
Abstract
This study examined the possibility to remove colour causing-compounds from synthetic effluent by indirect electrochemical oxidation using iridium oxide anode electrodes. Using a high concentration of chloride ions (17.1 mM) and various current densities, it was possible to produce high concentration of active chlorine with a specific production rate of 2.8 mg min−1 A−1. The best performance for acid methyl violet 2B dye (MV2B) decomposition was obtained using Ti/IrO2 anodes operated at a current density of 15 mA cm−2 during 40 min of treatment in the presence of 3.42 mM of chloride ions. Under these conditions, more than 99% of MV2B was removed (with a reaction rate apparent constant of 0.20 min−1), whereas COD and TOC removal were 51% and 75%, respectively. The electrolytic cell was then used for the degradation of three other synthetic dye solutions: Eosin yellowish (EOY), Trypan Blue (TRB), Acridine Orange (ACO). TRB was the most difficult dye to remove from solution with a value reaction rate constant of 0.12 min−1, compared to 0.19 min−1 and 0.24 min−1 recorded for ACO and EOY dyes, respectively. More than 99% of these dyes were removed by electrochemical oxidation.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
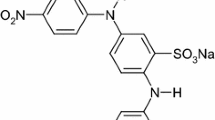
The effluents of many industries (textile, leather, pulp and paper, printing, photographs, cosmetics, pharmaceutical, and food) contain dyes, which represent an important environmental problem [1, 2]. These industries are major water consumers and are therefore a source of considerable pollution. Reactive dyes are hydrophilic, therefore, they have low affinity to adsorb to biomass and generally pass through conventional biological wastewater systems [3, 4]. Moreover, dye molecules are highly structured molecules that are potentially toxic to organisms [5]. Therefore, it is difficult to degrade them biologically [6]. With the discharge regulation becoming increasingly restrictive, conventional treatment systems and discharge into municipal sewers or natural water bodies would no longer be possible. For instance, methyl violet 2B is a triphenylmethane dye with a molecular mass of 380 Da (Fig. 1). Triphenylmethanes are extensively used in textile and paper mill industries to color nylon, wool, silk, and cotton [7]. This type of dye is hydrophilic, therefore, they have low affinity to adsorb to biomass and generally pass through conventional biological wastewater systems [3, 4].

Molecular structure of methyl violet 2B dye (MV2B)
Several methods have been used or proposed for color removal from wastewaters. This includes biological aerobic (e.g., activated sludge, SBR, biofilter) [8–10] or anaerobic (e.g., UASB) [1, 11] treatments, enzymatic biodegradation (actinomycetes, fungi) [12], chemical oxidation (H2O2/Fe2+ (Fenton), e.g., hypochlorite), [13, 14] or reduction (e.g., Fe0) [15], electrochemical oxidation (e.g., O3/UV, O3/H2O2, O3/UV/H2O2, H2O2/UV) [16, 17], photodegradation (e.g., TiO2/UV, photo-Fenton) [18, 19], adsorption (e.g., activated carbons, silica, biosorbents) [20, 21], membrane separation (e.g., microfiltration, ultrafiltration, nanofiltration) [22], chemical coagulation/flocculation (e.g., aluminum, iron, or calcium salts) [23], and electrolytic treatments, which include electroflocculation [24], and/or electrocoagulation [25–28], and electro-oxidation [29–31].
Electro-oxidation is an electrochemical technique of treating polluted effluent whereby the pollutant can be oxidized by using both direct and indirect effect of electrical current. Direct anodic oxidation, where the organics can be destroyed at the electrode surface, and indirect oxidation where a mediator (HClO, HBrO, H2O2, H2S2O8, and others) is electrochemically generated to carry out the oxidation [32, 33]. Two different ways can be followed in anodic oxidation: electrochemical conversion or electrochemical combustion [34]. Electrochemical conversion only transforms the non-biodegradable organic pollutants into biodegradable compounds, whereas electrochemical combustion yields water and carbon dioxide and no further treatment is then required. The indirect effect of electrolysis is also interesting to destroy recalcitrant organics. For instance, in the presence of chloride ions, these ions can be oxidized at the anode electrodes and formed hypochlorous acid (HClO) in solution. HClO is a powerful oxidant capable of oxidizing and modifying the structure of organic molecules [35, 36] and leading to more oxidized and less toxic compounds. In particular, in situ generation of active chlorine acid finds wide application in treatment of sewage [37, 38] for e.g., water onboard ship [37], textile dye and finishing wastewaters [39], tannery wastewater [37]. The process of electrochemical degradation of organic pollutant using in situ active chlorine generation has several advantages over other more common process of water treatment. Compared with process such as chlorination by the use of gaseous chlorine or concentrated hypochlorite solution, no addition of chemicals is necessary. Additionally, the hazards in handling these chemicals are also avoided. Likewise, aqueous solutions of sodium hypochlorite are much safer to use than chlorine gas. Storage and transportation costs are relatively high for chlorine gas. The electrolytic production of chlorine is one of the most important industrial electrochemical reactions [39, 40] and is widely used for the disinfection of drinking water [41, 42]. In general, the oxidation of chloride ion can be observed on a variety of anodic materials [43]. However, in dilute chloride solution, a parallel reaction occurs on the anode concomitant with the formation of chlorine, i.e., the oxygen evolution reaction. As the solution to improve the method, generally dimensionally stable anodes (DSA) are used. These electrodes are thermally prepared oxide electrodes for which a titanium substrate is covered by metallic oxides such as TiO2, IrO2, RuO2, SnO2 for e.g., [44–46]. Among them, iridium-coated titanium electrode (IrO2/Ti) is one of the cheapest electrode widely used for active chlorine production for water or wastewater treatment.
The objectives of this research were to investigate the ability of a cylindrical electrolytic cell comprising circular IrO2/Ti anode electrodes to generate active chlorine by the electro-catalytic oxidation of chloride solution in the recirculating batch test. In addition, the feasibility of using the approach to degrade a methyl violet (MV2B) dye by indirect generation of chlorine was studied to determine if this method was suitable for treating such a dye. The effects of current density, supporting electrolyte, electrode material, initial MV2B dye concentration, and electrolyte flow rate on MV2B decomposition were examined. Then, the performance of the electrolytic cell in treating three other synthetic dye solutions (Eosin yellowish (EOY), Trypan blue (TRB), Acridine orange (ACO)) were evaluated.
2 Materials and methods
2.1 Electrolytic reactor set-up
The reactor unit used in this study was of 2 l capacity and made of PVC material with a dimension of 15 cm (height) × 14 cm (diameter). The electrolytic cell comprised two anode and two cathode electrodes in the form of expanded metal, each having a solid surface area of 65 cm2 and a void surface area of 45 cm2 (Fig. 2). The electrodes were circular discs (12 cm diameter × 0.1 cm thick) and titanium (Ti) was used as cathode. The circular anode electrodes (12 cm diameter × 0.1 cm thick) were respectively titanium coated with iridium oxide (Ti/IrO2) and titanium coated with tin (Ti/SnO2). The inter-electrode gap was 10 mm in the electrolytic cell. The electrodes were horizontally installed inside the electrolytic cell and each anode was immediately followed by a cathode. The electrodes were supplied by Electrolytica Inc. (Amherst, NY, USA).

Configuration of electrolytic cells using circular electrodes
2.2 Experimental unit
The assays were carried out in a closed loop, depicted schematically in Fig. 3. A one liter of PVC reservoir (4), a recycling pump (5) and the electrolytic cell (1) (fully detailed in Fig. 2) constitute the loop. The assays were conducted in batch recirculation mode with a flow of wastewater entering the bottom of the cell. The recycle flow rate (varying from 2 l min−1 to 6 l min−1) was measured using a flow-meter (6). A needle valve (2) installed before a manometer (3) allowed controlling the hydrostatic pressure inside the cell. The excess of gas generated during electrolysis was rejected out of the system by means of a venting pipe (11) fixed on the 1-liter PVC reservoir. At the start of each assay, the raw effluent was injected in the experimental unit by means of a funnel (7) installed in the pipe and connected to the recycling pump, which allowed adding a working volume of 3.5 l. The electrochemical cells were operated under galvanostatic conditions, with current densities imposed during a period of treatment ranging from 10 to 60 min. Current densities were imposed by means of a DC power source, Xantrex XFR40-70 (Aca Tmetrix, Mississauga, ON, Canada) with a maximum current rating of 70 A at an open circuit potential of 40 V. Current densities were calculated using a factor taking into account the area of the electrode meshes.

Schematic view of the electro-oxidation cell with a recirculation loop
2.3 Process and treatment description
During electrolysis of dye effluent containing chloride ions, chlorine was produced at the anode and hydrogen gas was produced at the cathode. Due to the dissociation of the generated chlorine in water, hypochlorous acid and chloride were produced as illustrated by Eq. 1.
In order to characterize the electrolytic cell (described above) in terms of hypochlorous of acid generation, preliminary assays were performed using distilled water enriched with chloride ions added in form of chloride sodium (NaCl, Fisher Scientific, ACS reagent). Electrolysis was conducted at a pH around the neutral value (pH 6.0–7.0) for various current densities. Samples were drawn periodically and analyzed to assess the performance of the system for active chlorine production.
Once characterized, the experimental unit was used for the treatment of a synthetic solution containing a methyl violet 2B dye (MV2B) (J.T. Baker Chemical, New Jersey, USA). These experiments consisted to test successively different operating parameters such as, current densities (3.8–30 mA cm−2), retention times (10–60 min), initial dye concentration (25–150 mg l−1), nature of anode electrode (Ti/IrO2 and Ti/SnO2), and concentration of electrolyte (1.7–17 mM Cl−) in order to determine the best conditions (reduce cost and increase effectiveness) in treating MV2B-containing solution. The electrochemical treatment of the synthetic solution was conducted in a batch process with continuous recirculation. During these assays, only the residual dye concentrations were measured to evaluate the performance of the experimental unit for MV2B dye degradation. Thereafter, the best conditions determined while treating MV2B-containing solution were applied for the degradation of three other synthetic dyes representing azoic molecules with one or many acid functions: eosin yellowish (J.T. Baker Chemical, New Jersey, USA), Trypan blue (Allied Chemical, New Jersey, USA), Acridine orange (Fisher Scientific, New Jersey, USA). The synthetic dye solutions were individually prepared using distilled water and subjected to electro-oxidation treatment. These dyes were selected because they represent the main families of dye agents which can be found in the textile, paper mills, and leather industries. During the second set of experiments, DOC, TOC, and total chlorine concentration were monitored, in addition to residual dye concentration to assess the efficiency of the process.
2.4 Analytical details
The pH was determined using a pH-meter (Fisher Acumet model 915) equipped with a double-junction Cole–Palmer electrode with Ag/AgCl reference cell. A conductivity meter (Oakton Model 510) was used to determine the ionic conductivity of the effluent. The total chlorine concentration was estimated by an iodometric method by titration with thiosulfate in the presence of starch [47]. Concentrations of initial and residual dyes were established by spectrophotometric method after preparing calibration curves. The dyes used for this purposes were Methyl violet 2B (MV2B, λmax = 585 nm), Eosin yellowish (EOY, λmax = 516 nm), Trypan blue (TRB, λmax = 596 nm), and Acridine orange (ACO, λmax = 492 nm). COD was measured by Hach COD method and a reading spectrophotometer Carry UV 50 (Varian Canada Inc.). TOC was measured using a Shimadzu TOC 5000A analyzer (Shimadzu Scientific Instruments Inc.) equipped with an auto sampler. The chemical oxygen demand method (Hach COD method) was used for the determination of the current efficiency for the oxidation of dyes. In this method, the COD was measured during electrolysis, and the instantaneous current efficiency was calculated using the relation:
where COD t and CODt+Δt are the chemical oxygen demands (in gram of O2 per cubic decimeter) at times t and t + Δt (in seconds), respectively; I is the current intensity (in ampere), F is the Faraday constant (96,500 Coulombs) V is the volume of the electrolyte (in cubic decimeters), and 8 is a dimensional factor for unit consistency [(32 g of O2 mol−1)/(4 mol of electron exchanged)].
3 Results and discussion
3.1 Active chlorine production
The primary objective of the preliminary tests was to evaluate the efficacy of the electrolytic cell for active chlorine production. It is worth noting that, the overall concentration of dissolved chlorine in water is termed the “active” chlorine and is the sum of the three possible species, Cl2, HClO, and ClO−. In the pH range 6–9, the active chlorine is almost entirely constituted by hypochlorous acid (HClO) and hypochlorite (ClO−). In the present study the concentration was measured as active chlorine in mol l−1 and converted into mg l−1, this conversion being based on the atomic weight of Cl (35.45 g mol−1). The dependence of the active chlorine production on chloride ion concentration for chloride concentrations up to 17.1 mM (1,000 mg NaCl l−1) with a current density of 15 mA cm−2 is shown in Fig. 4. It can be seen that, the active chlorine concentration, in the loop of the unit increased with elapse time. For both lower concentrations of chlorides imposed (1.71 mM and 3.42 mM), the active chlorine concentration tended toward a plateau from 30 min of treatment time compared to higher chloride concentration imposed. The active chlorine concentration increases with increasing chloride ion concentration. For instance, at a treatment time of 30 min, the chloride concentration imposed of 17.1 mM allowed producing 3.1 times as much active chlorine as a chloride concentration imposed of 1.71 mM (100 mg NaCl l−1).

Effect of initial chloride ion concentration on active chlorine production during electrolysis (current density: 15 mA cm−2; recycling flow rate: 2 l min−1)
The rate of active chlorine production was proportional to current density (Fig. 5), which is in agreement with the result obtained by Kraft et al. [42]. They observed the same dependence on current density of the production rate of chlorine and the slope recorded was 1.0 mg min−1 A−1, IrO2 being used as anode with a 150 mg l−1 chloride concentration. By comparison, the specific chlorine production rate recorded in the present study was 2.8 mg min−1 A−1 with 121 mg l−1 of chloride concentration. This discrepancy can be mainly attributed to the design of the electrolytic cell which can be greatly influenced the mass transfer inside the reactor. In fact, all experiments of Kraft et al. [42] were carried out using divided electrode compartments to prevent the reduction of produced hypochlorite at cathode, anolyte, and catholyte being separated by a Nafion 450 cation exchanged membrane. Anolyte and catholyte were circulated at 0.3 l min−1 using ProMinent® gamma pumps. By comparison, in the present study the assays were carried out using a single-cell process (without ion exchanged membrane) and the electrolyte circulated in a closed loop at 3 l min−1. The liquid flow rate was 10 times higher than that imposed by Kraft et al. [42]. Indeed, this relatively high flow rate created turbulence effect inside the electrolytic cell so that chloride ions were easily transferred toward the electrode and easily oxidized at the anode.

Influence of the current density on active chlorine production (recycling flow rate: 2 l min−1; chloride ion concentration: 17.1 mM)
The performance of the electrolytic cell in terms of chlorine production was also evaluated by calculating the Faradayic efficiency against current density imposed for different retention time (Fig. 6). The values of Faradayic efficiencies for chlorine evolution reaction were calculated using the following equation:
where V is the volume of solution (l), F is Faraday’s constant (96,500 Coulombs), \( C_{{\text{Cl}}_{ 2}} \) is the concentration of active chlorine (g l−1), I is the applied current intensity (A), n e is the number of electrons transferred in the reaction at the electrode, \( M_{{\text{Cl}}_{ 2}} \) is the molecular weight (g mol−1) and t is the electrolysis time (s). In Fig. 6, Faradayic efficiencies for chlorine evolution are plotted against current density. For 20, 40, and 60 min of retention time the Faradayic efficiencies increase rapidly from 0 to 15 mA cm−2, and then decreased slowly. The maximum values of η recorded at 15 mA cm−2 decreased while increasing the retention time imposed, 17, 13, and 11% were measured for 20, 40, and 60 min of retention time. The limit of the diffusion current, corresponding to the oxidation of chloride into active chlorine was lower than the imposed current. The overall reaction was then limited by diffusion and small fraction of the overall energy was used to oxidize chloride into active chlorine. Moreover, the Faradayic yield is low compared to the other electrochemical processes; for example, 0.8 was obtained with an electrochemical water softener [48]. Parasitic reactions probably take place, such as water oxidation in oxygen, active chlorine reduction, or active chlorine decomposition. Kraft et al. [42], who produced active chlorine by electrolysis of a 150 mg l−1 chloride solution (anode in IrO2) obtained a current efficiency of 41.5%, twice to four time higher than that recorded in the present study. However, the electrolytic cell proposed has an advantage of producing active chlorine with a relatively high flow rate. In the following study concerning the application to dye removal, the electrolytic cell was operated for retention of 40 min since the Faradayic yield decreased while increasing the period of treatment time.

Variation of current efficiency in relation to current density for different retention times (recycling flow rate: 2 l min−1; chloride ion concentration: 17.1 mM)
3.2 Electrochemical oxidation of methyl violet (MV2B) dye-containing solution
3.2.1 Influence of applied current density
One of the main factors affecting the electrochemical oxidation efficiency is the current density. Current density is one of the most important parameters that can affect organic removal. The effect of current density on the electro-oxidation of MV2B dye solution was evaluated by comparing the rates of color removal (by measuring the residual MV2B concentration) at current densities of 3.8, 7.6,15, 23, and 30 mA cm−2 in 17.1 mM of chloride (1.0 g NaCl l−1). Figure 7 shows time course changes in the normalized concentration of MV2B. The initial MV2B concentration imposed during these assays was 50 mg l−1. With the exception of current density of 3.8 mA cm−2, 100% degradation of MV2B was reached for all current density imposed. At the higher current density of 30 mA cm−2, faster degradation was obtained as shown in Fig. 7. After only 5 min of electrolysis at the higher current density, there was complete MV2B degradation from the solution, compared to 15 and 30 min required for current densities of 15 and 7.6 mA cm−2, respectively. While imposing the lower current density, the yield of MV2B degradation reached 80% at the end of the treatment (40 min). As discussed above, the rate of active chlorine production was proportional to current density. Generally, the higher the concentration of active chlorine in the bulk solution, the more effective the oxidation is for organic pollutant degradation. These results are consistent with those obtained by Oliveira et al. (2007) while oxidizing acid red dye (4.0 × 10−5 mol l−1) using an electrolytic cell on titanium capable of generating active chlorine on (Ti) recovered with Sn(1−x)IrxO2 electrode. They obtained 100% color removal from solution in 12 min of electrolysis while imposing the higher current density of 25 mA cm−2.

Influence of the current density on MV2B dye degradation during electrolysis (recycling flow rate: 2 l min−1; chloride ion concentration: 17.1 mM)
The reaction order and the apparent rate constant of color removal can be determined by plotting Ln (C/C 0) against time (t) (Fig. 8). The experimental data are well fitted to first-order kinetics (with correlation coefficients ranging from 0.89 to 0.99) predicting a linear variation with elapsed time (t) of the –Ln (C/C 0):
where “C 0” and “C” represent respectively the initial and residual dye concentration in the bulk solution and “k” is the first-order rate coefficient. The fist-order reaction rate apparent constant of MV2B dye degradation reaction was measured at various current density from 3.8 to 30 mA cm−2 at a chloride concentration of 17.1 mM, initial MV2B of 25 mg l−1 and a recirculation flow rate of 2 l min−1. As shown in Fig. 9, the reaction rate apparent constants for MV2B degradation increased linearly (from 0.039 to 0.25 min−1) over the current density of 15 mA cm−2 and then remained quite stable beyond this value. It is interesting to compare the reaction rate apparent constants of MV2B degradation in synthetic solution with those obtained under different experimental condition. The reaction rate apparent constant of organic degradation has been determined by Kim et al. [49] while studying electrochemical oxidation of polyvinyl alcohol (PVA) using titanium coated with ruthenium oxide (Ti/RuO2). The apparent constant rate decreased from 0.0162 min−1 to 0.0021 min−1 while increasing initial PVA concentration from 33 to 2,400 mg l−1 at the condition of 1.34 mA cm−2 current density, 17.1 mM Cl− concentration and 0.7 l min−1 flow rate. The smaller the initial PVA concentration, the faster it could be removed from solution by anodic oxidation. It can be seen that, the kinetic rate apparent constants determined in the present study were 2 to 100 times higher than those measured by Kim et al. [49]. This discrepancy can be attributed to two major factors. First, in the present study a current density varying from 3.8 to 23 mA cm−2 was imposed, whereas Kim et al. [49] imposed a current density of 1.34 mA cm−2, which was 2 to 17 times lower. Secondly, polyvinyl alcohol was probably more difficult to oxidize than MV2B. Figure 9 presents also the change in energy consumption as a function of current density. The energy consumption varied in a linear fashion between 3.8 and 23 mA cm−2, from 0.35 to 4.8 kWh m−3. Since the reaction rate apparent constant of MV2B degradation increased between 3.8 and 15.0 mA cm−2, the energy consumption could be reduced by selecting the current density of 15 mA cm−2.

First-order relationship of MV2B degradation by electrochemical oxidation (current density: 15 mA cm−2; recycling flow rate: 2 l min−1; chloride ion concentration: 17.1 mM)

Effect of current density on reaction rate constants of MV2B dye removal and power consumption (recycling flow rate: 2.0 l min−1; chloride ion concentration: 17.1 mM; initial MV2B concentration: 50 mg l−1)
3.2.2 Effect of supporting electrolyte
The addition of an electrolyte in solution during electrolysis can influence the treatment since it modifies the conductivity of the effluent and facilitates the passage of the electrical current. The effect of the electrolyte concentration, particularly the effect of the chloride concentration, on the kinetic of electrochemical oxidation was determined. The first-order oxidation rate apparent constants of MV2B dye decomposition reaction were measured at various chloride concentrations ranging from 1.71 mM Cl− to 17.1 mM Cl− at a current density of 15.2 mA cm−2, initial MV2B of 50 mg l−1. As shown in Fig. 10, the reaction rate apparent constants for MV2B dye decomposition increased over this range, while the power consumption values decreased. The MV2B reaction rate apparent constant increased from 0.14 to 0.32 min−1. Power consumption decreased from 10.7 kWh m−3 to 2.82 kWh m−3 as the concentration of chloride was increased from 1.71 mM Cl− to 17.1 mM Cl−. The power consumption value at 1.71 mM Cl− was relatively higher than the value at other chloride concentrations. This is consistent with the results of Kim et al. [49] while oxidizing polyvinyl alcohol using Ti/RuO2 anode in the presence of chloride concentration varying from 6.0 mM Cl− to 85.6 mM Cl−. At a cost of 0.06 USD kWh−1 of energy and 60 USD t−1 of sodium chloride (NaCl), the electrochemical treatment involved a total cost of 0.65, 0.43, 0.26, and 0.23 USD per cubic meter of treated dye effluent while imposing 1.71, 3.42, 8.56. and 17.1 mM NaCl, respectively. The addition of the electrolyte modified the conductivity of the solution and facilitated the passage of the electrical current, so that the treatment cost (including only energy and electrolyte cost) was reduced to 0.23 USD m−3 while adding for instance 17.1 mM NaCl in dye synthetic solution, compared to 0.65 USD m−3 recorded for 1.71 mM of NaCl. Consequently, the addition of electrolyte had an effect on the treatment cost.

Effect of chloride concentration on reaction rate constants of MV2B degradation and power consumption (current density: 15 mA cm−2, recycling flow rate: 2.0 l min−1; initial MV2B concentration: 50 mg l−1)
The degradation of MV2B dye occurred due to the generation of active chlorine that is powerful oxidizing specie. On the other hand, OH° radical should also be generated on the catalytic anode (such as Ti/IrO2). In fact, during electrolysis the organics in the solution can be decomposed by both direct anodic electrochemical oxidation (by means of OH°) and indirect electrochemical oxidation via mediators, such as hypochlorite ion and hypochlorous acid. Both situations would lead to the formation of powerful oxidizing agents capable of degrading the MV2B dye. Likewise, hydroxyl radical production could be greatly influenced by anode material. In order to verify these hypotheses, complementary experiments using two types of supporting electrolyte (NaCl and Na2SO4) and two type of anode material (Ti/IrO2 and Ti/SnO2) were carried out. During these assays, a current density of 15 mA cm−2 was imposed in the presence of 1.0 g NaCl l−1 (17.1 mM NaCl) and 1.0 g Na2SO4 l−1 (7.0 mM Na2SO4), respectively. Residual MV2B dye concentrations were monitored and are shown in Fig. 11. The control assay consisted only in circulating the MV2B dye solution in the experimental unit without imposing any current density.The results indicate that MV2B dye removal occurred slowly in the sulfate media using either Ti/IrO2 or Ti/SnO2, reaching a maximum discoloration of only 20% after 90 min of electrolysis. The decrease in MV2B dye concentration during the control assay was probably attributed to the deposition of a small fraction of dye on the electrolytic tank or on the pipes of the experiment unit. Considering the possible deposition of MV2B dye, the real contribution of direct anodic electrochemical oxidation can be obtained by subtracting the yields of MV2B removal (while imposing current density) from the yields recorded without current density. Thus, in our experimental conditions, the real yield of MV2B degradation by direct oxidation was around 10%. By comparison, a remarkable difference in the MV2B dye decomposition was observed when sodium chloride was used as supporting electrolyte. Indirect electrochemical oxidation contributed to more than 80% in the MV2B dye decomposition using either Ti/IrO2 or Ti/SnO2 anode, Ti/IrO2 being more effective than Ti/SnO2. Finally, Ti/IrO2 anode and NaCl used as electrolyte support was retained for the next step of this study. A concentration of 3.42 mM NaCl was selected (rather than 17.1 mM NaCl) in order to minimize chlorine gas production during electrolysis.

Influence of anode material and supporting electrolyte on the degradation rate of MV2B (current density: 15 mA cm−2, recycling flow rate: 2.0 l min−1; chloride concentration: 17.1 mM; initial MV2B concentration: 50 mg l−1)
3.2.3 Effect of initial MV2B dye concentration
The effects of initial MV2B concentration on electrochemical oxidation are shown in Fig. 12. MV2B dye concentration varied from 25 to 150 mg l−1 at the condition of 15 mA cm−2 current density, 3.42 mM Cl− concentration, 2 l min−1 flow rate, and Ti/RuO2 anode material. As the initial concentration of MV2B dye decreased in solution, the depurative efficiency increased. For instance, after 10 min period of treatment, 40%, 60%, 90%, and 99% of dye removal were recorded while the initial MV2B dye concentrations of 150, 100, 50, and 25 mg l−1 were, respectively imposed. Likewise, as the initial MV2B concentration increased, the reaction rate apparent constants of MV2B decreased from 0.28 min−1 to 0.09 min−1. The smaller the initial MV2B concentration, the faster it could be removed from solution by indirect electrochemical oxidation. This is in agreement with Abdo et al. [29] and Kim et al. [49] studies which argued that the electrochemical oxidation process of dye solution was diffusion controlled process. The first reaction rate constant at 25 mg MV2B l−1 was three times higher than that of 150 mg l−1. In other words, if the initial MV2B concentration was low, indirect electrochemical oxidation could be more effectively conducted. From Fig. 12, it can also be seen that, power consumption decreased from 6.86 to 6.10 kWh m−3 as MV2B dye concentration increased. It was believed that the presence of MV2B dye contributed to increase the conductivity of the effluent and facilitate the passage of the electrical current.

Variation of normalized dye concentration with reaction time for different initial MV2B concentrations and effect of MV2B dye concentration on reaction rate constants of MV2B degradation (current density: 15 mA cm−2; recycling flow rate: 2.0 l min−1; chloride concentration: 3.42 mM)
3.2.4 Influence of recycling flow rate
The performance and the behaviors of the electrochemical oxidation of MV2B dye at various flow rates (2.0, 3.0, and 6.0 l min−1) were studied at a current density of 15 mA cm−2, Cl− concentration of 3.42 mM, and initial MV2B of 50 mg l−1. The influence of electrolyte flow rate on MV2B degradation is shown in Fig. 13. The reaction rate apparent constants for MV2B decomposition decreased slightly (from 0.18 min−1 to 0.13 min−1), as recycling flow rate increased from 2.0 to 6.0 l min−1. These results can be compared with those obtained by Nagata et al. [50] while treating different effluents containing endocrine disrupting chemicals (17β-estradiol, biphenol, pentachlorophenol, dichlorophenol, etc.,) using electro-oxidation process with a three-dimensional electrode system. They observed that, removal efficiency increased from 60 to 90% as the recycling flow rate increased from 0.1 to 0.8 l min−1. Similar results were also recorded by Kim et al. [49] while studying electrochemical oxidation of polyvinyl alcohol (PVA) using titanium coated with ruthenium oxide (Ti/RuO2). The reaction rate apparent constants for PVA removal increased steadily from 0.0036 min−1 to 0.0053 min−1 as the flow rates were increased from 0.1 l min−1 to 1.0 l min−1. However, in our experiment conditions, while increasing the recirculation rate to 6.0 l min−1, degradation efficiency decreased. It is worth noting that an increase in the recirculation rate is accompanied by higher velocity in the cell. For instance, a linear velocity of 0.58 cm s−1 was imposed for 6.0 l min−1 compared to 0.19 cm s−1 measured for 2.0 l min−1. It is worth noting that, during electrolysis, two possible extreme limitation processes can be considered: mass transfer control and electrochemical reaction control. In indirect electrooxidation process, the electro active specie (chloride ion) must be transported to the electrode surface first, and then be oxidized there into active chlorine (electrochemical reaction side), followed by its transportation in solution where the pollutant is oxidized by the electro-generated oxidant. Regarding the influence of recycling flow rate, it is believed that the process is not under mass transfer control, but it is probably limited by the electrochemical reaction contrary to Kim et al. [49] and Nagata et al. [50] studies. Indeed, in the present study, while increasing the recycling flow rate, the liquid arrived rapidly on anode electrodes so that the chloride ions cannot be efficiently oxidized to produce active chlorine required for dye oxidation. Thus, a recycling flow rate of 2.0 l min−1 was selected for the next step of the experiments. On the other hand, power consumption behavior was not significantly affected by the flow rate of solution.

Variation of normalized dye concentration with reaction time for different recycling flow rates and effect of recycling flow rate on reaction rate constants of MV2B degradation (current density: 15 mA cm−2; chloride concentration: 3.42 mM; initial MV2B concentration: 50 mg l−1)
3.2.5 Active chlorine requirement for MV2B degradation
In order to evaluate active chlorine required for MV2B dye decomposition during electrolysis, the best experimental condition was repeated using current density of 15 mA cm−2, initial MV2B of 50 mg l−1, Cl− concentration of 3.42 mM, 2.0 l min−1 of recycling flow rate, and a Ti/IrO2 anode. During electrolysis run, 20 ml samples were drawn at 10 min intervals in order to follow the residual active chlorine concentrations in solution, which were compared to the values recorded while electrolysing a synthetic effluent carried out in the same condition but without MV2B addition. The results are presented in Fig. 14. The residual MV2B concentration decreased rapidly over the first 15 min of the treatment and then remained quite stable until the end of experiment. It can also be seen that, once nearly all dye has been oxidized (after 15–20 min of treatment time), active chlorine concentration can linearly increase similarly to the case where there is no dye (with a slope of 0.48 mg l−1 min−1). By comparison, active chlorine concentration linearly increased from 0 to 50 mg l−1 with a relatively high slope (1.81 mg l−1 min−1) during electrolysis of synthetic solution without any addition of MV2B. As it can be seen from Fig. 14, treatment required 20 min to completely decompose MV2B dye. Considering this period of treatment, a concentration of 8.0 mg l−1 of active chlorine was measured during electrolysis of MV2B dye solution, whereas 34 mg l−1 of active chlorine was recorded in the absence of MV2B dye. Thus, the active chlorine dose required for complete MV2B degradation can be obtained by subtracting the residual active chlorine concentration measured in the absence of MV2B from the initial value recorded in the presence of MV2B dye. Thus, a minimum dose of chlorine concentration of 26 mg l−1 was required for complete MV2B decomposition.

Analogy between MV2B degradation and active chlorine consumption during electrolysis of MV2B solution (current density: 15 mA cm−2, recycling flow rate: 2.0 l min−1; chloride concentration: 3.42 mM; initial MV2B concentration: 50 mg l−1)
3.3 Application to the treatment of other synthetic dyes effluents
The electrolytic cell was then used for the degradation of three other synthetic dye solutions representing azoic molecules with one or many acid functions: Eosin yellowish (EOY), Trypan blue (TRB), Acridine orange (ACO). The best experimental conditions determined while treating MV2B-containing solution were applied. The results are summarized in Table 1. Additional information (such as COD and TOC) about MV2B dye decomposition was indicated in this table. The residual concentration COD and TOC recorded at the end of electro-oxidation of MV2B dye solution were 15.7 ± 0.0 mg COD l−1 and 19.8 ± 0.6 mg TOC l−1, respectively. By comparison 62.7 ± 3.7 mg l−1 and 40.6 ± 4.5 mg l−1 were measured respectively in the untreated-MV2B dye solution. The yield of TOC removal was 51% compared to 75% of COD removal for MV2B. It is believed that only a fraction of MV2B dye was completely oxidized into water and carbon dioxide, the majority of MV2B dye being transformed into small molecules that reduce the oxygen demand in the treated-effluent. In fact, the electrolytic cell broke the double bonds producing smaller molecules. It is worth noting that, during electrolysis, organic pollutants can be subjected to two different paths in anodic oxidation: electrochemical conversion or electrochemical combustion [32, 34, 51]. Electrochemical conversion only transforms the non-biodegradable organic pollutants into biodegradable compounds, whereas electrochemical combustion yields water and carbon dioxide and no further treatment is then required. In the present study, electrochemical conversion was the predominant reaction. The current efficiency recorded during electrolysis of MV2B dye solution was 41.3 ± 3.2%. It was calculated from Eq. 2. This value was relatively high compared to those measured (2 to 18%) by Panizza and Cerisola [52] while treating a synthetic wastewater containing acid blue 22 dye (C32H25N3O9S3Na2) using a boron-doped diamond electrode in the solution of 0.5 M Na2SO4. Table 1 compares also the performance of the electrolytic cell in treating three synthetic dye effluents (EOY, TRB, and ACO dyes). Considering the reaction rate constants, it can be seen that, TRB was the most difficult dye to remove from solution with a value of 0.12 min−1, compared to 0.19 min−1 and 0.24 min−1 recorded for ACO and EOY dyes, respectively. The current efficiencies were 17.2%, 16.1%, and 5.7% for EOY, ACO, and TRB, respectively, confirming that TRB remained the most difficult dye to remove. However, the current efficiencies for EOY and ACO were relatively low compared to the values (41.3%) recorded for MV2B dye although the reaction rates constants for the three dyes (EOY, ACO, and MV2B) were quite similar. This discrepancy can be attributed to the initial COD concentrations in EOY and ACO synthetic effluents, which were 1.8 to 2.8 times lower than that measured in MV2B effluent. In all cases, the current efficiencies were below 100% indicating that in our experimental condition the electrochemical oxidation was carried out at a current density higher than the stochiometrically required minimum to oxidize wastewater organic content, and that the process was under mass-transport control.
4 Conclusion
Active chlorine production in dilute chloride solution was electrochemically generated at a high specific rate (2.8 mg min−1 A−1) with a recirculating batch electrolytic cell. The rate of active chlorine production was proportional to current density. Faradayic yield was low, ranging between 0.11 and 0.17. A series of experiments using the electrolytic cell were performed in order to investigate the performances and behaviours of electrochemical of MV2B dye effluent. MV2B dye decomposition exhibited behaviours of first-order reactions. The decomposition of MV2B by electrochemical oxidation was affected by the operating conditions such as chloride concentration, MV2B concentration, current density, flow rate, and anode material. The efficiencies of the electrochemical oxidation of MV2B were proportional to chloride concentration, current density and disproportional to MV2B concentration and flow rate, respectively. The best performance for MV2B-dye decomposition was obtained using iridium oxide electrodes operated at a current density of 15 mA cm−2 during 40 min of treatment in the presence of 3.42 mM of chloride ions. When the concentration of the initial MV2B was low, the electrochemical oxidation was especially effective. Under these conditions, more than 99% of MV2B was removed (with a reaction rate apparent constant of 0.20 min−1), whereas COD and TOC removal were 51% and 75%, respectively. The electrolytic cell was then used for the degradation of three other synthetic dye solutions: Eosin yellowish (EOY), Trypan blue (TRB), Acridine orange (ACO). TRB was the most difficult dye to remove from solution with a value of 0.12 min−1, compared to 0.19 min−1 and 0.24 min−1 recorded for ACO and EOY dyes. The next step should be the study to determine a mathematic correlation between apparent constant kd and both parameters (namely, initial MV2B and active chlorine concentrations) by analyzing the active chorine consumption in function of each initial MV2B concentration imposed.
Abbreviations
- MV2B:
-
Methyl violet 2b
- EOY:
-
Eosin yellowish
- TRB:
-
Trypan blue
- ACO:
-
Acridine orange
- COD:
-
Chemical oxygen demand
- TOC:
-
Total organic carbon
- DSA:
-
Dimensionally stable anodes
- ICE:
-
Instantaneous current efficiency
- USD:
-
US dollar
- kd :
-
Dye decomposition rate apparent constant
- C0 :
-
Initial concentration of dye
- C:
-
Concentration of dye at time t
References
Bell J, Buckley CA (2003) Water SA 29:129
McMullan G, Meehan C, Conneely A et al (2001) Appl Microbiol Biotechnol 56:81
Carliell CM, Barclay SJ, Naidooo N et al (1995) Water SA 21:61
Bell CB (1998) Biological decolourisation of textile effluent in a nutrient removal system. M.Sc. Eng. Thesis, School of Chem. Eng., University of Natal, Durban, South Africa
Parac-Osterman D, Grancaric AM, Sutlovic A (2004) Influence of chemical structure of dyes on decolourization effects. AIC 2004 Color and Paints, Interim Meeting of the International Color Association, pp 179–182
Banat IM, Nigam P, Singh D et al (1996) Bioresour Technol 58:217
Zawlotzki Guivarch E (2004) Traitement des polluants organiques en milieux aqueux par procédé électrochimique d’oxydation avancée “Electro-Fenton”. Application à la minéralisation des colorants synthétiques. Ph.D. Thesis, Université Marne la Vallée, France
Chandra R (2001) J Environ Biol 22:23
Kodam KM, Soojhawon I, Lokhande PD et al (2005) World J Microbiol Biotechnol 21:367
Lourenco ND, Novais JM, Pinheiro HM (2000) Water Sci Technol 42:321
M′endez-Paz D, Omil F, Lema JM (2005) Enzym Microb Technol 36:264
Ramsay JA, Goode C (2004) Biotechnol Lett 26:197
Sevimli MF, Kinaci C (2002) Water Sci Technol 45:279
Shah V, Bhatt M, Stopka P et al (2005) Asian J Water Environ Pollut 2:61
Chang MC, Shu HY, Yu HH et al (2006) J Chem Technol Biotechnol 81:1259
Al-Kdasi A, Idris A, Saed K et al (2004) Global Nest Int J 6:222
Arslan-Alaton I (2003) Color Technol 119:345
El-Ghazi I, Elamrani MK, Mansour M (2004) Toxicol Environ Chem 85:1
Torrades F, Garcia-Montano J, Garcia-Hortal JA et al (2004) Coloration Technol 120:188
Ahmed MN, Ram RN (1992) Environ Pollut 77:79
Ben Tahar F, Ben Cheikh R, Blais JF (2004) J Environ Eng Sci 3:269
Mutlu SH, Yetis U, Gurkan T et al (2002) Water Res 36:609
Lin SH, Peng CF (1996) Water Res 30:587
Ciardelli G, Ranieri N (2001) Water Res 35:567
Golder AK, Hridaya N, Samanta AN et al (2005) J Hazard Mater B127:134
Ibanez JG, Singh MM, Szafran Z (1998) J Chem Educ 75:1040
Kashefialasl M, Khosravi M, Marandi R et al (2006) Int J Environ Sci Technol 2:365
Kobya M, Demirbas E, Can OT et al (2006) J Hazard Mater B132:183
Abdo MSE, Al-Ameeri RS (1987) J Environ Sci Health A22:27
Jia L, Liao J, Wang W et al (1999) Water Res 33:881
Kuperferle MJ, Galal A, Bishop PL (2004) J Environ Eng Sci 3:223
Grimm J, Bessarabov D, Sanderson R (1998) Desalination 115:285
Comninellis C, Pulgarin C (1993) J Appl Electrochem 23:108
Pulgarin C, Adler N, Peringer P et al (1994) Water Res 24:887
Drogui P, Blais JF, Mercier G (2007) Recent Patent Eng 1:257
Canizares P, Lobato J, Paz R et al (2005) Water Res 39:2687
Vijayaraghavan K, Ramanujam TK, Balasubramanian N (1998) J Environ Eng 124:887
Drogui P, Bureau MA, Blais JF et al. (2006) Electrochemical stabilization and preconditioning process for municipal and industrial sludge. Canada patent pending No. CA 2,511,091
Oliveira FH, Osugi ME, Paschoal FMM et al (2007) J Appl Electrochem 35:583
Xie YF (2004) Disinfection byproducts in drinking water, formation, analysis and control. CRC Press LLC, Boca Raton, Florida
Rudolf M, Rousar I, Krisa J (1995) J Appl Electrochem 25:155
Kraft A, Stadelmann M, Blaschke M et al (1999) J Appl Electrochem 26:861
Matskevich ES, Slipchenko AV (1993) Role of surface oxygen complexes on different anodes in the electrolysis of aqueous solutions. Zh Prikl Khim 66:1493
Feng YJ, Cui YH, Sun LX et al (2004) Harbin Gongye Daxue Xuebao 36:450
Zhang XD, Li WS, Lin Y (2007) Diandu Yu Tushi 26:51
Mousty C, Foti G, Comninellis C et al (1999) Electrochimica Acta 45:451
Rodier J, Bazin C, Broutin JP et al (1996) L’analyse de l’eau, eaux naturelles eaux résiduaires, eau de mer, 8th edn. Donud, Paris, France
Bannoud AH, Persin F, Rumeau M (1993) Water Res 27:1385
Kim S, Kim T, Park C et al (2003) Desalination 155:49
Nagata R, Prosnansky M, Sakakibara Y (2006) J Adv Oxid Technol 9:97
Drogui P, Elmaleh S, Rumeau M et al (2001) J Appl Electrochem 31:877
Panizza M, Cerisola G (2008) J Hazard Mater 153:83
Acknowledgments
Sincere thanks are extended to the National Sciences and Engineering Research Council of Canada for their financial contribution to this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zaviska, F., Drogui, P., Blais, JF. et al. In situ active chlorine generation for the treatment of dye-containing effluents. J Appl Electrochem 39, 2397–2408 (2009). https://doi.org/10.1007/s10800-009-9927-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-009-9927-x