
Abstract
Information related to geographical distribution and local strain composition is essential to an understanding of the dynamics of harmful algae in the environment. Previously, we identified a highly variable segment on the mitochondrial genome of Heterosigma akashiwo, a bloom-forming noxious unicellular algal species. Here, we assessed the utility of the mitochondrial hypervariable region for the phylogeographic study of the alga for different distance ranges. The sequences of H. akashiwo strains obtained from different geographic origins were successfully amplified and sequenced. We found differences among the sequences of the strains obtained from high-latitude regions of Northern Pacific/Atlantic; lower latitude regions of North America West Coast; and other regions including Brazil, Japan, Singapore, and North America East Coast. On the other hand, no strong geographic patterns for the sequences among Japanese strains were observed. Therefore, the hypervariable segment may be useful to distinguish H. akashiwo strains originated from distant regions (Atlantic/Pacific, high/low latitudes), rather than regions separated by shorter distances. The sequence contains an open reading frame coding for a protein with unknown function, and the transcription of the gene was confirmed by RNA-seq analysis. Despite the sequence variations observed among H. akashiwo strains originating in different parts of the world, three domains of the protein were highly conserved among all of the strains, suggesting that they may be important to the function of the protein.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Several algal species are known to form harmful algal blooms (HABs) that negatively impact the ecosystem and industry in the area, especially the fishery (Hallegraeff 1993; Maso and Garces 2006; Armbrust 2009; Fu et al. 2012). Because of the potential damage to the environment and the economy of an area, the population dynamics of HAB-related species are of great interest and importance (Hallegraeff 1993; Maso and Garces 2006; Armbrust 2009; Fu et al. 2012). Heterosigma akashiwo is one such HAB species that belongs to the class Raphidophyceae (Honjo 1993; Smayda 1997). It was originally regarded as a temperate species (Lackey and Lackey 1963; Throndsen 1969; Rojas de Mendiola 1979; Rensel et al. 1989; Chang et al. 1990; Black et al. 1991; Mackenzie 1991; Honjo 1993; Taylor 1993; Tseng et al. 1993; O’Halloran et al. 2006). However, recent studies revealed that the species also inhabits in arctic and tropical areas, including the Pacific Rim area, Oceania, and the North and the South Atlantic oceans (Engesmo et al. 2016). The identification of the alga over a wider area may be merely the result of recent exhaustive surveillances. Alternatively, the species may have been recently introduced to these areas. Recent global climate changes, including temperature changes and ocean stream shifts, may have resulted in the short- and long-distance dispersal of HAB-causing species (Hallegraeff and Bolch 1991; Smayda 2002; Fu et al. 2012). In addition, human-assisted dispersion, typically by ship ballast water (Hallegraeff and Bolch 1991, 1992; Elbrachter 1998; Bizsel and Bizsel 2002; Han et al. 2002; Burkholder et al. 2007; Drake et al. 2007; Butron et al. 2011), and the commercial transfer of live fish and spats (Matsuyama and Nagai 2010) may widen the geographic distribution of the species. To monitor H. akashiwo distribution at both the species and strain levels, an easy-to-use monitoring method is required. Previously, we identified a hypervariable segment of mitochondrial genome (MtDNA) with a length of ∼1.5 kbp by comparing the full-length MtDNA sequences obtained from seven different H. akashiwo strains (Ogura et al. 2016). Based on the information, we successfully designed a primer set, two primers for amplification, and three primers for sequencing, to amplify and sequence the segment to genotype H. akashiwo strains (unpublished).
In this study, we evaluated the utility of this mitochondrial hypervariable ORF (MtORFvar) as a molecular tool for monitoring H. akashiwo dynamics and found out that the sequences showed links to the geographic origins of H. akashiwo strains at certain degree. In addition, we confirmed the expression of the gene and examined the potential function of the MtORFvar product.
Materials and methods
Algal strains
Novel strains, HA series (isolated from Harima Nada, Hyogo Prefecture, Japan), AIC series (isolated from Mikawa Bay, Aichi Prefecture, Japan), AR series (isolated from Ariake Bay, Nagasaki Prefecture, Japan), Mie series (isolated from Ago Bay, Mie Prefecture, Japan), OF series (Ofunato Bay, Fukushima Prefecture, Japan), FUN series (Funka Bay, Hokkaido Prefecture, Japan), and RJ series (Guanabara Bay in Rio de Janeiro, Brazil) were established for this study. The HA-, AIC-, AR-, and Mie-strains were established by isolating single-algal cells from H. akashiwo bloom samples observed at the areas of origin. The strains of OF and FUN series were established by isolating single-algal cells germinated from saline sediment collected in April 2016. The RJ series were isolated by the same procedure from the saline sediment sampled in November 2016. Several clones were obtained from each region, and numbers were used as unique identifiers to distinguish the clones. The strains CCMP1595, CCMP1680, and CCMP2967 were obtained from Bigelow Laboratory for Ocean Sciences (East Boothbay, Maine, USA), and the strain CCAP934/3 was obtained from Culture Collection of Algae and Protozoa of Scottish Marine Institute (Argyll, UK). The strains were maintained in modified SWM3 medium in an environment-controlled chamber with a photoperiod (12 h of 100 μmol photons m−2 s−1 light/12 h dark) at 25 °C. For analysis, cells were collected by centrifugation at 5000 rpm for 5 min, snap frozen in liquid nitrogen, and stored at −80 °C until the analysis.
Amplification and sequencing of MtORFvar and 28S rRNA
Total DNA was extracted from the H. akashiwo cells using the CTAB method (Kamikawa et al. 2006). To amplify MtDNAvar, polymerase chain reaction (PCR) was performed using TaKaRa LA Taq according to the manufacturer’s instructions, on a thermal cycler (GeneAtlas 482, ASTEC) in a reaction mixture (20 μL) containing 0.5 ng of template and 5 μM of the previously designed primers, 5′-GGAGGCGTACAAAGGTAGGT-3′ and 5′-GCTGACGAAGAATCCGCAAC-3′ (manuscript under review). PCR conditions were 3 min at 95 °C, 35 cycles of 15 s at 95 °C and 4 min at 72 °C, and a final elongation of 10 min. After PCR, the yield of the product was checked by DNA electrophoresis on 1% agarose gel, and the products were treated by Exostar (Illumina) according to the manufacturer’s instructions to remove the remaining PCR primers in the reaction. The amplified MtDNAvar sequence was identified using Big Dye Terminator ver3 (Applied Biosystems) on an ABI3100 DNA sequencer according to the manufacturer’s instructions (Applied Biosystems) using three sequence primers, 4283-nt (5′-GTCAACATCATTTCGGGTTTG-3′), 5479-nt (5′- CGCTGATTTGCTTCAAACTCTTG-3′), and 6022-nt (5′-AAAGCCTGAATATAGGTTTTGTATTC-3′; primer positions are shown in Supplemental Fig. 1).
To confirm species of the strains, partial 28S rDNA sequences including the D1–D2 region were amplified and sequenced as previously described (Engesmo et al. 2016).
Nucleotide and protein sequence analysis
MtORFvar regions in MtDNAs of seven H. akashiwo strains, namely, H93616, Ha00_17, HaTj 01, NEPCC522, CCMP452, NIES293, and Y, were previously identified (Table 1). MtORFvar regions of EHU01/02, CCMP1596, Haek9505-1, CCAP934/7, CCAP934/9, CCMP1870, and CCMP2274 were previously sequenced (Table 1). For multiple sequence alignments and its visualization, Muscle software version 3.8.31 (http://www.drive5.com/muscle/) (Edgar 2004a, 2004b) and the BoxShade server (http://www.ch.embnet.org/software/BOX_form.html) were utilized.
Alignment and phylogenetic reconstructions were performed using the function “build” of ETE3 v3.0.0b32 (Huerta-Cepas et al. 2016) as implemented on the GenomeNet (http://www.genome.jp/tools/ete/). The sequences were aligned with MAFFT v6.861b (Katoh and Standley 2013), and the resulting alignments were manually trimmed. The best nucleotide model was selected using maximum-likelihood tree inference using pmodeltest v1.4., and the tree was inferred using RAxML v8.1.20 ran with model GTRGAMMAI and default parameters (Stamatakis 2014). Branch supports were computed out of 100 bootstrapped trees.
For protein sequence analysis, PSI-BLAST and BLASTX searches of the NCBI NR database were conducted with cut-off E-values of <10−8 and <10−5, respectively, to identify homologs of MtORFvar products from different species with defined functions. To find conserved domain in MtORFvar, CD-search (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and Pfam search (http://pfam.xfam.org) were conducted with cut-off E-value of <10–5.
Transcriptome analyses
Total RNA was extracted from ∼5 × 105 cells of H. akashiwo strain H93616 and HaTj01 using a PureLink RNA Mini kit (Ambion/Thermo Fisher Science). PolyA(+)-selected libraries were prepared using a SureSelect Strand-Specific RNA library preparation kit, and ∼71 million reads were generated by HiSeq2500 using the 100 bp paired-end mode. The reads from H93616 and HaTj01 were mapped to their MtDNA genomic sequences, accession numbers KU561547 and KU561550, respectively, using BWA software (http://bio-bwa.sourceforge.net) (Li and Durbin 2009). The reads per genomic nucleotide were counted by the R samtools pileup function (http://bioconductor.org/packages/devel/bioc/html/Rsamtools.html), and visualized using ArcWithColors software that was bundled in the GenomeMatcher package (http://www.ige.tohoku.ac.jp/joho/gmProject/gmdownloadJP.html) (Ohtsubo et al. 2008).
Results and discussion
MtORFvar sequence diversity among H. akashiwo strains isolated from different regions
In this study, we aimed to analyze the utility of MtORFvar as a phylogeographic marker to distinguish strains from origins separated by different distances. The primer set to amplify and sequence MtORFvar was previously designed based on the whole MtDNA sequences obtained from seven strains to date (manuscript under review, Supplemental Fig. 1a). MtORFvar segments were specifically amplified from H. akashiwo strains by the primers, demonstrating the adaptability of the primer set for further study (Supplemental Fig. 1b).
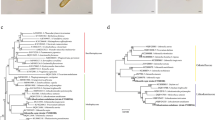
Analysis of MtORFvar sequences revealed that two to eight independent H. akashiwo strains were obtained from Funka Bay (2 strains), Ofunato (5 strains), Aichi (6 strains), Mie (6 strains), Harima (8 strains), and Ariake (6 strains) and two strains from Guanabara Bay in Rio de Janeiro, Brazil (Fig. 1a, Table 1, and Supplemental Fig. 2). The 28S D1-D2 of these strains were >99.8% identical to the previously published H. akashiwo 28S D1-D2 sequences (cf. GenBank accession numbers KP702886 and KP702887, in Engesmo et al. 2016), confirming that these strains belong to H. akashiwo (Table 1). MtORFvar of these strains and MtORFvar of the strains obtained from different regions of the world, including the East/West coasts of North America, Europe, and Singapore, were aligned for comparison (Fig. 1a and Table 1; Supplemental Fig. 2) In these strains, both single nucleotide polymorphisms and indels of up to 66 nt were observed, confirming that MtORFvar sequence is highly variable (Supplemental Fig. 2). To gain insight into potential links between MtORFvar sequences and origin of the strains, phylogenetic relationship among MtORFvar of the strains was analyzed (Fig. 1b, c). When sequences of MtORFvar of strains obtained from different regions of the world were analyzed, they were classified to four groups (Fig. 1b). Group 1 consisted of the strains obtained from the Northern Atlantic regions, mostly being originated from latitudes higher than 40°N (the latitude of the origin of CCMP452 was 41°N 73°W; Fig. 1b). Group 2 contained three strains obtained from the high-latitude area of the Pacific coast of North America (>47.6°N; Fig. 1b). Group 3 consisted of two strains obtained from different regions of California, USA (Fig. 1b). Finally, group 4 included Atlantic strains, mostly from the area of their latitude lower than 40°N. Group 4 also included strains originated in Japan, Brazil, and Singapore (Fig. 1b). These results indicate that MtORFvar may serve as a useful marker to distinguish group 1∼3 regions. To further test whether MtORFvar can be utilized for phylogeographic marker for higher resolution, we also analyzed the strains obtained from Japanese coastal water and two strains originated from the areas of latitudes >40°, NEPCC522 (group 2) and CCMP452 (group 1), as the outgroup (Fig. 1a, c). Our results revealed that the MtORFvar sequences of the strains originated from different regions of Japan did not show clear geographic pattern (Fig. 1c). These results indicate that MtORFvar sequences showed links to the origins of the strains, while the marker may be more useful to identify the origins separated by long distances, i.e., different oceanic areas or continents. Previously, two out of three identified polymorphic regions of H. akashiwo 18S showed links with geographic origins of the strains, particularly for the strains originated in the Atlantic side of North America (Engesmo et al. 2016). With more nucleotide substitutions (Supplemental Fig. 2), MtORFvar serves as a phylogeographic marker with more potential to distinguish the strain origins. We also observed that Haek9505-1 originated from Florida, USA associated with group 1, and CCMP1595 originated from Rhode Island associated with group 4, while most of the strains contained in the clades were from different regions (Fig. 1a, c). One possible reason for the association of these strains with the “unexpected” groups may be the long distance transport of the strains originated from different regions to the points of their isolations. For the long-distance or domestic dispersion processes, human-assisted dispersion, typically by ship ballast water (Hallegraeff and Bolch 1991, 1992; Elbrachter 1998; Bizsel and Bizsel 2002; Han et al. 2002; Burkholder et al. 2007; Drake et al. 2007; Butron et al. 2011), the commercial transfer of oyster spats and live fish (Matsuyama and Nagai 2010), and by seawater currents (Nagai et al. 2009), could be involved. These possibilities should be further tested by analyzing population structures of H. akashiwo in the regions by MtORFvar and adopting other polymorphic markers, such as microsatellites (Nagai et al. 2006).

Phylogeographic analysis of MtORFvar of H. akashiwo strains obtained from different regions of the world. a Origins of the strains used in this study. The circles represent the origins of the strains whose MtORFvar segments were sequenced, and the triangles represent the origins of the strains of which the whole mitohcondorial genome was sequenced previously. The position of the origin was manually plotted on the blank world map provided by Wikimedia Commons (https://commons.wikimedia.org/wiki/Maps_of_the_world#/media/File:BlankMap-World6.svg). WA Washington state, BC British Columbia, SF San Francisco, LA Los Angels, RI Rhode Island, NY New York, NJ New Jersey, FL Florida, RJ Rio de Janeiro, FR France, ESP Spain, SG Singapore. In inset, enlarged map of Japan and the origins of the strains are shown. FUN Funka Bay, ONG Onagawa, OF Ofunato, AIC Mikawa Bay, Mie Ise Bay, HA Harima Bay, TJ Tajiri port, Y Bingo-nada, Hiroshima, KCH Kochi, FK Fukuoka, AR Ariake Bay. For more detailed information for strain origins, see Table 1. b Phylogenetic analysis of the MtORFvar sequences originating from different regions of the world. c Phylogenetic analysis MtORFvar of H. akashiwo strains from North America and Japan Note that the H. akashiwo strains from different regions in Japan did not show apparent segregation depending on their origins whereas these strains and strains originating from North America clearly segregated from the rest. The bootstrap values that are >50% are shown at the nodes. The open circles show the positions of mid-point roots
Comparison of MtORFvar with other MtDNA-based strain markers
To date, parts of MtDNA have been utilized for species and/or strain identification and the discrimination of populations in many instances. For example, cytochrome oxidase I (COI) genes from different species were used as “DNA barcodes” to evaluate species or strain variations (e.g., Robba et al. 2006; Liu et al. 2011; Yasuda et al. 2012; Hodgkinson et al. 2014; Stoeckle and Thaler 2014; Tamburus and Mantelatto 2016). The analysis of the COI sequences obtained from the full-length MtDNA sequence of the seven H. akashiwo strains revealed that the variations observed in the COI coding region are much less than that in MtORFvar (Supplemental Fig. 3). Particularly, the differences between strains isolated from Japanese coastal waters and from North America are less clear, not showing specific nucleotide substitutions (Supplemental Fig. 2). The MtDNA control regions, or D-loops, in several vertebrate species are another segment of MtDNA that is reported to be rich in variations in many species (e.g., see Fujii and Nishida 1997; Bicknell et al. 2012; Remon et al. 2013; Terencio et al. 2013; Hadas et al. 2015; Wang et al. 2015; Huo et al. 2016; Hu and Gao 2016; Patra et al. 2016; Ramos et al. 2016). To date, the MtDNA control regions have not been determined in H. akashiwo MtDNA. The D-loop of MtDNA is a non-coding region that contains the origin of replication of the organelle genome and transcription promoters for both strands (Anderson et al. 1981; L’Abbe et al. 1991; Martinez-Diez et al. 2006; Fonseca et al. 2008; Pereira et al. 2008; Li et al. 2015). Because MtORFvar is located between two genes, cox3 and nad7, on H. akashiwo MtDNA, which are transcribed in the same direction (Fig. 2), MtORFvar is not likely to contain the D-loop.

Mapping of the MtDNA transcriptome. MtORFvar-coding regions are indicated by red, and other MtDNA genes on the forward and reverse strains are shown by green and blue, respectively (outside, HaTj01; inside, H93616). The light blue and orange curves in the concentric circles represent the read coverage for MtDNAs of HaTj01and H93616, respectively. Scales are numbered clockwise. Scale for read depth is indicated on the bottom right. rbsL large ribosomal RNA subunit, rbsS small ribosomal RNA subunit, cox3 cytochrome c oxidase subunit 3, nad7 NADH dehydrogenase subunit 7
While partial mitochondrial sequences, such as COI and D-loop sequences, are used to study intraspecies variations, study about variations of whole mitochondrial genome sequences in single species are still limited. Such variations are best studied in Homo sapiens (Thaler and Stoeckle 2016), while there are some information available for other organisms, including walking catfish (Clarias batrachus, (Kushwaha et al. 2015), brown brocket deer (Mazama gouazoubira, (Caparroz et al. 2015)), and Antarctic krill (Euphausia superba, (Johansson et al. 2012). While D-loop and COI sequences were widely adopted for many studies, SNPs in MtDNA were found to be distributed across many protein-encoding regions in these species (Johansson et al. 2012; Caparroz et al. 2015; Kushwaha et al. 2015). Although the size of the dataset was small, variations among H. akashiwo MtDNA are concentrated in MtORFvar, exhibiting unique patterns in the accumulation of mutations (Ogura et al. 2016).
Among the seven H. akashiwo strains with which the whole mitochondrial genome sequence information is available, six strains possessed MtORFvar orthologs and strain Y (Masuda et al. 2011) codes for the truncated version of the protein. Similarly, strains AIC41, AIC43, HA20, HA5 Mie16, and Mie 17 code for truncated MtORFvar (Supplemental Fig. 2). The comparison of the MtORFvar at the nucleotide level revealed one or more frame shifts because of single-nucleotide deletions in the coding regions that resulted in the appearance of stop codons, yielding truncated proteins (Supplemental Fig. 2). The deletions were observed within three or four mononucleotide repeats that are expected to be prone to generate errors during PCR amplification and/or sequencing (Supplemental Fig. 2). In addition, nucleotide sequences after the deletions are highly homologous to the corresponding sequences in the other strains (Supplemental Fig. 2). Therefore, observed frame shift in the sequences of these strains are highly likely to be due to artifactual errors during either PCR amplifications or the sequencing of the product.
H. akashiwo MtDNA transcriptome analysis
To further evaluate whether the MtORFvar sequence is actually a gene with coding capacity, we conducted transcriptome analyses of two H. akashiwo strains, H93616 and HaTj01 (Fig. 2). Approximately 0.05 and 0.03% of the total reads obtained from H93616 and HaTj01-derived poly(A+)-containing mRNA, respectively, were mapped to their MtDNA. The read depth at each base shows correlation with the existence of ORF at the position, implying that the genes were predicted correctly. One notable exception was the region flanked by large and small ribosomal RNA coding sequences: the region preceding the large subunit of ribosomal RNA coding sequences was transcribed at high levels although there is no predicted gene with the length. Importantly, a substantial number of reads were mapped to MtORFvar in both H93616 and HaTj01, with the adjacent regions on both sides, presumably the non-coding regions, exhibiting notably low expressions (Fig. 2). These data indicate that MtORFvar is likely to code for a functional protein as predicted.
Proteins coded by MtORFvar of various strains
Next, we attempted to infer the function of the MtORFvar-encoded protein based on sequence information. We conducted PSI-BLAST and BLASTP searches of the NCBI NR database to identify homologs of MtORFvar products from different species with defined functions. There were no hits other than the H. akashiwo MtDNA-encoded proteins homologous to MtORFvar products. To date, the full-length MtDNA sequence of Chattonella marina var. marina, another Raphidophyceae species, has been characterized. However, MtORFvar did not show homology to the C. marina var. marina MtDNA-encoded genes. These results indicate that MtORFvar is a unique gene. No relevant hits were obtained by searching the Pfam database or CD database either, indicating that the protein does not contain a domain or motif with any defined function to date. To gain insight into the functional domain(s) of the protein, the protein sequences of MtORFvar were aligned (Supplemental Fig. 4) to find the conserved regions (Fig. 3). As the differences in nucleotide sequences suggest, the sequences of the Northern Pacific/Atlantic strains that belonged to groups 1 and 2 (Fig. 1b) were distinctively different from isolates associated with two other groups. An extra N-terminal domain was observed in the group 1/2 strains, and several amino acid substitutions were observed between groups 1/2 and 3/4 (Fig. 3 and Supplemental Fig. 4). Regardless of the sequence variations among the strains, parts of the proteins are relatively well preserved (Fig. 3 and Supplemental Fig. 4). The domain may be relevant to the protein function, which remains to be characterized by future analysis.

Schematic representation of domain structure of MtORFvar-encoded proteins based on the sequence variations. The green boxes represent segments that are conserved at >80% identity of all strains, while yellow/red/cyan boxes represent segments with lower identities. The segment that contains indels in some strains are represented with red, and the N-terminal sequence that are only found in groups 1 and 2 in Fig. 1b are represented with cyan
MtDNAs are generally known to be compact, coding for proteins with functions vital for their hosts and with short stretches of non-coding regions. The preservation of MtORFvar as an open reading frame with this extent of sequence variations, especially those observed between strains originated from higher-latitude regions (groups 1 and 2 in Fig. 1b) and from other regions (groups 3 and 4) may imply its vital functions in adapting to distinctive geographic regions. The functional relevance of MtORFvar products in H. akashiwo physiology as well as their variation depending on the region of origin remains to be further analyzed in future studies.
References
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (1981) Sequence and organization of the human mitochondrial genome. Nature 290:457–465
Armbrust EV (2009) The life of diatoms in the world’s oceans. Nature 459:185–192
Bicknell AW, Knight ME, Bilton D, Reid JB, Burke T, Votier SC (2012) Population genetic structure and long-distance dispersal among seabird populations: implications for colony persistence. Mol Ecol 21:2863–2876
Bizsel N, Bizsel KC (2002) New records of toxic algae Heterosigma cf. akashiwo and Gymnodinium cf. mikimotoi in the hypereutrophic Izmir Bay (Aegean Sea): coupling between organisms and water quality parameters. Isr J Plant Sci 50:33–44
Black EA, Whyte JNC, Bagshaw JW, Ginther NG (1991) The effects of Heterosigma akashiwo on juvenile Oncorhynchus tshawytscha and its implications for fish culture. J Appl Ichthyol 7:168–175
Burkholder JM, Hallegraeff GM, Melia G, Cohen A, Bowers HA, Oldach DW, Parrow MW, Sullivan MJ, Zimba PV, Allen EH, Kinder CA, Mallin MA (2007) Phytoplankton and bacterial assemblages in ballast water of US military ships as a function of port of origin, voyage time, and ocean exchange practices. Harmful Algae 6:486–518
Butron A, Orive E, Madariaga I (2011) Potential risk of harmful algae transport by ballast waters: the case of Bilbao harbour. Mar Poll Bull 62:747–757
Caparroz R, Mantellatto AM, Bertioli DJ, Figueiredo MG, Duarte JM (2015) Characterization of the complete mitochondrial genome and a set of polymorphic microsatellite markers through next-generation sequencing for the brown brocket deer Mazama gouazoubira. Genet Mol Biol 38:338–345
Chang FH, Anderson C, Boustead NC (1990) First record of a Heterosigma (Raphidophyceae) bloom with associated mortality of cage-reared salmon in big Glory Bay, New Zealand. N Z J Mar Freshwat Res 24:461–469
Drake LA, Doblin MA, Dobbs FC (2007) Potential microbial bioinvasions via ships’ ballast water, sediment, and biofilm. Mar Poll Bull 55:333–341
Edgar RC (2004a) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi:10.1186/1471-2105-5-113
Edgar RC (2004b) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Elbrachter M (1998) Exotic flagellates of coastal North Sea waters. Helgol Meeresunters 52:235–242
Engesmo A, Eikrem W, Seoane S, Smith K, Edvardsen B, Hofgaard A, Tomas CR (2016) New insights into the morphology and phylogeny of Heterosigma akashiwo (Raphidophyceae), with the description of Heterosigma minor sp. nov. Phycologia 55:279–294
Fonseca MM, Posada D, Harris DJ (2008) Inverted replication of vertebrate mitochondria. Mol Biol Evol 25:805–808
Fu FX, Tatters AO, Hutchins DA (2012) Global change and the future of harmful algal blooms in the ocean. Mar Ecol Prog Ser 470:207–233
Fujii T, Nishida M (1997) High sequence variability in the mitochondrial DNA control region of the Japanese flounder Paralichthys olivaceus. Fish Sci 63:906–910
Hadas L, Hermon D, Boldo A, Arieli G, Gafny R, King R, Bar-Gal GK (2015) Wild gazelles of the southern Levant: genetic profiling defines new conservation priorities. PLoS One 10(3):e0116401
Hallegraeff G (1993) A review of harmful algal blooms and their apparent global increase. Phycologia 32:79–99
Hallegraeff GM, Bolch CJ (1991) Transport of toxic dinoflagellate cysts via ships ballast water. Mar Poll Bull 22:27–30
Hallegraeff GM, Bolch CJ (1992) Transport of diatom and dinoflagellate resting spores in ships ballast water—implications for plankton biogeography and aquaculture. J Plankton Res 14:1067–1084
Han MS, Kim YP, Cattolico RA (2002) Heterosigma akashiwo (Raphidophyceae) resting cell formation in batch culture: strain identity versus physiological response. J Phycol 38:304–317
Hodgkinson A, Idaghdour Y, Gbeha E, Grenier JC, Hip-Ki E, Bruat V, Goulet JP, de Malliard T, Awadalla P (2014) High-resolution genomic analysis of human mitochondrial RNA sequence variation. Science 344:413–415
Honjo T (1993) Overview on bloom dynamics and physiological ecology of Heterosigma akashiwo. In: Smayda TJ, Shimizu Y (eds) Toxic phytoplankton blooms in the sea. Elsevier, Amsterdam, pp 33–41
Hu XD, Gao LZ (2016) The complete mitochondrial genome of eastern lowland gorilla, Gorilla beringei graueri, and comparative mitochondrial genomics of Gorilla species. Mitochondrial DNA A DNA Mapp Seq Anal 27:1484–1485
Huerta-Cepas J, Serra F, Bork P (2016) ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol Biol Evol 33:1635–1638
Huo JH, Wei QP, Wan MC, Liu LX, Hu LF, Zhou QY, Xiong LG, Yang Q, Wu YP (2016) Population phylogenomic analysis and origin of mitochondrial DNA in Chinese domestic pig. Mitochondrial DNA A DNA Mapp Seq Anal 27:892–895
Johansson M, Duda E, Sremba A, Banks M, Peterson W (2012) Assessing population-level variation in the mitochondrial genome of Euphausia superba using 454 next-generation sequencing. Mol Biol Rep 39:5755–5760
Kamikawa R, Asai J, Miyahara T, Murata K, Oyama K, Yoshimatsu S, Yoshida T, Sako Y (2006) Application of a real-time PCR assay to a comprehensive method of monitoring harmful hlgae. Microb Environ 21:163–173
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Kushwaha B, Kumar R, Agarwal S, Pandey M, Nagpure NS, Singh M, Srivastava S, Joshi CG, Das P, Sahoo L, Jayasankar P, Meher PK, Shah TM, Patel AB, Patel N, Koringa P, Das SP, Patnaik S, Bit A, Sarika IMA, Kumar D, Jena JK (2015) Assembly and variation analyses of Clarias batrachus mitogenome retrieved from WGS data and its phylogenetic relationship with other catfishes. Meta Gene 5:105–114
L’Abbe D, Duhaime JF, Lang BF, Morais R (1991) The transcription of DNA in chicken mitochondria initiates from one major bidirectional promoter. J Biol Chem 266:10844–10850
Lackey JB, Lackey E (1963) Microscopic algae and protozoa in the waters near Plymouth in August 1962. J Mar Biol Ass UK 43:797–805
Li H, Durbin R (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25:1754–1760
Li DH, Shi W, Munroe TA, Gong L, Kong XY (2015) Concerted evolution of duplicate control regions in the mitochondria of species of the flatfish family Bothidae (Teleostei: Pleuronectiformes). PLoS One 10(8):e0134580
Liu M, Gao T, Sakurai Y, Jia N, Zhao L, Du X, Jiang Q, Lu Z (2011) Mitochondrial DNA control region diversity and poplation structure of Pacific herring (Clupea pallasii) in the Yellow Sea and the Sea of Japan. Chin J Oceanol Limnol 29:317–325
Mackenzie L (1991) Toxic and noxious phytoplankton in Big Glory Bay, Stewart Island, New Zealand. J Appl Phycol 3:19–34
Martinez-Diez M, Santamaria G, Ortega AD, Cuezva JM (2006) Biogenesis and dynamics of mitochondria during the cell cycle: significance of 3'UTRs. PLoS One 1:e107
Maso M, Garces E (2006) Harmful microalgae blooms (HAB); problematic and conditions that induce them. Mar Poll Bull 53:620–630
Masuda I, Kamikawa R, Ueda M, Oyama K, Yoshimatsu S, Inagaki Y, Sako Y (2011) Mitochondrial genomes from two red tide forming raphidophycean algae Heterosigma akashiwo and Chattonella marina var. marina. Harmful Algae 10:130–137
Matsuyama Y NG, Nagai S, 2010 Direct detection of harmful algae from the oyster spat and live fish transporting trailers. In: Kin-Chung Ho M, J, Zhou and Y. Z. Qi (ed) Proceedings of XIII International Conference on Harmful algae, Hong Kong. pp185–189.
Nagai S, Yamaguchi S, Lian CL, Matsuyama Y, Itakura S (2006) Development of microsatellite markers in the noxious red tide-causing algae Heterosigma akashiwo (Raphidophyceae). Mol Ecol Notes 6:477–479
Nagai S, Nishitani G, Sakamoto S, Sugaya T, Lee CK, Kim CH, Itakura S, Yamaguchi M (2009) Genetic structuring and transfer of marine dinoflagellate Cochlodinium polykrikoides in Japanese and Korean coastal waters revealed by microsatellites. Mol Ecol 18:2337–2352
Ogura Y, Nakayama N, Hayashi T, Ueki S (2016) Mitochondrial genome sequences of four strains of the bloom-forming raphidophyte Heterosigma akashiwo. Genome Announcements 4(6):e01288–e01216
O’Halloran C, Silver MW, Holman TR, Scholin CA (2006) Heterosigma akashiwo in central California waters. Harmful Algae 5:124–132
Ohtsubo Y, Ikeda-Ohtsubo W, Nagata Y, Tsuda M (2008) GenomeMatcher: a graphical user interface for DNA sequence comparison. BMC Bioinformatics 9:376. doi:10.1186/1471-2105-9-376
Patra AK, Kwon YM, Kang SG, Fujiwara Y, Kim SJ (2016) The complete mitochondrial genome sequence of the tubeworm Lamellibrachia satsuma and structural conservation in the mitochondrial genome control regions of order Sabellida. Mar Genomics 26:63–71
Pereira F, Soares P, Carneiro J, Pereira L, Richards MB, Samuels DC, Amorim A (2008) Evidence for variable selective pressures at a large secondary structure of the human mitochondrial DNA control region. Mol Biol Evol 25:2759–2770
Ramos R, Song G, Navarro J, Zhang R, Symes CT, Forero MG, Lei F (2016) Population genetic structure and long-distance dispersal of a recently expanding migratory bird. Mol Phylogenet Evol 99:194–203
Remon N, Galan P, Vila M, Arribas O, Naveira H (2013) Causes and evolutionary consequences of population subdivision of an Iberian mountain lizard, Iberolacerta monticola. PLoS One 8(6):e66034
Rensel J, Homer RA, Postel LE (1989) Effects of phytoplankton blooms on salmon aquaculture in Puget Sound, Washington: initial research. Northwest Environ J 5:53–59
Robba L, Russell SJ, Barker GL, Brodie J (2006) Assessing the use of the mitochondrial cox1 marker for use in DNA barcoding of red algae (Rhodophyta). Am J Bot 93:1101–1108
Rojas de Mendiola B (1979) Red tide along the Peruvian coast. In: Taylor DL, Seliger HH (eds) Toxic dinoflagellate blooms. Elsevier, Amsterdam, pp 183–190
Smayda TJ (1997) Harmful algal blooms: their ecophysiology and general relevance to phytoplankton blooms in the sea. Limnol Oceanogr 42:1137–1153
Smayda TJ (2002) Adaptive ecology, growth strategies and the global bloom expansion of dinoflagellates. J Oceaogr 58:281–294
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Stoeckle MY, Thaler DS (2014) DNA barcoding works in practice but not in (neutral) theory. PLoS One 9(7):e100755
Tamburus AF, Mantelatto FL (2016) Taxonomic and biogeographical studies of three species of the spider crabs of genus Acanthonyx Latreille, 1828 (Jajoidea:Epialtidae) as determined by DNA barcoding and morphological anlaysis along the Western Atlantic. Zool Stud 55. d
Taylor FJR (1993) Current problems with harmful phytoplankton blooms in British Colombia waters. In: Smayda TJ, Shimizu Y (eds) Toxic phytoplankton blooms in the sea. Elsevier, Amsterdam, pp 699–703
Terencio ML, Schneider CH, Gross MC, Feldberg E, Porto JI (2013) Structure and organization of the mitochondrial DNA control region with tandemly repeated sequence in the Amazon ornamental fish. Mitochondrial DNA 24:74–82
Thaler DS, Stoeckle MY (2016) Bridging two scholarly islands enriches both: COI DNA barcodes for species identification versus human mitochondrial variation for the study of migrations and pathologies. Ecol Evol 6:6824–6835
Throndsen J (1969) Flagellates of Norwegian coastal waters. Nytt Mag Bot 16:161–216
Tseng CK, Zhou MJ, Zou JZ (1993) Toxic phytoplankton studies in China. In: Shimizu Y (ed) Smayda TJ. Toxic Phytoplankton Blooms in the Sea. Elsevier, Amsterdam, pp 347–352
Wang X, Liu N, Zhang H, Yang XJ, Huang Y, Lei F (2015) Extreme variation in patterns of tandem repeats in mitochondrial control region of yellow-browed tits (Sylviparus modestus, Paridae). Sci Rep 5:13227
Yasuda N, Taquet C, Nagai S, Fortes M, Suharsono SHA, Phongsuwan N, Nadaoka K (2012) Genetic structure of Culcita sp. pincusion starfish in the coral triangle. In: 12th international coral reef symposium. Cairns, Australia, pp 9–13
Acknowledgements
This work was supported by Ohara Foundation for Agricultural Research, KAKENHI (26291092, 16H06449a8, and 15H01263), and Priority Areas “Comprehensive Genomics” (No. 221S0002) provided by the Ministry of Education, Culture, Sports, Science, and Technology of Japan to SU. The collaboration between SSP and SU was supported by Supporting Program for Interaction-based Initiative Team Studies of Kyoto University. We would like to thank Dr. Natsuko Nakayama (National Research and Development Agency, Japan Fisheries Research and Education Agency) for testing the primers on the H. akashiwo strains.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(PDF 55 kb).
Rights and permissions
About this article

{kind=link}
Cite this article
Higashi, A., Nagai, S., Salomon, P.S. et al. A unique, highly variable mitochondrial gene with coding capacity of Heterosigma akashiwo, class Raphidophyceae. J Appl Phycol 29, 2961–2969 (2017). https://doi.org/10.1007/s10811-017-1142-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-017-1142-2