
Abstract
Cobalt/graphitic carbon catalysts were prepared from Co-MOF crystals, which were synthesized by a solvothermal method in a Teflon-lined autoclave. The MOF crystals were calcined for 2 h to produce Co-MOF-derived catalysts. X-ray diffraction and X-ray photoelectron spectroscopy analysis showed that calcination of the Co-MOF at 600 °C favors the formation of Co3O4, while calcination at 700 °C produced a catalyst composes of metallic Co embedded in a carbon matrix. Raman spectra showed the carbon layer on the Co particles has graphitic properties. The Co-MOF calcined at 700 °C was observed to have the highest surface area and catalytic activity for oxygen reduction reaction (ORR) and oxygen evolution reaction (OER), respectively. During charge/discharge cycling of the Co-MOF-derived catalyst (N700), the potentials were changed within 10–50 mV from the initial potential after 250 h. This catalyst showed excellent stability for ORR and OER, which were superior to these kinds of catalysts reported in the literatures, because graphitic carbon layers covered on the cobalt nanoparticles.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The rechargeable Zn–air battery has been attracting attention as an energy conversion and storage device due to its high energy density, stability, low cost, and environmentally benign properties. However, the practical application of rechargeable Zn–air batteries has been limited, mainly because of the dendritic formation of zinc at the anode, and the sluggish oxygen reaction kinetics at the cathode. An approach to enhance the performance of the rechargeable Zn–air battery is to find a catalyst with good catalytic activity for both the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER). Such a catalyst is called a bifunctional catalyst. Pt–Ru-based catalysts are considered to have excellent catalytic activity for ORR and OER. However, their scarcity, high cost, and stability issues have hindered their commercial application. Hence, in order to mass-produce rechargeable Zn–air batteries, it is necessary to find a low-cost bifunctional catalyst with good stability and high catalytic activity for ORR and OER. Several studies have been conducted to find a good non-precious metal-based bifunctional catalyst for rechargeable Zn–air batteries. Most of the catalysts investigated are transition metal (Fe, Ni, Mn, and Co) oxides, such as spinels and perovskites, because of their stability, ORR and OER catalytic activities, and simple preparation techniques. In general, transition metal-based catalysts are synthesized by calcination of precursors that are usually prepared by a sol–gel, electrospinning, or solvothermal method [1,2,3,4,5,6,7,8].
Recently, the use of a metal–organic framework (MOF) as a precursor for the preparation of catalysts has been investigated [9,10,11]. MOFs, or porous coordination polymers (PCPs), are crystalline materials that exist in one-, two-, or three-dimensional structures composed of highly organized central metal-containing units, or secondary building units (SBUs), and polyfunctional organic ligands. The structural versatility of MOFs allows control of their physical characteristics, such as particle size and porosity, and is highly influenced by the kind of metal ion and the organic ligand, which acts as a linker bridging the SBUs to form the MOF structure [10,11,12,13,14,15,16,17,18,19,20]. Several metals, in the form of nitrates or oxides, have been utilized in MOFs, depending on the intended application [14], whereas the organic linkers used commonly are carboxylates, because of their ability to adapt to different bridging modes [13].
Many studies of MOF-derived bifunctional catalysts for the ORR and OER in alkaline solution have been conducted. Some works dealt with the effect of composites with graphene or carbon nanotubes (CNTs) [21,22,23]. Most research focused on the catalytic evaluation of MOF-derived materials using the rotating disk electrode (RDE) test [24,25,26]. In real Zn–air rechargeable batteries, the catalysts in the gas diffusion electrode should be evaluated for long-term durability (cycling) because the chemical stabilities and capacity retention of electrode components, including the catalyst, at relatively high current and in high concentration alkaline solution (~ 30%) are very important. Only a few studies have reported this kind of test [27, 28]. However, nobody has showed that these kinds of catalysts are stable during OER and ORR cycling under hard condition (20 mA cm−2 for 4 h per cycle) over 200 h (Supplementary Table s1).
In this study, Co/graphitic carbon derived from Co-MOF was investigated for their bifunctional catalytic activity and long-term stability for the ORR and OER in the rechargeable Zn–air battery. The Co-MOF precursor was synthesized by a simple solvothermal method. Co-MOF-derived catalysts were obtained through calcination in nitrogen. The efficiency of the Co-MOF-derived catalysts as bifunctional catalysts using the gas diffusion electrode was determined through their physical and electrochemical characteristics, and one of the catalysts showed that the potentials were changed within 10–50 mV from the initial potential after 250 h.
2 Experimental
The preparation of the electrocatalyst consisted of two parts: the first part was the synthesis of Co-MOF, which was performed by a solvothermal method, and the second part was the synthesis of catalysts from the prepared Co-MOF by calcination. The preparation of the Co-MOF was performed by dissolving 21.9 mmol of cobalt (II) nitrate hexahydrate and 18 mmol of 1,4-benzenedicarboxylic acid (BDC) in 15 mL N,N-dimethylformamide (DMF). Then, the mixture was sealed and heated in a Teflon-lined autoclave at 100 °C for 3 days, followed by cooling to room temperature. The process resulted in the formation of MOF crystals, which were washed with ethanol for several times, and then dried under vacuum at room temperature [15, 18, 29].
The second part of the electrocatalyst preparation was the heat-treatment of MOF crystals. The obtained MOF crystals were pulverized using a mortar and pestle prior to their calcination for 2 h in a nitrogen atmosphere. The calcination temperature of the MOF powder was varied from 500 to 900 °C, with a heating rate of 10 °C min−1.
Imaging of the synthesized Co-MOF crystals and Co-MOF-derived electrocatalysts was performed using scanning electron microscopy (SEM, Hitachi S-4800) and high-resolution-transmission electron microscopy (HR-TEM, JEM-2010) while the specific surface area was measured by BELSORP-max. The crystallinity of the materials was observed by XRD (EMPYRAN HR–XRD using Cu Kα radiation). XPS was performed with MultiLab 2000, using Al Kα radiation. Thermal gravimetric analysis (TGA) was performed to measure weight loss during calcination, and elemental analysis (EA) was also conducted to measure the weight percent of carbon and oxygen. In addition, Raman spectra of the synthesized Co-MOF-derived catalysts were measured with an objective of × 100 and an excitation wavelength of 532 nm to identify the nature of the carbon formed by the carbonization of organic species.
The working electrode was prepared by attaching the catalytic active layer (CAL) to the gas diffusion layer (GDL), which was a carbon paper. The CAL is composed of the Co-MOF-derived catalyst (20 wt%), Ni powder (60 wt%, NICO 255, Vale), carbon black (CB, 10 wt%, Vulcan XC-72, Cabot), and polytetrafluoroethylene solution (PTFE, 10 wt%, Aldrich), which acts as the binder. CAL was prepared by a dispersion method, wherein the catalyst, Ni powder, and carbon black were added to a mixed solution of deionized water and ethanol. Afterwards, PTFE was added, and the mixture was stirred for a few minutes. The mixture was dried overnight at 60 °C to evaporate the solvent completely. The resulting powder mixture was rolled to form a sheet after adding isopropyl alcohol, and was then attached to a carbon paper (P50T, Ballard) using a hot press at 350 °C for 1 min [4, 5]. For comparison, Co3O4 and Co metal powder (Aldrich Co) were used as catalysts. The electrochemical performance of the PtRu/C electrode (2.0 mg cm−2, Fuel Cell Store) was also measured. To identify the electrocatalytic activities for the ORR and OER of the Co-MOF-derived catalysts, linear sweep voltammetry (LSV) was performed in a three-electrode cell system using a potentiostat/galvanostat (WBCS 3000, WonATech) for 20 cycles with a voltage range of 0.5–2.4 V (vs. Zn wire) and at a scan rate of 1 mV s−1. The three-electrode cell system used 8 M KOH solution as the electrolyte, while Pt mesh and Zn wire were used as the counter and reference electrodes, respectively. A cycling test for charge/discharge was conducted in the three-electrode cell system, using Pt wire as the reference electrode instead of Zn to exclude Zn dissolution. The currents for the ORR and OER were induced in the electrodes at 20 mA cm−2 for 2 h per step with 10-min rest time, while measuring the voltage change in the process.
3 Results and discussion
The Co-MOF, a three-dimensional (3D) metal-carboxylate framework with the formula [Me2NH2]2[Co3(1,4-bdc)4]·4DMF (Me2NH2 = protonated dimethylamine), was prepared from Co(NO3)2·6H2O and 1,4-bdcH2 in DMF by a solvothermal reaction. The crystal structure of the Co-MOF is constructed from the tri-nuclear SBUs (secondary building units) Co3(RCOO)8, which contain two crystallographically equivalent six-coordinate terminal Co(II) centers and a six-coordinate central Co(II) center. Each of the SBUs of the Co-MOF framework is linked to eight separate building blocks through carboxyl oxygens in the bdc units, generating a rare bcg net (a uninodal eight-connected net) [29].
Scheme 1 shows the synthesis of cobalt-containing carbon composites from the heat-treatment of the Co-MOF. The resultant metal/carbon composite catalysts have metal nanoparticles covered by a porous carbon layer. The porous carbon layer can prevent an increase in the particle size of the metal during heat-treatment and create a path for the gas to reach the catalyst surface for the oxygen reaction.

Diagram for the synthesis of cobalt-containing carbon composites for oxygen reaction from Co-MOF
The calcination of the synthesized MOF powder under varying temperature and gas atmosphere yielded six samples, which were named according to their calcination conditions in Table 1.
Figure 1a shows the result of TGA under nitrogen of the Co-MOF synthesized by a solvothermal method. The TGA curve shows that the weight loss can be divided into three major regions: (1) weight loss at less than 200 °C, which can be due to the evaporation of non-coordinated and coordinated water molecules; (2) weight loss between 201 and 300 °C, which is attributed to the decomposition of residual nitrates and solvent; (3) weight loss at temperatures greater than 400 °C, which is attributed to the degradation of the organic framework [20, 29]. On the other hand, Fig. 1b shows the results of TGA in air of the catalysts derived from Co-MOF by calcination. The TGA curves show that all the catalysts, aside from N600, gained weight at a certain point between the beginning of the analysis and 500 °C. This can be explained through the formation of cobalt oxide compounds by the oxidation reaction with oxygen. In addition, the drastic drop in weight percent before reaching 600 °C can be explained through the decomposition of carbon compounds formed by the carbonization of organic compounds of MOFs under nitrogen. The weight percent of Co in catalysts was calculated from TGA results, assuming that the final product was cobalt oxide (Co3O4) after 600 °C. Table 1 lists the weight percentages of elements in the catalysts analyzed by energy dispersive X-ray spectroscopy (EDS), EA and TGA. The weight percent of carbon measured by EDS was relatively higher than that measured by TGA. This means that carbon materials were coated on the surface of cobalt particles.

TGA curves of a synthesized Co-MOF under nitrogen and b Co-MOF-derived catalysts under air
In Table 1, the surface area of the catalyst prepared at 700 °C is the highest among the catalysts, and decreases with increasing calcination temperature.
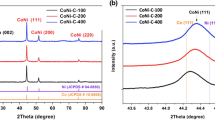
The XRD patterns of the catalysts derived from calcination of the Co-MOF at different temperatures are shown in Fig. 2. It can be observed that catalysts N500 and N600 have similar peaks. Likewise, catalysts N700, N800, and N900 have parallel peaks. However, it is evident that the peak of N900 is more prominent. The peak patterns of N500 and N600 match the XRD pattern JCPDS 78-1970, which corresponds to a cubic Co3O4 spinel at 2θ = 19.14 (111), 31.43 (220), 36.99 (311), 38.7 (222), 44.97 (400), 59.50 (511), and 65.40 (440) with their corresponding Miller indices [30, 31]. The peaks at 2θ = 44.42 (111), 51.67 (200) and 75.81 (220) of N700, N800, and N900 are the characteristic peaks of metallic Co (JCPDS 15-0806), while the peak at 2θ = 26.41 (002) of N800 and N900 indicates the formation of graphitic carbon due to the catalytic effect of Co at high temperature [10, 31, 32].

XRD pattern of Co-MOF-derived catalysts calcined in various temperatures
Figure 3 shows the surface morphology of Co-MOF (a) crystals and (b–f) Co-MOF-derived catalysts that were calcined at different temperatures. The Co-MOF crystals synthesized by the solvothermal method, shown in Fig. 3a, have a smooth surface with a sheet shape and with particle size > 100 μm. Figure 3b–f is the image of Co-MOF-derived catalysts that were calcined under a nitrogen atmosphere at a final temperature of 500–900 °C, respectively. Calcination of the synthesized Co-MOF powder at different temperatures produced a wide range of particle sizes and random shapes. However, unlike the Co-MOF crystal and powder (Fig. 3a), the surface of the Co-MOF-derived catalysts is rough, indicating that the particles are agglomerates of smaller particles.

SEM images of a Co-MOF, b N500, c N600, d N700, e N800, f N900
Figure 4 shows the EDS image of the catalyst prepared at 700 °C and the variation in the weight percent of elements for catalysts calcined at different temperatures. The carbon content continuously increased and the oxygen content sharply decreased to zero over 700 °C. The cobalt content relatively decreased, although the weight of the catalyst did not change after 600 °C, as shown in Fig. 1a. The weight change of cobalt in Fig. 4b may be a result of the increase of carbon coating on the surface of cobalt by the agglomeration of metal particles.

a EDS image of the catalyst, Co-MOF.N700, and b weight percent of each element for catalysts calcined at different temperatures
TEM images of the catalysts derived from the Co-MOF by calcination under nitrogen atmosphere at various temperatures are shown in Fig. 5, respectively. In the figures, it can be observed that particle aggregation increases with increasing calcination temperature. However, the distribution of the Co species (the dark particles embedded in the carbon matrix, which can be either Co3O4 or metallic Co as indicated by the XRD analysis) decreases with increasing calcination temperature by the agglomeration. These particles were covered by a graphitic carbon layer [33, 34], as shown in Fig. 5f.

TEM images of Co-MOF-derived catalysts calcined at different temperatures a N500, b N600, c N700, d N800, e N900, and f N700 (high-resolution)
To identify the chemical state of elements in the Co-MOF-derived catalysts calcined at different temperatures, XPS was carried out and the results are shown in Fig. 6. The overall XPS survey spectrum for the catalysts, shown in Fig. 6a, contains the characteristic peaks for C 1s, O 1s, and Co 2p. The intensity of C 1s peak was amplified with increasing calcination temperature, while the effect on O 1s and Co 2p peak intensity is the opposite. In Table 2, the binding energies and atomic concentrations for the C, Co, and O atoms are listed. The C peak for the catalysts calcined between 700 and 900 °C, observed at the binding energy of 285 eV, and shown in Fig. 6b, indicates the presence of graphitic C in a mixture of sp3 or sp2 hybridized states, while the C peak for the catalysts calcined at 500 and 600 °C suggests the presence of C atoms in different chemical environments, such as C–C, C=O [2, 34, 35]. The Co 2p scan of the Co-MOF-derived catalysts, shown in Fig. 6c, indicates the smaller percentage of Co atoms on the surface of catalysts calcined at higher temperatures. This phenomenon can be explained by the agglomeration of Co particles at higher temperatures, which allows more C atoms on the surface of the catalysts, as observed in the TEM images in Fig. 5. Furthermore, the variation of the binding energy for Co 2p and C 1s from previous studies can be a result of several factors, such as the matrix effect, because the Co species were embedded in a carbon matrix (Fig. 5) [36]. Thus, for the Co 2p spectra of N500 and N600, the peaks for Co 2p3/2 and Co 2p1/2 are observed at a binding energy of 781.8 and 796.1 eV, which can be assigned to the presence of Co3O4. The peak for the Co 2p3/2 of the catalysts calcined at 700 °C and higher, which was observed at 780.5 eV, can be assigned to the Co metal species [3, 37,38,39].

XPS spectra of Co-MOF-derived catalysts a survey spectrum, b C 1s scan, c Co 2p scan, and d O 1s scan
Figure 7 shows the Raman spectra of the catalysts derived from the calcination of Co-MOF. The peaks observed at the Raman shifts ~ 480 and 685 cm−1 represent the Eg and the A1g phonon modes of Co3O4 [40, 41]. This indicates that N900 also contains a traceable amount of Co3O4, like in N500. Furthermore, all the catalysts calcined under nitrogen atmosphere contain graphitic carbon, as suggested by the peaks at ~ 1340 and ~ 1580 cm−1, which represent the D and G peaks from the A1g and E2g symmetries for nanocrystalline graphitic carbon. In addition, the presence of the D peak can be attributed to the disorder caused primarily by the presence of metallic Co, which is observed in the XRD and XPS data (Figs. 2, 6) [41,42,43]. As listed in Table 2, the ratio of ID and IG decreased with increasing heat-treatment temperature. This means that the graphitic property of the carbon coated on the surface increased at a higher temperature [44,45,46].

Raman Spectra of Co-MOF-derived catalysts calcined at various temperatures in N2-atmosphere and fitting curves for catalyst prepared at 700 °C
The LSV results for the Co-MOF-derived catalysts, Co3O4, Co metal powder and PtRu/C, shown in Fig. 8a and b. Figure 8c and d shows average values of apparent current density (CD) based on electrode area and mass activity (MA) on the weight of catalyst in a 1 cm2 electrode from more than five cell tests. Co-MOF-derived catalysts, N500 and N600, which consisted mainly of cobalt oxide compounds, showed higher performance than Co3O4 catalyst. Also, the cell performances of N700, N800, and N900 were higher than that of Co metal catalyst. These indicate that the catalyst prepared from the MOF had high catalytic activity because of its small size and high surface area. Among the Co-MOF-derived catalysts, N700 is the mostly active catalyst for the ORR at 0.5 V (vs. Zn), followed by N500, N800, N900, and N600. Similarly, for the OER at 2.4 V (vs. Zn), N700 has the highest current density followed by N800, N500, N900, and N600. N700 and N800 showed higher performance than PtRu/C for both the ORR and the OER. The notable performance for the ORR and the OER of N700 catalyst can be attributed to its high specific surface area, wherein the calcination temperature is one of the major distinguishing factors. Among the Co-MOF-derived catalysts, N700 showed the highest surface area, as shown in Table 1. The large difference in the surface areas of N700 and N800 from those of N500 and N600 can be explained by the XRD pattern and TEM images, in Figs. 2 and 5. The XRD peaks and TEM images indicate the presence of graphitic carbon, which might largely contribute to their high surface area [2, 31, 39].

Linear sweep voltammetry (a, b), current density(CD) (mA cm−2), and mass activity(MA) (×10−1 mA mg-cat−1) (c, d) at 0.5 and 2.4 V of Co-MOF-derived catalysts, Co3O4, Co and PtRu/C for ORR (c) and OER (d) in 8M KOH solution
N700 has shown an impressive catalytic activity for both the ORR and the OER, and to further determine its potential use as a bifunctional catalyst for a rechargeable Zn–air battery, its cycle durability in alkaline electrolyte was determined. Figure 9a shows the cycle test of N700 for charge/discharge at 20 mA cm−2. At the beginning of the test, the potential was − 0.20 and 0.61 V (vs. Pt) for the ORR and the OER, respectively. After 250 h (almost 60 cycles) of continuous charge and discharge, the potentials were changed to − 0.26 and 0.60 V, respectively. After the cycle test, peaks for Co (catalyst) or Ni metal (conductive additive) were found and no other peaks were detected in the XRD of the electrode, as shown in Fig. 9b. Only H. Dai’s group has reported charge/discharge profiles cycled at 20 mA cm−2 or higher with 10 h per step for ~ 200 h, which showed a negligible voltage change at the end, using CoO/carbon hybrid catalyst and a three-electrode configuration (two cathodes and one anode) [47]. Except for Dai’s report, we have not seen charge/discharge results at 20 mA cm−2 with more than 2 h per step and stable voltage retention for over 200 h (Supplementary Table s1).

Charge/discharge profile a at 20 mA cm−2 with 2 h per step and XRD patterns b before and after cycle test of N700 electrode
Finally, Co-MOF-derived catalysts had high surface area, around 200 m2 g−1, and Co particles, ~ 50 nm in size, and were covered by a graphitic carbon layer, although the Co content in the catalyst powder was around 60%. Heat-treatment over 700 °C converted Co oxide compound to Co metal, which increased the catalytic activity of Co. Nanosize and highly porous Co particles improved the catalytic activities for the OER and the ORR, and the graphitic carbon layer on Co particles increased their chemical resistance in alkaline solution during the long-term cycle durability test. It has been confirmed that the graphitic carbon layer does not have any catalytic activity because the layer does not contain any nitrogen [9, 48, 49]. The synthesis of Co-based catalysts through MOFs and calcination may be a simple way to produce highly porous and stable catalysts for oxygen reactions in alkaline solution.
4 Conclusions
The preparation of electrocatalysts by combined solvothermal synthesis of metal–organic frameworks and calcination is an effective method to produce catalysts with a large surface area, which in turn results in a higher catalytic activity. XPS and Raman spectra showed that Co-MOF-derived catalysts contain traceable amounts of Co3O4, metallic Co and graphitic carbon, which varies with the calcination temperature. Thus, the calcination temperature of Co-MOF-derived catalysts greatly influences the kind of Co species that were embedded in a carbon matrix. Calcination at 600 °C and below favored the formation of Co3O4 over metallic Co, while calcination at 700 °C and higher produced more metallic cobalt than Co3O4. Furthermore, calcination at higher temperature produced significant amounts of graphitic carbon, which enhanced the catalytic activity of N700 and N800. During the cycle test at 20 mA cm−2 with 2 h per step, the catalyst showed stable voltage retention for 250 h because the graphitic carbon layer on the Co particles increased their chemical resistance in alkaline solution.
References
Lee DU, Park MG, Park HW, Seo MH, Ismayilov V, Ahmed R, Chen Z (2015) Highly active Co-doped LaMnO3 perovskite oxide and N-doped carbon nanotube hybrid bi-functional catalyst for rechargeable zinc–air batteries. Electrochem Commun 60:38–41
Prabu M, Ramakrishnan P, Shanmugam S (2014) CoMn2O4 nanoparticles anchored on nitrogen-doped graphene nanosheets as bifunctional electrocatalyst for rechargeable zinc–air battery. Electrochem Commun 41:59–63
Song J, Zhu C, Fu S, Song Y, Du D, Lin Y (2016) Optimization of cobalt/nitrogen embedded carbon nanotubes as an efficient bifunctional oxygen electrode for rechargeable zinc–air batteries. J Mater Chem A 4:4864–4870
Lopez K, Park G, Sun HJ, An JC, Eom S, Shim J (2015) Electrochemical characterizations of LaMO3 (M = Co, Mn, Fe, and Ni) and partially substituted LaNixM1–xO3 (x = 0.25 or 0.5) for oxygen reduction and evolution in alkaline solution. J Appl Electrochem 45:313–323
Lopez K, Sun HJ, Park G, Eom S, Shim J (2015) Electrochemical characterization of electrospun LaCoO3 perovskite nanofibers prepared at different temperature for oxygen reduction and evolution in alkaline solution. Trans Korean Hydrogen New Energy Soc 26:148–155
Hardin WG, Slanac DA, Wang X, Dai S, Johnston KP, Stevenson KJ (2013) Highly active, nonprecious metal perovskite electrocatalysts for bifunctional metal—air battery electrodes. J Phys Chem Lett 4:1254–1259
Shen Q, Yang J, Chen KL, Wang H, Liu JB, Yan H (2016) Co3O4 nanorods–graphene composites as catalysts for rechargeable zinc-air battery. J Solid State Electrochem 20:3331–3336
Su Y, Zhu Y, Jiang H, Shen J, Yang X, Zou W, Chen J, Li C (2014) Cobalt nanoparticles embedded in N-doped carbon as an efficient bifunctional electrocatalyst for oxygen reduction and evolution reactions. Nanoscale 6:15080–15089
Zhao S, Yin H, Du L, He L, Zhao K, Chang L, Yin G, Zhao H, Liu S, Tang Z (2014) Carbonized nanoscale metal–organic frameworks as high performance electrocatalyst for oxygen reduction reaction. ACS Nano 8:12660–12668
Wang X, Zhou J, Fu H, Li W, Fan X, Xin G, Zhen J, Li X (2014) MOF derived catalysts for electrochemical oxygen reduction. J Mater Chem A 2:14064–14070
Qian Y, Hu Z, Ge X, Yang S, Peng Y, Kang Z, Liu Z, Lee JY, Zhao D (2017) A metal-free ORR/OER bifunctional electrocatalyst derived from metal-organic frameworks for rechargeable Zn-air batteries. Carbon 111:641–650
Furukawa H, Cordova KE, O’Keeffe M, Yaghi OM (2013) The chemistry and applications of metal-organic frameworks. Science 341:1230444,1–12
Park G, Kim H, Lee GH, Park SK, Kim K (2006) Solvothermal synthesis, crystal structure, and magnetic properties of [Co3(SDA)3(DMF)2]; 2-D layered metal-organic framework derived from 4,4′-stilbenedicarboxylic acid (H2SDA). Bull Korean Chem Soc 27:443–446
Dikio ED, Farah AM (2013) Synthesis, characterization and comparative study of copper and zinc metal organic frameworks. Chem Sci Trans 2:1386–1394
Nguyen VH, Nguyen NPT, Nguyen TTN, Le TTT, Le VN, Nguyen QC, Ton TQ, Nguyen TH, Nguyen TPT (2011) Synthesis and characterization of zinc-organic frameworks with 1,4-benzenedicarboxylic acid and azobenzene-4,4′-dicarboxylic acid. Adv Nat Sci: Nanosci Nanotechnol 2:025008–025012
Morozan A, Jaouen F (2012) Metal organic frameworks for electrochemical applications. Energy Environ Sci 5:9269–9290
Cui Y, Li B, He H, Zhou W, Chen B, Qian G (2016) Metal–organic frameworks as platforms for functional materials. Acc Chem Res 49:483–493
Hu X, Hu H, Li C, Lou X, Chen Q, Hu B (2016) Cobalt-based metal organic framework with superior lithium anodic performance. J Solid State Chem 242:71–76
Zhao Y, Li K, Li J (2010) Solvothermal synthesis of multifunctional coordination polymers. Z Naturforsch 65:976–998
Sargazi G, Afzali D, Ghafainazari A, Saravani H (2014) Rapid synthesis of cobalt metal organic framework. J Inorg Organomet Polym 24:786–790
Hou Y, Wen Z, Cui S, Ci S, Mao S, Chen J (2015) An advanced nitrogen-doped graphene/cobalt-embedded porous carbon polyhedron hybrid for efficient catalysis of oxygen reduction and water splitting. Adv Funct Mater 25:872–882
Liu S, Zhang H, Zhao Q, Zhang X, Liu R, Ge X, Wang G, Zhao H, Cai W (2016) Metal-organic framework derived nitrogen-doped porous carbon@graphene sandwich-like structured composites as bifunctional electrocatalysts for oxygen reduction and evolution reactions. Carbon 106:74–83
Jiao L, Zhou YX, Jiang HL (2016) Metal–organic framework-based CoP/reduced graphene oxide: high-performance bifunctional electrocatalyst for overall water splitting. Chem Sci 7:1690–1695
Chaikittisilp W, Torad NL, Li C, Imura M, Suzuki N, Ishihara S, Ariga K, Yamauchi Y (2014) Synthesis of nanoporous carbon–cobalt oxide hybrid electrocatalysts by thermal conversion of metal–organic frameworks. Chem Eur J 20:4217–4221
Gadipelli S, Zhao T, Shevlin SA, Guo Z (2016) Switching effective oxygen reduction and evolution performance by controlled graphitization of a cobalt–nitrogen–carbon framework system. Energy Environ Sci 9:1661–1667
Xia BY, Yan Y, Li N, Wu HB, Lou XW, Wang X (2015) A metal–organic framework-derived bifunctional oxygen electrocatalyst. Nature Energy 1:15006
Chen Z, Yu A, Higgins D, Li H, Wang H, Chen Z (2012) Highly active and durable core—corona structured bifunctional catalyst for rechargeable metal—air battery application. Nano Lett 12:1946–1952
Liu S, Zhang X, Wang G, Zhang Y, Zhang H (2017) High-efficiency Co/CoxSy@S,N-codoped porous carbon electrocatalysts fabricated from controllably grown S, N including Co-based MOFs for rechargeable zinc-air batteries. ACS Appl Mater Interfaces 9:34269–34278
Wang X, Zhang Y, Xue W, Qi X, Chen X (2010) Two temperature-induced isomers of metal-carboxylate frameworks based on different linear trinuclear Co3(RCOO)8 clusters exhibiting different magnetic behaviours. CrystEngComm 12:3834–3839
Patil V, Joshi P, Chougule M, Sen S (2012) Synthesis and characterization of Co3O4 Thin Film. Soft Nanosci Lett 2:1–7
Liu Y, Zhang X (2009) Effect of calcination temperature on the morphology and electrochemical properties of Co3O4 for lithium-ion battery. Electrochim Acta 54:4180–4185
Xia W, Zou R, An L, Xia D, Guo S (2015) A metal–organic framework route to in situ encapsulation of Co@Co3O4@C core@bishell nanoparticles into a highly ordered porous carbon matrix for oxygen reduction. Energy Environ Sci 8:568–576
Aijaz A, Masa J, Rçsler C, Xia W, Weide P, Botz AJR, Fischer RA, Schuhmann W, Muhler M (2016) Co@Co3O4 encapsulated in carbon nanotube-grafted nitrogen doped carbon polyhedra as an advanced bifunctional oxygen electrode. Angew Chem Int Ed 55:4087–4091
Wu MS, Hsu WH (2015) Nickel nanoparticles embedded in partially graphitic porous carbon fabricated by direct carbonization of nickel-organic framework for high-performance supercapacitors. J Power Sources 274:1055–1062
Baek JH, Park JY, Hwang AR, Kang YC (2012) Spectroscopic and morphological investigation of Co3O4 microfibers produced by electrospinning process. Bull Korean Chem Soc 33:1242–1246
Kim KS, Winograd N (1975) X-ray photoelectron spectroscopic binding energy shifts due to matrix in alloys and small supported metal particles. Chem Phys Lett 30(1):91–95
Biesinger MC, Payne BP, Grosvenor AP, Lau LWM, Gerson AR, Smart RSC (2011) Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl Surf Sci 257:2717–2730
Lee LM, Kim JW, Lim JS, Kim TJ, Kim SD, Park SJ, Lee YS (2007) X-ray photoelectron spectroscopy study of cobalt supported multi-walled carbon nanotubes prepared by different precursors. Carbon Sci 8:120–126
Park J, Shen X, Wang G (2009) Solvothermal synthesis and gas-sensing performance of Co3O4 hollow nanospheres. Sens Actuators B 136:494–498
Hadjiev VG, Iliev MN, Vergilov IV (1988) The Raman spectra of Co3O4. J Phys C 21:L199–L201
Lorite I, Romero JJ, Fernandez JF (2012) Isolated nanoparticle raman spectroscopy. J Raman Spectrosc 43:889–894
Tembre A, Henocque J, Clin M (2011) Infrared and raman spectroscopic study of carbon-cobalt composites. Int J Spectrosc. https://doi.org/10.1155/2011/186471
Filik J, May PW, Pearce SRJ, Wild RK, Hallam KR (2003) XPS and laser Raman analysis of hydrogenated amorphous carbon films. Diamond Relat Mater 12:974–978
Lee HS, Baek GY, Hwang IT, Jung CH, Choi JH (2017) Preparation of porous carbon films from polyacrylonitrile by proton irradiation and carbonization. Rad Phys Chem 141:369–374
Yoo SH, Joh HI, Lee SH (2017) Synthesis of porous carbon nanofiber with bamboo-like carbon nanofiber branches by one-step carbonization process. Appl Surf Sci 402:456–462
Zhang Z, Zhang Y, Mu X, Du J, Wang H, Huang B, Zhou J, Pan X, Xie E (2017) The carbonization temperature effect on the electrochemical performance of nitrogen-doped carbon monoliths. Electrochim Acta 242:100–106
Li Y, Gong M, Liang Y, Feng J, Kim JE, Wang H, Hong G, Zhang B, Dai H (2013) Advanced zinc-air batteries based on high-performance hybrid electrocatalysts. Nat Commun 4:1805–1811
Zhang P, Sun F, Xiang ZH, Shen ZG, Yun J, Cao DP (2014) ZIF-derived in situ nitrogen-doped porous carbons as efficient metal-free electrocatalysts for oxygen reduction reaction. Energy Environ Sci 7:442–450
Silva R, Voiry D, Chhowalla M, Asefa T (2013) Efficient metal-free electrocatalysts for oxygen reduction: polyaniline-derived N- and O-doped mesoporous carbons. J Am Chem Soc 135:7823–7826
Acknowledgements
This work was supported by the National Research Foundation of Korea Grant funded by the Korean Government (MEST) (NRF-2012-M1A2A2-029538 & 2017-R1D1A3B03035157).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kim, K., Lopez, K.J., Sun, HJ. et al. Electrochemical performance of bifunctional Co/graphitic carbon catalysts prepared from metal–organic frameworks for oxygen reduction and evolution reactions in alkaline solution. J Appl Electrochem 48, 1231–1241 (2018). https://doi.org/10.1007/s10800-018-1245-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-018-1245-8