAT was localized on M. incisa plasma membrane, and this was further supported by immunocytochemical observations. A di-lysine motif present at the C-terminus implying that MiLPEAT was an endoplasmic reticulum resident, how MiLPEAT was transported to the microalgal plasma membrane was therefore discussed. This study will lay a foundation to understand that how M. incisa uses ArA to synthesize TAG.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Myrmecia incisa Reisigl H4301 is a unicellular microalga, which belongs to the class Trebouxiophyceae in Chlorophyta (Reisigl 1964). Its cells are spherical or oval in shape, and they are often gathered together to form cell aggregates similar to a non-coenobium. The most impressive characteristic of this microalga is the accumulated arachidonic acid (20:4Δ5,8,11,14, ArA) (Ouyang et al. 2013b), an essential polyunsaturated fatty acid (PUFA) in human growth and development despite some side effects in nutrition. While under the stress of nitrogen starvation for growth, the content of ArA can reach nearly 7% of cell dry weight of this microalga (Ouyang et al. 2013b), suggesting that this microalga is a representative organism with high ArA content and hence has a potential development and utilization value as suggested by Bigogno et al. (2002). Using gas chromatography (GC)-mass spectrometry (MS) to detect, Ouyang et al. (2016b) calculated that ArA accounted for 25.23% of total fatty acids in this microalga whereas it accounted for 68.0% of the total fatty acids in the extracted triacylglycerol (TAG) from this microalga by thin-layer chromatography (TLC)(Supplementary Table 1). Once the microalga was cultured under the nitrogen starvation for 4 days, the percentage of ArA continued to increase and reached 75.6% of the total fatty acids in TAG (Ouyang et al. 2016b). This percentage is comparable to that of α-eleostearic acid (Δ9,11trans, 13trans-octadecantaenoic acid, 18:3Δ9,11t,13t, 77.2%) in Vernicia fordii seed oil (Zhang et al. 2014) but slightly lower than that of ricinoleic acid (12-hydroxy-Δ9-octadecanedenoic acid, 12OH 18:1Δ9, 89%) in Ricinus communis seed oil (Ogunniyi 2006). Similar to these unusual fatty acids produced in higher plants as reviewed by Napier (2007), how ArA is used for TAG biosynthesis in M. incisa is of interest to be investigated.
As suggested by Bates (2016), flux of various acyl groups to the fatty acid modification was a key factor to affect the biosynthesis of TAG with unusual fatty acids. Acyl modifications, for example, the extra-plastidial desaturation reactions of fatty acids, occurred at sn-2 glycerol backbone of phospholipids such as phosphatidylcholine (PC) and phosphatidylethanolamine (PE)(Stymne and Appelqvis 1978; Sperling et al. 1993), whereas the chain elongation ones of fatty acids to long-chain PUFAs occurred in acyl-CoA pool of endoplasmic reticulum (ER)(Dyer et al. 2008; Bates 2016). As a result, the deacylation and acylation reaction process, principally the Lands’ cycle (Lands 1958), should center round the phospholipids, for example, PC and PE. That is to say, under catalysis of hydrolytic enzymes such as phospholipase A2 (EC 3.1.1.4), the de novo PC and PE will be deacylated at their sn-2 positions to lysophosphatidylcholine (LPC) and lysophosphatidylethanolamine (LPE), respectively (Bates 2016). The generated LPE will be acylated by acyl-CoA: lysophosphatidylethanolamine acyltransferase (LPEAT, EC 2.3.1.n7). In this way, the special fatty acids such as long-chain PUFAs from the acyl-CoA pool might be esterified to the sn-2 hydroxyl of LPE to form a modified PE. Subsequently, the long-chain PUFAs in the modified PE could be transferred as an acyl donor to DAG by catalysis of phospholipid: DAG acyltransferase (PDAT, EC 2.3.1.158) (Liu et al. 2016) to synthesize TAG along an acyl-CoA-independent route (Napier 2007).
From the abovementioned analysis of acyl remodeling, it is inferred that LPEAT plays a very important role in the asymmetry and diversity of fatty acid composition of membrane phospholipids as reviewed by Shindou et al. (2013), thus resulting in the introduction of long-chain PUFAs and other special fatty acids in TAG as reviewed by Napier (2007). Up to date, most reports about LPEAT have been focused on Escherichia coli(Jackowski et al. 1994), Saccharomyces cerevisiae Riekhof et al. (2007), and animals including mammals such as human being as reviewed by both Hishikawa et al. (2014) and Yamashita et al. (2014). On the contrary, few reports on LPEAT of higher plants such as Arabidopsis thaliana (Stålberg et al. 2009; Jasieniecka-Gazarkiewicz et al. 2016, 2017, 2021) and Camelina sativa (Klińska et al. 2020, 2021) have been documented. In contrast to this, there is limited molecular information and subcellular localization about the LPEAT of cyanobacteria (Weier et al. 2005) and eukaryotic algae.
Based on the transcriptome data of M. incisa(Ouyang et al. 2013a) and BLASTx searching result from the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/) website, contig 8647 was found to have 57% identity with lysophosphatidic acid acyltransferase (LPAAT, EC 2.3.1.51) (GenBank accession No. XP_005646810) of Coccomyxa subellipsoidea C-169. In the present study, primers were designed on the basis of this contig sequence for full-length cDNA cloning of this gene from M. incisa by rapid amplification of cDNA ends (RACE). BLASTp searching result from NCBI website showed that the amino acid sequence as encoded by this cloned cDNA was homologous to LPEAT, so it was designated as MiLPEAT. In order to determine the subcellular location by immunogold electron microscopy, anti-MiLPEAT polyclonal antibody was prepared against the heterologously expressed MiLPEAT in E. coli. The plasmalemma-located MiLPEAT was further supported by immunocytochemical observation using this prepared antibody. TLC of products from in vitro reaction system with soluble recombinant MiLPEAT identified that MiLPEAT was able to acylate LPE to PE. This study will lay a foundation to understand that how M. incisa uses ArA to synthesize TAG and that how to enhance target PUFA in the storage TAG of this microalga or other related green ones.
Materials and methods
Algal species and culture conditions
Myrmecia incisa Reisigl H4301 was obtained from the Culture Collection of Algae of Charles University of Prague via Prof. C.-W. Zhang at Ji’nan University (Guangzhou, China). This microalga was inoculated in flasks containing BG-11 medium (Stanier et al. 1971), and the flasks were placed in a 25°C chamber for culture under the irradiance of 115 μmol quanta m-2s-1 with a light: dark regime of 12 h: 12 h (Yu et al. 2012). During the culture, the flasks were shaken several times per day at a regular interval. Algal cells were harvested at the late logarithmic growth phase (approximately 14 days) by centrifugation at 5,500 rpm for 10 min and washed three times with sterilized distilled H2O.
Nucleic acid extraction and cDNA synthesis
The freshly harvested M. incisa cells were ground into powder in liquid nitrogen. Genomic DNA was extracted according to the modified cetyltrimethyl ammonium bromide (CTAB) method as described previously (Hu and Zhou 2001). Extraction of total RNA was performed using TRIzol reagent according to the manufacturer’s instructions (Invitrogen, USA). Complementary DNA (cDNA) used as template for gene cloning was synthesized from the extracted total RNA using a PrimeScript RT Reagent Kit (TaKaRa, Japan). Complementary DNA used for 5′ RACE and 3′ RACE was synthesized using a SMART RACE cDNA kit (Clontech, USA) following the manufacturer’s protocol.
DNA and full-length cDNA cloning of LPEAT
From the transcriptome data of M. incisa(Ouyang et al. 2013a), contig 8647 was searched since it was 57% identity to LPAAT of Coccomyxa subellipsoideaC-169 (GenBank accession No. XP_005646810). According to this sequence, one pair of primers, 8647F and 8647R (Table 1), was online designed using the Primer3web server v. 4.1.0 (https://bioinfo.ut.ee/primer3/) for this contig calibration. Twenty five-microliter reaction volume contained 1 μL the synthesized cDNA, 12.5 μL 2×Pfu PCR MasterMix (Tiangen Biotech, China), 0.5 μL each forward and reverse primers 8647F and 8647R (10 μM) and 10.5 μL distilled deionized (dd) H2O. Polymerase chain reaction (PCR) was performed in a gradient Mastercycler (Eppendorf, Germany) and programmed as follows: 1 cycle of 5 min pre-denaturation at 94°C, then 30 cycles of 45 s denaturation at 94°C, 45 s annealing at 68°C, and 30 s extension at 72°C, and followed by 1 cycle of 10 min extension at 72°C.
The amplified product was recovered using an agarose gel purification and extraction kit (Aidlab, Beijing, China) and was ligated to pMD19-T vector (TaKaRa). The constructed vector was subsequently transformed into Escherichia coli DH5α competent cells (TaKaRa) by heat shock method (Hanahan 1983). Then, the liquid transformed bacteria was evenly spread on solid Luria-Bertani(LB) medium containing 100 μg mL-1 ampicillin, 20 μg mL-1 5-bromo-4-chloro-3-indol β-D-galactopyranoside (X-gal), and 50 μg mL-1isopropyl-β-D-1-thiogalactoside(IPTG) for blue-white screening. White or positive clones were selected for verification by PCR with the general primers RV-M and M13-20(TaKaRa) and sent to Sangon Biotech (Shanghai, China) for sequencing analysis using an automated DNA sequencer (ABI Prism 3730, USA).
According to the verified sequence of contig 8647, four gene-specific primers, 5′RACE1, 5′RACE1N, 3′RACE1, and 3′RACE1N, were designed for the 5′- and 3′-RACE PCR reactions. Of these primers, 5′RACE1N and 3′RACE1N were used in the second round of nested PCR reactions. The obtained 5′- and 3′-RACE ends were recovered and ligated to pMD19-T (TaKaRa), and the constructed vector was transformed separately into E. coli DH5α competent cells (TaKaRa) as described above. Afterwards, the positive clones were sent to Sangon Biotech (Shanghai) for sequencing. In combination with the sequenced 5′- and 3′-RACE ends and the verified contig 8647 fragment, full-length cDNA of MiLPEAT was assembled. The cDNA sequence was confirmed by PCR amplification using the re-designed primers, 1cDNAF and 1cDNAR (Table 1).
On the basis of the verified full-length cDNA sequence of MiLPEAT, three pairs of primers, DNA1F and DNA1R, DNA2F and DNA2R, and DNA3F and DNA3R (Table 1), were designed for DNA cloning of this gene by PCR with the extracted genomic DNA as template. Twenty five-μL reaction volume contained 1 μL genomic DNA, 12.5 μL 2×Pfu PCR MasterMix (Tiangen Biotech), 1 μL each forward and reverse primers and 9.5 μL ddH2O. PCR was programmed as pre-denaturation at 94°C for 3 min, and then pre-denaturation at 94°C for 45 s, annealing at the provided temperature (Table 1) for 45 s, and extension at 72°C for 2 min for 30 cycles, and finally extension at 72°C for 10 min. Upon obtaining the sequenced target products, DNA sequence of MiLPEAT was assembled to bring gene structure to light by comparing its corresponding cDNA using Splign program embedded in NCBI website (https://www.ncbi.nlm.nih.gov/sutils/splign/splign.cgi).
Bioinformatics and phylogeny analysis
ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/) was used to predict open reading frame (ORF) of MiLPEAT. The deduced amino acids from this gene were translated by the Primer Premier 5 (Premier Biosoft, San Francisco, USA) software. Isoelectric point (pI), molecular weight (MW), transmembrane region, hydrophobicity, signal peptide, transit peptide, structure domain, functional site, and secondary structure of this putative MiLPEAT were predicted and shown using the listed websites or servers in Supplementary Table 2.
Protein sequences of LPEAT were retrieved from NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi), and phylogenetic inference was constructed using MEGA 6.0 program (Tamura et al. 2013) by neighbor-joining(NJ) algorithm based on the amino acid sequences of LPEAT. Homologous sequences of LPEAT from different organisms were aligned by the Clustal_X software (Thompson et al. 1997) with default parameter settings.
Construction of prokaryotic expression vectors
Two prokaryotic expression plasmids, pET-28a(TaKaRa) and pMAL-c2X (Addgene, USA), were used in the present study for preparation of polyclonal antibody and assay of enzyme activity, respectively. The MiLPEAT ORF without its corresponding nucleotide sequence of 102 amino acids, which contained two predicted transmembrane regions from the N terminus on, was cloned with the pair of primers QIE1F and QIE1R or QIE2F and QIE2R, respectively (Table 1). The PCR reaction and program was the same to DNA cloning of MiLPEAT, except for the primers and the annealing temperature (Table 1). The amplified target product was recovered and ligated to pMD19-T vector (TaKaRa), and the resultant constructs were transformed to E. coli DH5α and sequenced as early described. The transformants harboring the correct orientation and reading frame of MiLPEAT were used for extraction of cloning plasmids. Both the extracted plasmids and the empty vectors pET-28a or pMAL-c2X were separately digested by two restriction endonucleases EcoRI and NotI or EcoRI and HindIII, respectively. The linearized target products were ligated to generate the prokaryotic expression plasmids pET28a-MiLPEAT and pMALc2X-MiLPEAT. The resulting constructs were transformed to E. coli DH5α for proliferation and sequence verification, and then transformed to E. coli BL21 for heterologous expression of MiLPEAT.
Expression and detection of recombinant MiLPEAT
Induced expression and extraction of recombinant MiLPEAT (rMiLPEAT) was performed as described by Bi et al. (2021). In brief, the transformed E. coli harboring the recombinant plasmid pET28a-MiLPEAT was cultured in liquid LB medium supplemented with 50 μg mL-1 kanamycin (Kan) on a 180-rpm orbital shaker at 37°C. Once optical density (OD) at 600 nm of the culture reached 0.6, IPTG was added to a final concentration of 1 mM. The culture was allowed to grow at 37°C for another 4 h and then harvested by centrifugation at 6,000 rpm for 5 min at 4°C. The collected transformants were washed and re-suspended in 1×phosphate-buffered saline (PBS) (0.137 M NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4). The re-suspended solution was treated by sonication using Scientz-IID Ultrasonic Homogenizer (Ningbo, China) at 4°C. Then, the supernatant and pellet fractions were separated by centrifugation at 12,000 rpm for 15 min at 4°C. Proteins in the both fractions, of the pellet which was added with the supernatant equal volume of 1×PBS, were quantified by a ND-2000c spectrophotometer (NanoDrop, USA) using the Bradford (1976) method. The proteins were fractioned by denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) according to Laemmli (1970). The transformed E. coli with empty pET-28a vector were taken as a negative control throughout this experiment.
Preparation of anti-MiLPEAT polyclonal antibody
Recombinant MiLPEAT expressed in inclusion bodies was extracted from the transformed E. coli carrying the construct pET28a-MiLPEAT. The crude extracts were solved in 8 M urea and then loaded onto Bio-Scale Mini Profinity IMAC Cartridges nickel columns (Bio-Rad, USA) for affinity chromatography purification of rMiLPEAT using the fused 6×His tag. The rMiLPEAT was eluted with different designed concentrations, i.e. 10 mM, 20 mM, 200 mM, and 500 mM, of imidazole, following the manufacturer’s protocol and the eluted fraction containing target product was collected after checking by SDS-PAGE.
The affinity-purified rMiLPEAT was dialyzed against decreased-gradients of urea from 4 M, 2 M, 1 M, to 0 M in a 20-mM Tis-HCl (pH 8.0) solution for 12 h each dialysis. Afterwards, the purified and dialyzed rMiLPEAT was employed to immunize New Zealand white rabbits. After three immunizations, antiserum was collected from the immunized rabbits for the preparation of anti-MiLPEAT antibody. The polyclonal antibody was purified using a CNBr-activated Sephrose 4B antigen affinity chromatography column (GE Healthcare, Sweden) following the manufacturer’s protocols. Antibody titer was determined by indirect ELISA detection. Specificity or quality of the purified anti-MiLPEAT polyclonal antibody was checked by Western blot analysis using the purified rMiLPEAT as well as crude proteins extracted from M. incisa using RIPA buffer (Thermo Scientific, USA) containing 1 mM phenylmethanesulfonyl fluoride as a protease inhibitor.
Western blot analysis
After SDS-PAGE, proteins on the gel were transferred electronically to a nitrocellulose membrane for Western blot analysis, which was performed as described in detail by Ye et al. (2014). Recombinant MiLPEAT heterologously expressed in E. coli and endogenous MiLPEAT in M. incisa were immuno-blotted with the commercially supplied either anti-6×His tag monoclonal antibody (Youke Biotech, China) or anti maltose-binding protein (MBP) monoclonal antibody (New England Biolabs, USA), and the purified anti-MiLPEAT polyclonal antibody, respectively. Immuno-blotting signal was amplified with horseradish peroxidase (HRP)-conjugated AffiniPure goat anti-rabbit IgG (Youke Biotech) as secondary antibody. Color reaction was visualized with a Diaminobenzidine (DAB) HRP Color Development Kit (Tiangen Biotech) following the manufacturer’s instructions. Western blot analysis was performed with gradient dilutions of an antibody until the blotting signals were strong but less background.
Immunoprecipitation of microalgal MiLPEAT
Five microliters of the purified anti-rMiLPEAT polyclonal antibody and 150 μL of crude extracts containing endogenous MiLPEAT from M. incisa were mixed with 200 μL of immunoprecipitation (IP) lysis and wash buffer (Pierce Classic IP Kit, Thermo Scientific) for overnight incubation at 4°C to form immune complex. At the same time, 20 μL protein A/G resin slurry was pipetted into a spin column and centrifuged at 1,000 rpm for 1 min. Subsequently, the antibody:antigen complexes were loaded to the spin column, and the non-target proteins was eluted and discarded according to the manufacturer’s instructions.
Approximately 50 μL of 2×non-reducing lane marker sample buffer containing 20 mM dithiothreitol (DTT) was added to the column and incubated at 100°C for 10 min. The prepared samples were then fractioned by the early described SDS-PAGE. The gel was stained with Coomassie brilliant blue R250 and the target band gel was excised and sent to Bioprofile Technology Co., Ltd. (Shanghai, China) for MS analysis.
MS analysis
Prior to MS analysis, the band gel was cut into pieces and treated according to the described procedures by Kussmann and Roepstorff (2000). Afterwards, 100 μL of trypsin (20 ng⋅μL-1) was added into the gel pieces followed by incubation overnight at 37°C for proteolytic cleavage. Protein digestion was quenched by adding 10% trifluoroacetic acid to adjust pH to 2. The supernatant was pipetted to a new Eppendorf tube, while the pellet was mixed with 100 μL of 60% acetonitrile containing 0.1% trifluoroacetic acid and sonicated for further extraction thrice. The supernatants were pooled, desalted by C18 Stage Tip column (Thermo Scientific), and lyophilized. The specimen was re-dissolved in appropriate volume of 0.1% formic acid.
An Easy-nLC 1200 system (Thermo Scientific) was used to analyze the digested peptide sequences. An aliquot of sample was loaded onto a trap column (100 μm×20 mm, 5 μm particle size, C18, Dr. Maisch GmbH, Germany) and washed for 6 min with gradient with Solvent A (0.1% formic acid) and Solvent B (water:acetonitrile=2:98, v/v, 0.1% formic acid) at a flow rate of 5 μL min-1. The peptides were separated on a capillary column (75 μm×150 mm, 3.5 μm particle size, C18, Dr. Maisch GmbH) at a flow rate of 300 nL min-1 with gradient at 2-35% Solvent B over 30 min, then from 35 to 90% over 10 min, followed by 90% Solvent B for 5 min, and finally 5% Solvent B for 15 min.
The separated peptides were analyzed using a Q Exactive Plus Orbitrap mass spectrometer (Thermo Scientific) in data-dependent acquisition mode. Full scan spectra are acquired at scan time of 0.3 s followed by MS/MS spectra acquisition at scan time of 0.1 s. Total run time was 60 min. The top 10 most intensity precursor ions were selected to perform MS/MS acquisition with the following parameters: resolution = 70,000 at 200 m/z, automatic gain control (AGC) targeted 1×106, maximum injection time = 50 ms, scan range from 300 to 1800 m/z, and spectrum data = profile. Detector settings for dd-MS2 of the product ions were as follows: micro scans = 1, resolution = 17,500 at 200 m/z, AGC targeted 1×105, maximum injection time = 50 ms, MS2 activation type = higher energy collision dissociation (HCD), isolation window = 2.0 Th, normalized collision energy = 27, and spectrum data = profile.
The raw tandem mass spectra were visualized and processed with the Xcalibur 3.0 package (Thermo Scientific). Peptide identification was performed by correlating the acquired experimental MS/MS spectra with theoretical spectra or searching them against protein sequence databases, for example, from the NCBI website, following the guidelines laid out by Nesvizhskii (2007). Only the best scoring peptide to spectrum match for each MS/MS spectrum was considered the potential peptide identification as suggested by Nesvizhskii (2007).
Immuno-electron microscopy
Subcellular localization of this MiLPEAT in M. incisa was investigated using immunogold electron microscopy as described by Ye et al. (2014). In brief, freshly harvested M. incisa cells were fixed with 1:1 ratio of 8% (w/v) paraformaldehyde to 2% glutaraldehyde in sterilized deionized H2O. The fixed specimens were dehydrated in ethanol:acetone series from 30% through 100% of ethanol, and then were infiltrated and embedded in epoxy resin E51 (Lingteng, China) as described by Ouyang et al. (2012). The specimen was cut into 70-nm thick thin-sections using a LKB Ultrotome 4802 ultracut microtome (Leica Microsystems, Germany).
The ultrathin sections used for immuno-electron microscopic observation were collected on 200-mesh nickel grids (SPI, USA) with a non-carbon coated Formvar-supporting film. The nickel grids carrying ultrathin sections were pre-incubated with 50 mM glycine, followed by etching with 1% (w/v) sodium metaperiodate dissolved in 1×PBS (Bendayan and Zollinger 1983). The sections were blocked by adding 5% (w/v) bovine serum albumin (BSA) dissolved in 1×PBS, and then incubated primarily with the purified anti-MiLPEAT polyclonal antibody at 4°C for 48 h or without this antibody as a negative control. After washes in 1×PBS and another 20-min incubation with 1% (w/v) BSA, the grids were incubated with the secondary antibody, anti-rabbit IgG conjugated to 10 nm gold particles (Sigma, USA) at room temperature for 2 h. Followed by sequential washes in 1×PBS thrice (15 min each time) and dehydration in air, the sections were observed in a Tecnai G2 Sprit BioTWIN transmission electron microscope (FEI, USA) at 80 kV.
In total, 9 images were taken for gold particle counts and density estimation. Numbers of gold particles in the region containing cell wall and plasma membrane, i.e. periplasmic space, as well as the remaining area of a cell, were calculated in each image. The percentages of particles that were present in periplasmic space and in the remaining area of the cell were easily calculated. In addition, labeling density was defined as the number of gold particles per area unit (μm2) as described by Bernal et al. (2007), and the area was estimated using the Adobe Photoshop software (ver. 3.0). The statistical analysis of subcellular distribution of MiLPEAT was carried out using the independent sample test in SPSS Statistics v. 22 program (IBM Corp., USA). Statistical significance of difference was tested using Student’s t test and indicated as P < 0.05 (*) or P < 0.01 (**).
Immuno-light microscopy
To further understand whether MiLPEAT located on plasma membranes or cell walls of M. incisa, plasmolyzed microalgal cells were used for immunocytochemical observations. Approximately 30 mL of microalgal culture was centrifuged at 3,000 rpm for 5 min at 4°C and washed with ddH2O thrice in the same way. The harvested microalgal cells were immersed in a 0.6 g mL-1 sucrose solution until the cytoplasm separated noticeably from cell walls. The plasmolyzed cells were harvested and fixed with 0.2% (w/v) paraformaldehyde and 0.5% glutaraldehyde dissolved in 1×PBS overnight at 4°C. The fixed samples were washed 3 times with 1 mM DTT dissolved in 1×PBS for 10 min each time. The specimens were placed in an equal volume ratio mixture of 20 g L-1 sucrose and Tissue-Tek optimal cutting temperature (OCT) compound mounting medium (Sakura Finetek Inc., USA) and soaked at room temperature for 2 h. The samples were transferred to and immersed in a new OCT compound mounting medium for 4 h at 4°C, while the medium was replaced once. Six hours later, the samples were stored in a sealed container at -80°C after plunging into liquid nitrogen.
The frozen specimens were cryostat-sectioned into 5-μm thick slices using a Leica CM1520 microtome (Leica Microsystems). Subsequently, the semi-ultrathin sections were mounted on poly-l-lysine coated slides and oven dried for 1 h at 37°C. The fixed sections were rinsed three times with 1×PBS for 10 min each time to remove the fixative solution and incubated with pre-cooled anhydrous methanol at -20°C for 5 min to eliminate endogenous peroxidase activity. The slides were incubated at 94°C for 20 min in the mixture of 0.1 M Tris and 10 mM EDTA at pH 9.0 for antigen retrieval (Krenacs et al. 2010). The specimens were blocked for 1 h by adding 2% (w/v) BSA dissolved in 1×PBS, and then incubated overnight primarily with the purified anti-MiLPEAT polyclonal antibody at 4°C or without this antibody as a negative control.
After rinsing thoroughly with 1×PBS, the specimens were incubated with the secondary antibody goat F(ab′)2 anti-rabbit IgG-H&L (Biotin, Abcam, Cambridge, UK) at room temperature for 1 h. Followed by washing in 1×PBS, color reaction was visualized with a rabbit specific HRP/DAB(ABC) Detection IHC Kit (Abcam) following the manufacturer’s instructions. Images were taken with a Leica DM4000 B LED epifluorescent microscope (Wetzlar, Germany) equipped with a different filter set and a digital camera.
Purification of soluble rMiLPEAT
Amylose resin (New England Biolabs) was used as an affinity matrix for purification of rMiLPEAT fused to MBP from E. coli harboring the constructed plasmid pMALc2X-MiLPEAT. While the bacterial cells were cultured in LB medium containing 0.1 mg⋅mL-1 ampicillin till the value of OD600 up to 0.5, target protein was induced to express by adding IPTG to a final concentration of 0.3 mM. Crude proteins were extracted from these cells by centrifugation at 12,000 rpm for 30 min at 4°C after the aforementioned sonication. The supernatant (crude extract) was loaded to an affinity chromatograph column filled with the Amylose resin, and the fusion MiLPEAT was eluted with column buffer consisting of 0.02 M Tris-HCl (pH 7.4), 1 mM EDTA, 0.2 M NaCl, and 10 mM β-mercaptoethanol plus 100 mM maltose. The target protein was collected and detected by SDS-PAGE and Western blot analysis as early described.
Qualitative assay of rMiLPEAT activity by TLC
Stålberg et al. (2009) documented that Arabidopsis thaliana LPEAT preferred both palmitoyl (16:0)-CoA as an acyl donor and 1-oleoyl-2-hydroxy-sn-glycero-3-phosphoethanolamine (18:1-LPE) as an acyl acceptor. These two chemicals were hence chosen in the present study as reactants to establish in vitro reaction system for enzyme activity detection of the fusion rMiLPEAT. The reaction system was composed of 400 μL purified rMiLPEAT, 30 μL 18:1-LPE (30 μg μL-1, Sigma), 10 μL 16:0-CoA (10 μg μL-1, Sigma), 50 μL Tris-HCl (25 mM, pH 7.6), and 50 μL KCl (0.4 M). KCl was added to avoid precipitation of acyl-CoA since cations could precipitate most of palmitoyl-CoA at mM concentration (Stålberg et al. 2009). LPE was dissolved in chloroform:methanol:acetic acid (95:5:5, v/v/v). To understand the substrate preference, 18:1-LPA (30 μg μL-1, Sigma) and 18:1-LPC (30 μg μL-1, Sigma) were used separately as a reactant in the same way. The reaction was started by adding the purified rMiLPEAT, incubated at 30°C for 10 min, and stopped by adding 1.33 mL methanol:chloroform:acetic acid (50:50:1, v/v/v) and vortexing. After addition of 133 μL H2O, the mixture was vortexed, centrifuged at 5,500 rpm for 15 min, and the lower phase was transferred to a new tube. The water phase was extracted once again by adding 660 μL chloroform. The pooled organic phases were evaporated under a stream of N2, dissolved in 20 μL chloroform, and spotted on a silica gel 60 F254TLC plate (Merck, Germany). The TLC plate was developed to 3/4 of the plate length in ethyl acetate:chloroform:dimethylcarbinol:methanol:0.25% KCl (25:25:25:10:9, v/v/v/v/v) (Olsen and Henderson 1989). After development, the TLC plate was air dried for 5 min, and sprayed by 1% primuline solution. Five minutes later, the phospholipids were identified under an ultraviolet light by comparing known standards on the same TLC plate. Both the purified products from E. coli harboring the empty plasmid pMAL-c2X and the reaction buffer instead of the purified rMiLPEAT were considered negative controls.
Results
Full-length cDNA and DNA cloning of MiLPEAT
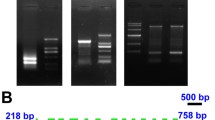
In the transcriptome data of Myrmecia incisa(Ouyang et al. 2013a), a 252-bp fragment (contig 8647) was found to be 57% identity to LPAAT of Coccomyxa subellipsoideaC-169 (GenBank accession No. XP_005646810). To calibrate the contig sequence by PCR amplification, one pair of primers 8647F and 8647R (Table 1) was designed on the basis of contig 8647 sequence. Based upon the verified sequence of contig 8647, several primers 5′RACE1, 5′RACE1N, 3′RACE1, and 3′RACE1N (Table 1) were designed to amplify 5′- and 3′-RACE end sequences of this gene by RACE technique. Amplified products after the second round of nested PCR were electrophoretically analyzed as shown by Fig. 1 (Lanes 1 and 2). Sequence analyses of the target products indicated that the 3′-RACE end sequence was 705 bp in length (Fig. 1, Lane 1) with a typical poly A tail of nuclear-coding genes in eukaryotes while the 5′-RACE one was 511 bp (Fig. 1, Lane 2).

Agarose gel electrophoretograms of PCR products for full-length cDNA and DNA cloning of MiLPEAT (A), and schematic diagram of the gene structure (B). In panel A, Lane M: DL2000 standard DNA marker (Tiangen Biotech, Beijing, China); Lane 1: amplified products of the second round of nested PCR with 3′RACE1N as a gene specific primer; Lane 2: amplified products of the second round of nested PCR with 5′RACE1N as a gene specific primer; Lane 3: PCR products with DNA1F and DNA1R as primers; Lane 4: PCR products with DNA2F and DNA2/R as primers; and Lane 5: PCR products with DNA3F and DNA3/R as primers. In panel B, grey lines represent 5′- or 3′-UTR; black boxes represent exons; black lines represent introns; black arrows denote the primers employed for gene cloning
The obtained 5′- and 3′-RACE end sequences were assembled by hand with the verified contig sequence into a new cDNA sequence and it was verified by PCR using a re-designed pair of primers 1cDNAF and 1cDNAR (Table 1) based upon the assembled sequence. The full-length cDNA of this gene was found to be 1,303 bp in length. Its ORF sequence was 981 bp, encoding 326 amino acids, in length as predicted by ORF finder. MW and pI of the gene product were 36.23 kD and 9.3, respectively, as predicted by Compute pI/MW tool (Supplementary Table 2). The 5′-untranslated region (UTR) of this gene was 129 bp in length while 3′-UTR was 193 bp (Fig. 1B).
Based upon the verified cDNA sequence, three pairs of primers, DNA1F and DNA1R, DNA2F and DNA2R, and DNA3F and DNA3R (Table 1), were designed for PCR amplification to clone DNA sequence with genomic DNA from M. incisa as template. Sequence analyses of the amplified products pointed out that the sizes of target fragments were 222 bp, 1,533 bp, and 200 bp (Fig. 1, Lanes 3, 4, and 5, respectively). After assembly and calibration by PCR, DNA sequence of this gene was obtained and it was 1,871 bp in length. Comparing the obtained DNA to its corresponding cDNA by Splign program, this gene was found to be separated by 6 introns, all of which were located in the coding region and were 80 bp, 84 bp, 84 bp, 82 bp, 138 bp, and 106 bp in length from 5′-UTR on as depicted in Fig. 1B. Sequence analyses showed that all these intron sequences complied with the “GT-AG” splicing rule.
BLASTx search result (data not shown) suggested that the protein encoded by this gene belonged to the superfamily of lysophospholipid acyltransferase (LPLAT), and it had nearly 40% identity with LPEAT from a lot of organisms. For example, it had 39.2% identity with LPEAT1 (GenBank accession No. NP_565249.1) that was functionally identified in Arabidopsis thaliana(Stålberg et al. 2009) and 43.15% identity with Setaria italica LPEAT1 (GenBank accession No. XP_004962349.1). It was worth noticed that although the BLASTx search result revealed that it had 47.43% identity with C. subellipsoideaC-169sn-glycerol-3-phosphate acyltransferase (GPAT, EC 2.3.1.15) (GenBank accession No. XP_005646810.1), the C. subellipsoidea GPAT gene was not yet identified functionally, and phylogenetic inference (see below for details) demonstrated that the obtained sequence was not clustered with GPAT. Therefore, the abovementioned gene cloned from M. incisa in this study was designated as MiLPEAT, and it was deposited in GenBank under the accession number MG558460.
Characterization of MiLPEAT gene
SMART search and InterPro analysis (Supplementary Table 2) disclosed that MiLPEAT had a typical phosphate acyltransferase domain PlsC (smart00563) that was located between 117A and 234G of its deduced amino acid sequence. In addition, MiLPEAT possessed a LPLAT_LPCAT1-like domain (cd07991) which was located between 86A and 308R, and an acyltransferase domain (Pfam01553) which was situated between 112P and 232K as searched by BLASTp. Multiple sequence alignment of MiLPEAT and several LPEATs from other organisms (Fig. 2) illustrated that MiLPEAT had four conserved motifs in this aforementioned region, and they were NH(x)4D, GCxYVxR, FPEGT, and PVxPVx as denoted in Fig. 2 whatever they were LPEAT1 or LPEAT2. This result coincided with a conclusion which was drawn by Lewin et al. (1999) from LPLAT including LPAAT and LPEAT. Since these four motifs were also found in GPAT, LPAAT, and LPEAT, the conserved amino acids in these motifs were proposed to be diagnostic for various acyltransferase reactions (Lewin et al. 1999). All these pieces of information suggested that MiLPEAT would be a phospholipid acyltransferase and its enzyme activity was determined by its carboxyl terminus rather than the amino one.

Amino acid sequence alignment of LPEATs from Myrmecia incisa and different species of higher plants. Amino acids conserved between LPEAT1 and LPEAT2 are shaded in black while they are done in grey only when they are conserved among LPEAT1 or LPEAT2. Four conserved acyltransferase motifs (motif I, NH(x)4D; motif II, GCxYVxR; motif III, FPEGT; and motif IV, PVxPVx) are illustrated. The predicted phosphate acyltransferase domain PlsC is underlined by a dotted line. Asterisks denote the residues that play a role in LPEAT catalysis. Triangles denote the residues which are important in binding the substrate lysophosphatidylethanolamine (LPE). Carboxyl-terminal dilysine (KK) motif of MiLPEAT is indicated as a box.
Secondary structure prediction by PredictProtein (Supplementary Table 2) showed that MiLPEAT contained nine α-helices. Of them, the longest α-helix was composed of 38 amino acids, and it was located between 31V and 68V of MiLPEAT which was consistent to that a transmembrane region might be generated by the residues from 37L to 63C (Supplementary Table 2) as predicted by Phobius and TMHMM Server v. 2.0. Since this α-helix was composed of longer than 20 amino acids, MiLPEAT was theoretically capable of spanning membrane as suggested by Bowie (1997). Hydrophobic amino acids in MiLPEAT account for 47.87% of the total amino acids as estimated by PredictProtein, and the hydrophobic percentage in the region between 37L and 63C increased to 62.96%. These hydrophobic amino acids were believed to make the secondary and even tertiary structure of MiLPEAT stable for subcellular localization. Accordingly, MiLPEAT had at least one transmembrane region between 37L and 63C and it would hence be an integral membrane protein. Another α-helix was located between 88A and 104D of MiLPEAT as predicted by TMpred Server (Supplementary Table 2), whether it spanned membrane or not remained unknown because it consisted of only 17 amino acids. In brief, these N-terminal hydrophobic amino acids of MiLPEAT were responsible for its membrane-localization as predicted by PredictProtein (Supplementary Table 2).
Since GPAT9 and LPEAT have the same acyl donor and similar acyl acceptor in function, GPAT9 was proposed by Shockey et al. (2016) to be an out-group for phylogenetic tree inference of LPEAT. Compared to the highly conservative GPAT9, LPEAT1 and LPEAT2 were quite divergent in sequence, and even the conservative motifs (Fig. 2), thus explaining why the functions of LPEAT1 and LPEAT2 were not completely the same to each other as examined by both Ståhl et al. (2008) and Stålberg et al. (2009). Besides, average length of the selected LPEAT1 (396.3±28.5 amino acids, n = 23) was significantly shorter than that of LPEAT2 (545.4±12.6 amino acids, n = 23) due to the absence of an approximately 160-residue C-terminal extension in LPEAT1 as revealed by multi-sequence alignment (data not shown). From the constructed NJ phylogeny tree (Fig. 3), the evolutionary difference between LPEAT1 and LPEAT2 was significant (bootstrap value reaches 99% and 99% while excluding the annotated but unidentified LPEAT-like one in Chlorella sorokiniana, respectively), suggesting their possibly independent origin. MiLPEAT was clearly clustered into LPEAT1 subclade (bootstrap value is 99%, Fig. 3) in this study, implying that it likely had the similar structure and function of LPEAT1.

Phylogenetic tree inferred from the deduced amino acid sequences of LPEAT genes. Branch lengths are proportional to the number of substitutions per site (see the scale bar). The numbers at the nodes indicate the neighbor-joining bootstrap proportion (BP) values (only values ≥50% are shown). All of the accession numbers are presented in the parentheses after the Latin names of each species
As predicted by SignalP, TargetP, and iPSORT, MiLPEAT did not have a signal peptide, but this signal sequence was predicted insignificantly (PProwler score = 0.64) to be present by Protein Prowler v. 1.2 (Supplementary Table 2). As for subcellular localization, MiLPEAT was predicted to be located either on plasma membrane by both MemLoci and CELLO v.2.5, or in nucleus by BaCelLo, or in ER by Cell-PLoc 2.0, or in other place except chloroplast and mitochondrion by both Predotar v. 1.04 and TargetP-2.0 Server (Supplementary Table 2). In addition, MiLPEAT could reside in chloroplast, vacuole, plasma membrane, and ER in the same possibility as predicted by WoLF PSORT (Supplementary Table 2). These inconsistently predicted results suggested that direct and reliable cytological evidence would be necessary for the subcellular localization determination of MiLPEAT.
Prokaryotic expression of MiLPEAT for antibody preparation
The abovementioned information suggested that MiLPEAT would be a membrane-bound protein. In order to heterologously express MiLPEAT in E. coli with success, its ORF sequence corresponding to the predicted transmembrane regions (102 amino acids in length from the N-terminus on) was deleted. Based on the remaining ORF sequence of MiLPEAT and the multiple cloning sites of the plasmid pET-28a, one pair of primers QIE1F and QIE1R carrying restriction recognition sites of EcoRI and NotI was designed (Table 1). With the designed primers and synthesized cDNA from M. incisa, a target product was amplified by PCR and fractioned electrophoretically as shown in Lane 1 of Supplementary Fig. 1. Cloning and subsequently sequencing results revealed that the length of this product was 689 bp which was composed of a 675-bp ORF of MiLPEAT and 14-bp nucleotides for restriction recognition. After double digestion with restriction enzymes EcoRI and NotI (Supplementary Fig. 1, Lane 2), this target fragment was sub-cloned into a digested pET-28a(Supplementary Fig. 1, Lane 3) to generate a construct pET28a-MiLPEAT. Double enzyme digestion products of this construct pET28a-MiLPEAT were separated by electrophoresis, showing the presence of two bands. The larger one was identical in size with the digested pET-28a (5,350 bp) (Supplementary Fig. 1, Lanes 4 vs. 3) while the smaller one was the same as the amplified target product (689 bp) (Supplementary Fig. 1, Lanes 4 vs. 2 and 1). This digestion pattern (Supplementary Fig. 1, Lane 4) and sequencing (data not shown) of this insert in the construct pET28a-MiLPEAT suggested the correct orientation and reading frame of MiLPEAT.
Escherichia coli strains harboring the recombinant plasmid pET28a-MiLPEAT and empty plasmid pET-28a were cultured independently in a 37°C chamber and induced by supplying with IPTG at a final concentration of 0.5 mM in the LB medium containing 50 μg mL-1 Kan. The bacterial cells were collected for detection of the target protein by SDS-PAGE. Compared to the strain carrying empty plasmid alone as a negative control (Supplementary Fig. 2, Lane 1), cell lysate of the transformant with the construct pET28a-MiLPEAT displayed a dark-color protein band of about 30 kD (Supplementary Fig. 2, Lane 2). The size of this band seemed close to that which was composed of both the target protein (24.62 kD, after removal of putative transmembrane regions from the amino terminus) and a 6×His tag plus a peptide (3.84 kD in total) as coded by multiple cloning site sequence upstream of MiLPEAT in pET28a-MiLPEAT. Therefore, this band was expected to be the recombinant MiLPEAT (rMiLPEAT).
To confirm this by Western blot analysis, the commercially supplied anti-6×His tag monoclonal antibody was employed since a 6×His tag was fused with the target protein during the construction of pET28a-MiLPEAT. Immunoblotting profile (Supplementary Fig. 2, Lane 5) of cell lysate from the transformant harboring the construct pET28a-MiLPEAT demonstrated that there was only a clear blotting signal around the 30-kD band. This immuno signal corresponded greatly to the fused MiLPEAT in size (Supplementary Fig. 2, Lanes 5 vs. 2), thus indicating clearly that the dark-color band at approximately 30 kD in Lane 2 of Supplementary Fig. 2 was the expected rMiLPEAT.
From the protein electrophoresis profiles of the supernatant (Supplementary Fig. 2, Lane 3) and precipitate (Supplementary Fig. 2, Lane 4) of the transformant carrying the plasmid pET28a-MiLPEAT, it was inferred that the 30-kD band mainly appeared in the precipitate. That is to say, rMiLPEAT was mainly expressed in the form of inclusion bodies in the transformed E. coli. Although soluble proteins were the first choice for antigen production, this form of induced rMiLPEAT could also be used for antibody preparation after purification as proposed by Rancour et al. (2010). The recombinant MiLPEAT could be purified by affinity chromatography with different concentrations of imidazole due to the presence of a 6×His tag in the fusion rMiLPEAT. Electrophoresis patterns of the eluted products (Supplementary Fig. 2, Lanes 8 through 11) showed that several bands were present in addition to the target one (about 30 kD in size) while 10 mM, 20 mM, and 200 mM of imidazole (Supplementary Fig. 2, Lanes 8, 9, and 10) were used. However, when 500 mM of imidazole was used for elution, only a single and clear band appeared at about 30 kD in the electrophoresis profile (Supplementary Fig. 2, Lane 11). This concentration of imidazole was thereby used to elute the target protein, and the purified rMiLPEAT accounted for 94.6% after SDS-PAGE as estimated by the ImageJ software, thus suggesting that the purity would meet the requirements for antibody preparation as proposed by Rancour et al. (2010). After purification, the concentration of rMiLPEAT was detected spectrophotometrically and it was approximately 2.0 mg⋅mL-1.
Following the affinity chromatography purification, rMiLPEAT was used to prepare anti-MiLPEAT polyclonal antibody by immunizing white rabbits. After immunization three times, about 30 mL of antiserum was collected from the immunized rabbits. The valence of anti-MiLPEAT polyclonal antibody in the antiserum was 1:80,000 by indirect ELISA detection (data not shown) with rMiLPEAT as an antigen, thus suggesting that it could reach the requirements of subsequent experiments. Prior to quality check, this prepared antibody was further purified using CNBr activated Sephrose 4B associated with the purified antigen rMiLPEAT to eliminate the possible negative effect.
To check the quality of the purified anti-MiLPEAT polyclonal antibody by Western blot analysis, crude proteins (Fig. 4, Lane 1) were extracted from M. incisa with the RIPA lysis and extraction buffer. Immunoblotting pattern (Fig, 4, Lane 2) demonstrated that a clear and single blotting signal appeared near 37.0 kD. The protein band corresponding to this signal seemed to be similar in size with MiLPEAT (36.23 kD). In order to ensure that the band was the target one, endogenous MiLPEAT was isolated from the crude extracts of M. incisa by immunoprecipitation with the purified anti-MiLPEAT polyclonal antibody. A total of seven peptide chains consisting of 58 amino acids were detected (Supplementary Fig. 3, lower panel), and MS spectrum of one, i.e. YGDMGQAR, of them was exampled in Supplementary Fig. 3 (upper panel). Although the detected amino acids only accounted for 17.79% of the total deduced ones, the sequences of the detected peptides completely matched the corresponding deduced ones of MiLPEAT (Supplementary Fig. 3, lower panel). It was worthy of note that the detected peptide YGDMGQAR matched the corresponding amino acid sequence exactly at MiLPEAT N-terminus(Supplementary Fig. 3) which was deleted during the construction of pET28a-MiLPEAT. This in part supported the prediction that MiLPEAT lacked a signal peptide (Supplementary Table 2), or else YGDMGQAR might be cleaved from MiLPEAT during the maturation processing of proteins. In combination with the Western blotting profile (Fig. 4), the MS analysis (Supplementary Fig. 3) after immunoprecipitation suggested the purified anti-MiLPEAT polyclonal antibody was of high quality for immuno-electron microscopic observation to determine the subcellular localization of MiLPEAT.

Electrophoresis (left) and Western blotting (right) profiles of the crude proteins extracted from Myrmecia incisa. Lane M: PageRuler Prestained Protein Ladder (Thermo Scientific, Rockford, USA); Lane 1: crude proteins extracted from Myrmecia incisa using RIPA buffer (Thermo Scientific, Rockford, USA); Lane 2: the blotting signal was stained with the prepared anti-MiLPEAT polyclonal antibody against the crude proteins extracted from M. incisa.
Subcellular localization of MiLPEAT
Prior to immuno-electron microscopic experiment, subcellular ultrastructure of M. incisa was observed using a transmission electron microscope. After several immuno-electron microscopic trails, the optimal dilution ratios of primary (i.e. anti-MiLPEAT polyclonal antibody) and secondary antibodies were determined, and they were 1:2,500 and 1:8,000, respectively. Ultrathin sections of M. incisa cells were incubated with the optimized concentrations of the purified anti-MiLPEAT polyclonal antibody and anti-rabbit IgG conjugated to 10-nm gold particles as the secondary antibody. Subcellular localization of MiLPEAT in M. incisa after washing was observed by a transmission electron microscope, and several micrographs were taken as examples in Fig. 5. A total of 220 immuno-labeling gold particles were counted from 9 selected micrographs. Of these gold particles, 208 particles (accounting for 94.55%) were estimated to decorate the region including the cell wall and plasma membrane of M. incisa cells, while 5.45% of particles (i.e. 12/220) were scattered in the other region. Meanwhile, the density of gold particles was measured after the area of different subcellular regions was estimated by the Adobe Photoshop software. The density of gold particles in the region of cell wall and cell membrane was estimated to be 12.36±8.89 particles μm-2, and it was significantly different (P = 0.0042 < 0.01) from that (0.45±1.05 particles μm-2) in the other region. Comparison of both percentage and density of gold particles in the selected images demonstrated that MiLPEAT might be localized in the region of cell wall and plasma membrane of M. incisa cells, especially close to the microalgal plasma membrane as shown in Figs. 5C and 5D, to perform its function.

Transmission electron micrographs showing the immunogold labeling distribution of MiLPEAT in the cells of Myrmecia incisa. A Ultrastructure of a cell of M. incisa; B, C, and D enlarged images corresponding to the marked areas 1, 2, and 3, respectively, in A; CW, cell wall; OB, oil body; S, starch; Thy, thylakoid.
In view of that MiLPEAT was a putative membrane-bound protein as mentioned above, this protein was speculated to be located on the plasma membrane rather than the cell wall of M. incisa. To prove this hypothesis, immunocytochemical observation on plasmolyzed cells of M. incisa was performed. Upon treatment with 0.6 g mL-1 sucrose, plasmolysis of M. incisa was observed under a light microscope (Fig. 6A). Afterwards, the plasmolyzed specimens were frozen and cryostat-sectioned into slices, and the plasma membrane of M. incisa was found to be still separated from the cell wall (Fig. 6B). When the sections were incubated sequentially with anti-MiLPEAT as primary antibody, biotin-labeled goat anti-rabbit IgG as secondary antibody plus HRP-labeled streptavidin as the tertiary antibody to further amplify the signal for color reaction, a nearly complete dark circle was found to be around the cytoplasm which was separated as well from the cell wall of M. incisa(Fig. 6C). By contrast, this phenomenon could not be observed (Fig. 6D) while the anti-MiLPEAT polyclonal antibody was absent from the sequentially incubated slice in the same way. This immunocytochemical observation provided direct evidence that MiLPEAT was a plasmalemma-located enzyme to work as it did.

Immunocytochemical images of MiLPEAT in Myrmecia incisa cells with the prepared anti-MiLPEAT polyclonal antibody. A Morphology of a plasmolyzed microalgal cell treated with 0.6 g mL-1 sucrose solution; B morphology of a plasmolyzed microalgal cell after cryosectioning; C immunocytochemical observation of a cryostat-sectioned plasmolyzed microalgal cell with the prepared anti-MiLPEAT polyclonal antibody; D immunocytochemical observation of a cryostat-sectioned plasmolyzed microalgal cell without the primary anti-MiLPEAT antibody as a negative control.
Prokaryotic expression of MiLPEAT for enzyme assay
As shown in Lane 4 of Supplementary Fig. 2, rMiLPEAT was expressed in inclusion bodies in E. coli while it was fused with a 6×His tag into the plasmid pET-28a. It has been understood that proteins expressed in inclusion bodies were often inactive for enzyme assay mainly due to mis-folding(Singh and Panda 2005). To obtain soluble rMiLPEAT for enzyme activity detection, the plasmid pMAL-c2X was employed in the present study as suggested by Walker et al. (2010). As mentioned above, the motifs for acyltransferase were located between 112P and 232K of MiLPEAT whereas two putative transmembrane regions ranged from 37L through 63C and from 88A through 104D (Supplementary Table 2, Supplementary Fig. 3). To avoid possible negative effects of transmembrane on MiLPEAT expression in E. coli, the ORF sequence corresponding to the N-terminal 102 amino acids was also deleted during the construction of pMALc2X-MiLPEAT.
Based on the sequences of remaining ORF of MiLPEAT and multiple cloning sites of pMAL-c2X, two primers QIE2F and QIE2R (Table 1) containing restriction recognition sites were designed. With these primers, the target sequence (Supplementary Fig. 4, Lane 1) was amplified by PCR and then ligated to pMD19-T to generate the construct pMD19T-MiLPEAT-s (the appended s is to distinguish this plasmid from the former pMD19T-MiLPEAT). After digestion with EcoRI and HindIII (Supplementary Fig. 4, Lanes 2 and 4), target sequence from pMD19T-MiLPEAT-s was ligated pMAL-c2X to generate the construct pMALc2X-MiLPEAT. Double-enzyme digestion pattern of pMALc2X-MiLPEAT showed that the larger band was in agreement with the digested pMAL-c2X(6,620 bp, Supplementary Fig. 4, Lanes 3 vs. 4) in size and the other one (687 bp, Supplementary Fig. 4, Lanes 3 vs. 2 and 1) was consistent with the cloned ORF of MiLPEAT minus 102-amino-acid corresponding cDNA plus 12-bp nucleotides for restriction recognition. In combination with sequencing analysis (data not shown), these results indicated that the construct pMALc2X-MiLPEAT was successfully generated as expected in correct orientation and reading frame. The transformants were screened and obtained by transforming pMALc2X-MiLPEAT into E. coli BL21.
The transformant harboring pMALc2X-MiLPEAT was cultured and induced for target protein expression in the same way as that carrying pET28a-MiLPEAT. SDS-PAGE profile showed that a conspicuous band of about 70 kD (Supplementary Fig. 5, Lane 2) emerged in the cell lysate of this transformant while comparing to that of one harboring the empty plasmid pMAL-c2X(Supplementary Fig. 5, Lane 1). This band seemed to be consistent in MW with the target protein (24.62 kD) plus a 40.34-kD MBP tag and a 2.64-kD peptide corresponding multiple cloning site sequence in the vector. This was supported by Western blotting analysis (Supplementary Fig. 5, Lanes 7 and 8) with the commercially supplied anti-MBP monoclonal antibody. In addition, the fusion protein extracted from the transformed E. coli with pMALc2X-MiLPEAT was expressed in both soluble (the supernatant, Supplementary Fig. 5, Lane 3) and insoluble (the inclusion bodies, Supplementary Fig. 5, Lane 4) fractions as fractionated by SDS-PAGE, but the amount expressed in the supernatant appeared more abundant (Supplementary Fig. 5, Lanes 3 vs. 4).
A gravity flow column filled with the affinity matrix Amylose resin was employed for the isolation of MiLPEAT fused to MBP. The impurities would flow naturally through the gravity flow column, once the supernatant extracted from the transformant harboring pMALc2X-MiLPEAT was loaded onto the column. The recombinant MiLPEAT, however, specifically bound to Amylose resin via the fused MBP, could be exchanged with maltose with stronger binding ability for purification. Although a few impurities were still present in the elution (Supplementary Fig. 5, Lanes 5 and 6) as analyzed by SDS-PAGE, the fusion MiLPEAT was purified greatly while comparing to the supernatant (Supplementary Fig. 5, Lanes 6 vs. 3), and the purified fusion MiLPEAT accounted for 52.5% as estimated by the ImageJ software. To confirm the fusion protein, Western blot analysis was employed with the aforementioned purified anti-MiLPEAT polyclonal antibody. Immunoblotting pattern (Supplementary Fig. 5, Lane 9) illustrated that only one signal was present in the same place as that obtained with anti-MBP antibody (Supplementary Fig. 5, Lanes 9 vs. 8), suggesting that the fusion protein was the target one consisting of two components MBP and carboxyl terminus of MiLPEAT.
According to Stålberg et al. (2009), the in vitro reaction system containing palmitoyl (16:0)-CoA as an acyl donor and 18:1-LPE as an acyl acceptor was established. The reaction was started by adding the purified MiLPEAT fused with MBP, and the products were extracted by chloroform and then analyzed by TLC. Corresponding to standard PE as shown by TLC plates (Fig. 7), a spot emerged in the reaction system while supplying with this purified fusion MiLPEAT. On the contrary, such a spot was not observed in the reaction system whatever either the purified extract from the transformant carrying empty plasmid pMAL-c2X or buffer alone was added (Fig. 7). In addition, when 18:1-LPE was substituted by 18:1-lysophosphatidic acid (LPA) or 18:1-LPC as an acyl acceptor, the corresponding product spot could hardly be found on the TLC plates (Fig. 7). However, while 18:1-LPE was added to the mixture of 18:1-LPA and 18:1-LPC, PE appeared again (Fig. 7). It hence came to the conclusion that the fusion MiLPEAT could transfer acyl group from palmitoyl-CoA to the sn-2 of 1-oleoyl-LPE rather than 1-oleoyl-LPA or 1-oleoyl-LPC to produce PE, thus identifying this microalgal gene MiLPEAT in function.

Thin-layer chromatography (TLC) patterns of the in vitro enzyme reaction products catalyzed by the recombinant MiLPEAT (rMiLPEAT) with various substrates. Controls 1 or 2: products of the reaction system supplemented with either the purified extract from the transformed Escherichia coli with the empty plasmid or reaction buffer instead of rMiLPEAT, respectively; rMiLPEAT: products of the reaction system supplemented with the purified rMiLPEAT; LPA, LPC, LPE, PA, PC, and PE: the commercially supplied lysophosphatidic acid (LPA), lysophosphatidylcholine (LPC), lysophosphatidylethanolamine (LPE), phosphatidic acid (PA), phosphatidylcholine (PC), and phosphatidylethanolamine (PE) (Sigma, St. Louis, USA), respectively, as references; LPA, LPC, and LPE above the images denoting the supplied substrates.
Discussion
Prior to the present study, several genes encoding the acyltransferase domain PlsC have been screened from the transcriptome database of Myrmecia incisa(Ouyang et al. 2013a). Of these genes, except for MiLPEAT which has been characterized as described above, two genes coding correspondingly for a chloroplast-localized GPAT and an ER-localized GPAT have been documented (Ouyang et al. 2016a; Sun et al. 2021). Through clustering analysis of these gene sequences cloned from this microalga (data not shown), the gene MiLPEAT is disclosed to be clustered with LPEAT from other species, thus showing its distinction from the reported GPAT. On this basis, more LPEAT data from different organisms are retrieved and then clustered. The phylogenetic inference (Fig. 3) shows that MiLPEAT has the closest relationship with LPEAT1 of higher plants but is different from LPEAT2, further supporting that LPEAT1 and LPEAT2 are quite divergent as found by Shockey et al. (2016). As well as shown in Fig. 2, MiLPEAT is more similar to LPEAT1 than LPEAT2 of higher plants in their both conserved motifs and length of amino acids. MiLPEAT is reasonably located at the root of the LPEAT1 subclade (Fig. 3), thus implying that LPEAT1 of higher plants might evolve from the green algal LPEAT. The algal sub-branch, however, consisting of only two analogues correspondingly from M. incisa and Chlorella sorokiniana in this study (Fig. 3), needs to be reconstructed, once the precise data on algal LPEAT are available. In that time, the phylogenetic relationship between every two of algal LPEAT, higher plant LPEAT1, and LPEAT2 is believed to be unveiled.
To identify MiLPEAT, first of all, the domains and conserved motifs, such as NH(x)4D, GCxYVxR, FPEGT, and PVxPVx (Fig. 2), are characterized. In the motif NH(x)4D, the residues Asp (D) and His (H) which are proposed to be important acyltransferase active centers by Lewin et al. (1999) and Yamashita et al. (2007) are highly conserved in both LPEAT1 and MiLPEAT. In this motif of LPEAT2, however, Asp is usually mutated to Glu (E) as shown in Fig. 2, thus suggesting that there is a probable difference in enzyme activity between LPEAT1 and LPEAT2. The FPEGT motif is highly conserved in all aligned sequences of LPEAT (Fig. 2), reflecting its importance. As found by Lewin et al. (1999) using site-directed mutagenesis experiments for GPAT motif analysis, Gly (G) in FPEGT motif and Pro (P) in PVxPVx motif constitute the important active center of acyltransferase, while Glu in the motif FPEGT and Arg (R) in the motif GCxYVxR are the binding sites of substrate or phosphorus in the substrate. All these important residues highly conserved in MiLPEAT (Fig. 2) ensure that the algal LPEAT has the possible activity of acyltransferase. As searched by BLASTp, MiLPEAT has a typical phosphate acyltransferase domain PlsC (from 117A through 234G), a LPLAT_LPCAT1-like domain (from 86A through 308R), and an acyltransferase domain (from 112P through 232K), thus suggesting that the carboxyl terminus of MiLPEAT might work as an acyltransferase.
The gene MiLPEAT cDNA corresponding to the carboxyl terminus containing the phosphate acyltransferase domain PlsC is thereby used to construct the recombinant vector pMALc2X-MiLPEAT(Supplementary Fig. 4) for obtainment of rMiLPEAT (Supplementary Fig. 5). The products catalyzed by rMiLPEAT are analyzed by TLC to judge whether this recombinant enzyme works or not. Based on the TLC profiles (Fig. 7), rMiLPEAT is able to acylate LPE to PE exclusively, thus experimentally not only identifying this gene MiLPEAT but also partially supporting the BLSATp and InterPro predication that the C-terminal phosphate acyltransferase domain PlsC is responsible for LPEAT. In vitro studies conducted by Jasieniecka-Gazarkiewicz et al. (2016) have suggested that Arabidopsis thaliana LPEAT2 had the abilities to acylate LPE to PE and act reversibly on this product PE. Meanwhile, the plant LPEAT2 showed the highest levels of reversibility towards PC and PE of all tested LPLAT (Jasieniecka-Gazarkiewicz et al. 2016). However, there is no data available on the reversible reaction catalyzed by LPEAT1 and a close relationship between LPEAT1 and MiLPEAT (Fig. 3), the catalytic reversibility by rMiLPEAT remains to be investigated. As for the substrate preference, rMiLPEAT seems to use LPE exclusively but LPA or LPC as one substrate (Fig. 7), whereas the other one, i.e. species of acyl-CoA, needs to be examined, too.
As predicted by SignalP, TargetP, and iPSORT, MiLPEAT lacks a signal peptide at its N-terminus(Supplementary Table 2). However, this does not mean that MiLPEAT cannot be transported in the secretory pathway. MiLPEAT possibly has two predicted transmembrane domains (TMDs, located between 37L and 63C, and 88A and 104D, Supplementary Table 2 and Supplementary Fig. 3), especially of the first which is proposed to act as a signal peptide by interacting with signal recognition particle (SRP)(for reviews, see Rapoport et al. 2004; Spiess et al. 2019). It is consequently proposed to be inserted into ER membrane by co-translational translocation via a SRP complex or Sec-mediated translocon as reviewed by Johnson and van Waes (1999), Osborne et al. (2005), and Akopian et al. (2013). All the genes coding for the components constituting the SRP complex have been screened from the transcriptome database of M. incisa(Ouyang et al. 2013a) and compiled in Supplementary Table 3. Once the first TMD of MiLPEAT enters the aqueous channel of translocon, the correct conformation of this protein will exit laterally into the lipid phase of ER membrane via this TMD (for reviews, see Johnson and van Waes 1999; Gemmer and Förster 2020). This TMD is therefore thought to be a signal-anchoring sequence, and the polypeptide chain continues to extend until the emergence of terminal code. The orientation of MiLPEAT is well determined during the co-translationally translocational insertion as reviewed by Osborne et al. (2005), and the di-lysine motif KKxx at its carboxyl terminus (Fig. 2) is predicted to be exposed to the cytoplasm of this algal cell (Supplementary Table 2).
It should be pointed out, however, that the cytoplasmic di-lysine motif KKxx functioning as an ER retention or retrieve signal (Nilsson et al. 1989) can usually lead MiLPEAT back to ER but not be transported to plasma membrane. How the membrane-bound proteins like MiLPEAT are destined to cell surface is not well known according to Jürgens (2004) and Wang et al. (2018). One mechanism has been posited by Letourneur et al. (1995) that the vesicles carrying these assembled membrane-bound proteins might be prevented from the retrograde trafficking from Golgi apparatus back to ER via binding their di-lysine motif with molecular chaperones. In this way, the vesicles carrying sorted and integrated MiLPEAT are suggested to be transported to the algal plasma membrane as shown in Figs. 5 and 6 along the proposed conventional trafficking route by both Goring and Di Sansebastiano (2018) and Wang et al. (2018). This is partially supported by the screened genes coding for the components related to the conventional vesicular transport in M. incisa (Supplementary Table 3). Although this mechanism remains to be evidenced experimentally, the present study facilitates explaining why Larsson et al. (2007) have detected the activity of LPCAT, an analogue of LPEAT, in the plasma membrane fractions extracted from the seedlings of Pisum sativum, Glycine max, and A. thaliana. In contrast to this, the ER-localizedA. thaliana LPEAT1 and Golgi apparatus-localizedA. thaliana LPEAT2, which have been observed by Jasieniecka-Gazarkiewicz et al. (2021) using transiently-transformedNicotiana tabacumBY-2 suspension-cultured cells, seem difficult to explain what Larsson et al. (2007) have found. Accordingly, the cytoplasmic di-lysine motif KKxx possibly contributes to the quality control of assembled proteins as suggested by Béthune et al. (2006). In other words, once MiLPEAT is incompletely or incorrectly folded, for example, it will be recruited from Golgi complex back to ER through the anterograde trafficking pathway.
In general, this is the first report about algal LPEAT so far. The present study reveals that a LPEAT from M. incisa is characterized by two (at least one) predicted TMDs (Supplementary Fig. 3) and a cytoplasmic di-lysine motif KKxx (Fig. 2) present at the amino and carboxyl termini, respectively. MiLPEAT is localized to the microalgal plasma membrane (Figs. 5 and 6), which is different from its counterparts from A. thaliana (Jasieniecka-Gazarkiewicz et al. 2021). Although the in vivo function of MiLPEAT will be disclosed during the algal growth, especially under some stress conditions for autophagosome formation during autophagy as reported in A. thaliana (Jasieniecka-Gazarkiewicz et al. 2021), MiLPEAT is identified at least as a catalyst to acylate LPE to PE (Fig. 7) in the acyl remodeling of phospholipid via the Lands’ cycle (Lands 1958). The elongation of fatty acids has been suggested by Bates (2016) to occur in acyl-CoA pool of ER. In that case, the produced long-chain fatty acids such as ArA in M. incisa can be esterified to the sn-2 hydroxyl of LPE to form a modified PE by MiLPEAT. One possible result is that ArA in the modified PE can be donated to DAG as catalyzed by another documented acyltransferase PDAT by Liu et al. (2016) to generate ArA-rich TAG. The present study, therefore, lays a foundation for the investigation into the biosynthesis route of ArA-enriched TAG in M. incisa in addition to the Kennedy pathway as documented in detail by Chen et al. (2015), Liu et al. (2021) and Sun et al. (2021).
Data availability
The datasets generated during and/or analyzed during the current study are available in the GenBank repository [https://www.ncbi.nlm.nih.gov/nuccore/mg558460].
References
Akopian D, Shen K, Zhang X, Shan S (2013) Signal recognition particle: an essential protein-targeting machine. Annu Rev Biochem 82:693–721
Bates PD (2016) Understanding the control of acyl flux through the lipid metabolic network of plant oil biosynthesis. Biochim Biophys Acta 1861:1214–1225
Bendayan M, Zollinger M (1983) Ultrastructural localization of antigenic sites on osmium-fixed tissues applying the protein A-gold technique. J Histochem Cytochem 31:101–109
Bernal M, Testillano PS, Alfonso M, del Carmen RM, Picorel R, Yruela I (2007) Identification and subcellular localization of the soybean copper P1B-ATPase GmHMA8 transporter. J Struct Biol 158:46–58
Béthune J, Wieland F, Moelleken J (2006)COPI-mediated transport. J Membr Biol 211:65–79
Bi Y-H, Qiao Y-M, Wang Z, Zhou Z-G(2021) Identification and characterization of a periplasmic α-carbonic anhydrase (CA) in the gametophytes of Saccharina japonica (Phaeophyceae). J Phycol 57:295–310
Bigogno C, Khozin-Goldberg I, Boussiba S, Vonshak A, Cohen Z (2002) Lipid and fatty acid composition of the green oleaginous alga Parietochloris incisa, the richest plant source of arachidonic acid. Phytochemistry 60:497–503
Bowie JU (1997) Helix packing in membrane proteins. J Mol Biol 272:780–789
Bradford MM (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chen C-X, Sun Z, Cao H-S, Fang F-L, Ouyang L-L, Zhou Z-G(2015) Identification and characterization of three genes encoding acyl-CoA: diacylglycerol acyltransferase (DGAT) from the microalga Myrmecia incisa Reisigl. Algal Res 12:280–288
Dyer JM, Stymne S, Green AG, Carlsson AS (2008)High-value oils from plants. Plant J 54:640–655
Gemmer M, Förster F (2020) A clearer picture of the ER translocon complex. J Cell Sci 133:jcs231340
Goring DR, Di Sansebastiano GP (2018) Protein and membrane trafficking routes in plants: conventional or unconventional? J Exp Bot 69:1–5
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580
Hishikawa D, Hashidate T, Shimizu T, Shindou H (2014) Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J Lipid Res 55:799–807
Hu Y-J, Zhou Z-G(2001) Extraction of RAPD-friendly DNA from Laminaria japonica(Phaeophyta) after enzymatic dissociation of the frozen sporophyte tissue. J Appl Phycol 13:415–422
Jackowski S, Jackson PD, Rock CO (1994) Sequence and function of the aas gene in Escherichia coli. J Biol Chem 269:2921–2928
Jasieniecka-Gazarkiewicz K, Demski K, Gidda SK, Klińska S, Niedojadło J, Lager I, Carlsson AS, Minina EA, Mullen RT, Bozhkov PV, Stymne S, Banaś A (2021) Subcellular localization of acyl-CoA: lysophosphatidylethanolamine acyltransferases (LPEATs) and the effects of knocking-out and overexpression of their genes on autophagy markers level and life span of A. thaliana. Int J Mol Sci 22:3006
Jasieniecka-Gazarkiewicz K, Demski K, Lager I, Stymne S, Banaś A (2016) Possible role of different yeast and plant lysophospholipid: acyl-CoA acyltransferases (LPLAT) in acyl remodeling of phospholipids. Lipids 51:15–32
Jasieniecka-Gazarkiewicz K, Lager I, Carlsson AS, Gutbrod K, Peisker H, Dörmann P, Stymne S, Banaś A (2017) Acyl-CoA: lysophosphatidylethanolamine acyltransferase activity regulates growth of Arabidopsis. Plant Physiol 174:986–998
Johnson AE, van Waes MA (1999) The translocon: A dynamic gateway at the ER membrane. Annu Rev Cell Dev Biol 15:799–842
Jürgens G (2004) Membrane trafficking in plants. Annu Rev Cell Dev Biol 20:481–504
Klińska S, Demski K, Jasieniecka-Gazarkiewicz K, Banaś A (2021) LPEATs tailor plant phospholipid composition through adjusting substrate preferences to temperature. Int J Mol Sci 22:8137
Klińska S, Jasieniecka-Gazarkiewicz K, Demski K, Banaś A (2020) Editing of phosphatidic acid and phosphatidylethanolamine by acyl-CoA: lysophospholipid acyltransferases in developing Camelina sativa seeds. Planta 252:4
Krenacs L, Krenacs T, Stelkovics E, Raffeld M (2010)Heat-induced antigen retrieval for immunohistochemical reactions in routinely processed paraffin sections. Methods Mol Biol 588:103–119
Kussmann M, Roepstorff P (2000) Sample preparation techniques for peptides and proteins analyzed by MALDI-MS. Methods Mol Biol 146:405–424
Laemmli UR (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lands WEM (1958) Metabolism of glycerolipides: a comparison of lecithin and triglyceride synthesis. J Biol Chem 231:883–888
Larsson KE, Kjellberg JM, Tjellström H, Sandelius AS (2007) LysoPC acyltransferase/PC transacylase activities in plant plasma membrane and plasma membrane-associated endoplasmic reticulum. BMC Plant Biol 7:64
Letourneur F, Hennecke S, Démollière C, Cosson P (1995) Steric masking of a dilysine endoplasmic reticulum retention motif during assembly of the human high affinity receptor for immunoglobulin E. J Cell Biol 129:971–978
Lewin TM, Wang P, Coleman RA (1999) Analysis of amino acid motifs diagnostic for the sn-glycerol-3-phosphate acyltransferase reaction. Biochemistry 38:5764–5771
Liu W, Sun Z, Chen C-X, Liu J-G, Zhou Z-G(2021) Novel insights into type-2 diacylglycerol acyltransferases in microalga Myrmecia incisa. J Appl Phycol 33:25–35
Liu X-Y, Ouyang L-L, Zhou Z-G(2016) Phospholipid: diacylglycerol acyltransferase contributes to the conversion of membrane lipids into triacylglycerol in Myrmecia incisa during the nitrogen starvation stress. Sci Rep 6:26610
Napier JA (2007) The production of unusual fatty acids in transgenic plants. Annu Rev Plant Biol 58:295–319
Nesvizhskii AI (2007) Protein identification by tandem mass spectrometry and sequence database searching. Methods Mol Biol 367:87–119
Nilsson T, Jackson M, Peterson PA (1989) Short cytoplasmic sequences serve as retention signals for transmembrane proteins in the endoplasmic reticulum. Cell 58:707–718
Ogunniyi DS (2006) Castor oil: a vital industrial raw material. Bioresour Technol 97:1086–1091
Olsen RE, Henderson RJ (1989) The rapid analysis of neutral and polar marine lipids using double-development HPTLC and scanning densitometry. J Exp Mar Biol Ecol 129:189–197
Osborne AR, Rapoport TA, van den Berg B (2005) Protein translocation by the Sec61/SecY channel. Annu Rev Cell Dev Biol 21:529–550
Ouyang L-L, Chen S-H, Li Y, Zhou Z-G(2013a) Transcriptome analysis reveals unique C4-like photosynthesis and oil body formation in an arachidonic acid-rich microalga Myrmecia incisa Reisigl H4301. BMC Genomics 14:396
Ouyang L-L, Du D-H, Yu S-Y, Li C-Y, Zhang C-W, Gao H-J, Zhou Z-G(2012) Expressed sequence tags analysis revealing the taxonomic position and fatty acid biosynthesis in an oleaginous green microalga, Myrmecia incisa Reisigl (Trebouxiophyceae, Chlorophyta). Chin Sci Bull 57:3342–3352
Ouyang L-L, Li H, Liu F, Tong M, Yu S-Y, Zhou Z-G(2013b) Accumulation of arachidonic acid in a green microalga, Myrmecia incisa, enhanced by nitrogen starvation and its molecular regulation mechanisms. In: Dumancas GG, Murdianti BS, Lucas EA (eds) Arachidonic Acid: Dietary Sources and General Functions. Nova Science Publishers, New York, pp 1–20
Ouyang L-L, Li H, Yan X-J, Xu J-L, Zhou Z-G(2016a)Site-directed mutagenesis from Arg195 to His of a microalgal putatively chloroplastidial glycerol-3-phosphate acyltransferase causes an increase of phospholipid level in yeast. Front Plant Sci 7:286
Ouyang L-L, Li X-L, Zhou Z-G(2016b) Alternations of photosynthetic membrane lipids and triacylglycerol and their fatty acids in Myrmecia incisa grown in a shift from nitrogen starvation to replenishment. Mar Fish 38:653–662 (in Chinese with English abstract)
Rancour DM, Backues SK, Bednarek SY (2010) Protein antigen expression in Escherichia coli for antibody production. Methods Mol Biol 657:3–20
Rapoport TA, Goder V, Heinrich SU, Matlack KES (2004)Membrane-protein integration and the role of the translocation channel. Trends Cell Biol 14:568–575
Reisigl H (1964) Zur Systematik und Ökologie alpiner Bodenalgen. Österr Bot Z 111:402–499
Riekhof WR, Wu J, Jones JL, Voelker DR (2007) Identification and characterization of the major lysophosphatidylethanolamine acyltransferase in Saccharomyces cerevisiae. J Biol Chem 282:28344–28352
Shindou H, Hishikawa D, Harayama T, Eto M, Shimizu T (2013) Generation of membrane diversity by lysophospholipid acyltransferases. J Biochem 154:21–28
Shockey J, Regmi A, Cotton K, Adhikari N, Browse J, Bates PD (2016) Identification of Arabidopsis GPAT9 (At5g60620) as an essential gene involved in triacylglycerol biosynthesis. Plant Physiol 170:163–179
Singh SM, Panda AK (2005) Solubilization and refolding of bacterial inclusion body proteins. J Biosci Bioeng 99:303–310
Sperling P, Linscheid M, Stöcker S, Mühlbach HP, Heinz E (1993)In vivo desaturation of cis-Δ-9-monounsaturated to cis-Δ9,12-diunsaturated alkenylether glycerolipides. J Biol Chem 268:26935–26940
Spiess M, Junne T, Janoschke M (2019) Membrane protein integration and topogenesis at the ER. Protein J 38:306–316
Stanier RY, Kunisawa R, Mandel M, Cohen-Bazir G (1971) Purification and properties of unicellular blue-green algae (Order Chlorococcales). Bacteriol Rev 35:171–205
Ståhl U, Stålberg K, Stymne S, Ronne H (2008) A family of eukaryotic lysophospholipid acyltransferases with broad specificity. FEBS Lett 582:305–309
Stålberg K, Ståhl U, Stymne S, Ohlrogge J (2009) Characterization of two Arabidopsis thaliana acyltransferases with preference for lysophosphatidylethanolamine. BMC Plant Biol 9:60
Stymne S, Appelqvist L-Å (1978) Biosynthesis of linoleate from oleoyl-CoA via oleoyl-phosphatidylcholine in microsomes of developing safflower seeds. Eur J Biochem 90:223–229
Sun L-P, Ouyang L-L, Bao H, Liu J-G, Sun Z, Zhou Z-G(2021) Comparison between two isoforms of glycerol-3-phosphate acyltransferase in microalga Myrmecia incisa: subcellular localization and role in triacylglycerol synthesis. Algal Res 54:102172
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Walker IH, Hsieh P-c, Riggs PD (2010) Mutations in maltose-binding protein that alter affinity and solubility properties. Appl Microbiol Biotechnol 88:187–197
Wang X, Chung KP, Lin W, Jiang L (2018) Protein secretion in plants: conventional and unconventional pathways and new techniques. J Exp Bot 69:21–37
Weier D, Müller C, Gaspers C, Frentzen M (2005) Characterisation of acyltransferases from Synechocystis sp. PCC6803. Biochem Biophys Res Commun 334:1127–1134
Yamashita A, Hayashi Y, Nemoto-Sasaki Y, Ito M, Oka S, Tanikawa T, Waku K, Sugiura T (2014) Acyltransferases and transacylases that determine the fatty acid composition of glycerolipids and the metabolism of bioactive lipid mediators in mammalian cells and model organisms. Prog Lipid Res 53:18–81
Yamashita A, Nakanishi H, Suzuki H, Kamata R, Tanaka K, Waku K, Sugiura T (2007) Topology of acyltransferase motifs and substrate specificity and accessibility in 1-acyl-sn-glycero-3-phosphate acyltransferase 1. Biochim Biophys Acta 1771:1202–1215
Ye R-X, Yu Z, Shi W-W, Gao H-J, Bi Y-H, Zhou Z-G(2014) Characterization of α-type carbonic anhydrase (CA) gene and subcellular localization of α-CA in the gametophytes of Saccharina japonica. J Appl Phycol 26:881–890
Yu S-Y, Li H, Tong M, Ouyang L-L, Zhou Z-G(2012) Identification of a Δ6 fatty acid elongase gene for arachidonic acid biosynthesis localized to the endoplasmic reticulum in the green microalga Myrmecia incisa Reisigl. Gene 493:219–227
Zhang L, Jia B, Tan X, Thammina CS, Long H, Liu M, Wen S, Song X, Cao H (2014) Fatty acid profile and unigene-derived simple sequence repeat markers in tung tree (Vernicia fordii). PLoS One 9:e105298
Funding
This work was supported by the National Natural Science Foundation of China (grant No. 31772821 to Z.-G. Z.) and the State Double First-class Discipline Project of Aquaculture (to Z.-G. Z.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, Y., Bao, H., Zhu, ML. et al. Subcellular localization and identification of acyl-CoA: lysophosphatidylethanolamine acyltransferase (LPEAT) in the arachidonic acid-rich green microalga, Myrmecia incisa Reisigl. J Appl Phycol 34, 837–855 (2022). https://doi.org/10.1007/s10811-021-02681-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-021-02681-z