
Abstract
Purpose
To explore the protective effect of probucol on human retinal Müller cells cultured in high glucose.
Methods
Primary Müller cells from human retinas were cultured in complete DMEM. Third-generation Müller cells were identified using glutamine synthetase (GS) antibody and randomly divided into three groups: normoglycemia (NG, 5.5 mmol/L); hyperglycemia (HG, 30 mmol/L); and hyperglycemia (30 mmol/L) with probucol (10 μmol/L; HGPB). After a 24-h intervention, cell proliferation, apoptosis, and cellular reactive oxygen species (ROS) were measured with a CCK-8 kit, flow cytometry, and DCFH-DA probe, respectively. Kelch-like ECH-associated protein 1 (Keap1), NF-E2-related factor 2 (Nrf2), and glutamate cysteine ligase catalytic subunit (GCLC) protein expression were detected by immunofluorescence staining.
Results
For NG, HG, and HGPB, optical density (OD) values for cell proliferation were 0.98 ± 0.23, 0.58 ± 0.11, and 0.73 ± 0.11; apoptotic rates were 2.79 ± 0.52%, 7.70 ± 0.44%, and 4.00 ± 0.95%; and intracellular ROS were 20.89 ± 5.14, 55.17 ± 14.07, and 26.28 ± 4.73, respectively. Compared to NG, OD was markedly decreased (P < 0.01), apoptosis was increased (P < 0.001), and intracellular ROS level was significantly higher than in HG (P < 0.01). Compared to HG, OD was markedly increased (P < 0.01), apoptosis was meaningfully decreased (P < 0.01), and intracellular ROS level was significantly lower than in HGPB (P < 0.01). GS, Keap1, Nrf2, and GCLC had positive expression.
Conclusions
Probucol could inhibit intracellular ROS generation, promote proliferation, and decrease apoptosis of human retinal Müller cells cultured in high glucose. This might also be associated with Keap1/Nrf2/ARE oxidative stress signaling pathway activation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetic retinopathy (DR) is considered one of the most common and severe microvascular complications of diabetes and has become the leading cause of blindness. High blood pressure, hyperglycemia, inflammation, and some pro-angiogenic factors are involved in its pathological process, which ultimately harms retinal ganglion cells, vascular endothelial cells, and glial cells. Müller cells are important glial cells in the retina, which play a significant role in the regulation of neuron cell function and the maintenance of the blood–retina barrier. Before the disorder of endothelial cells and pericytes, Müller cells are notably reduced and act dysfunctional in the early process of DR [1,2,3]. Vascular endothelial growth factor (VEFG) is the main protein that promotes the formation of neovascularization, and is mostly produced by Müller cells in DR. High glucose levels stimulate Müller cells to activate endoplasmic reticulum stress (ERS) and produce high levels of VEGF. Increasing evidence suggests that Müller cell-derived VEGF critically contributes to retinal vascular leakage and neovascularization in DR [4,5,6]. Exploring the functional changes of Müller cells in DR is thus of great significance.
Although mechanisms of DR are still unclear, several classical molecular mechanisms are widely recognized to be involved in the pathogenesis of DR, namely the polyol pathway, elevation of advanced glycation end-product (AGE) formation, protein kinase C (KPC) activation, the amino-hexose pathway, the metalloproteinase pathway, and oxidative stress. Oxidative stress is considered to be the final pathway of in these mechanisms and is closely associated with reactive oxygen species (ROS) induced by hyperglycemia [7,8,9,10,11,12,13]. In normal conditions, ROS are generated continuously as natural products and play crucial roles in many biochemical reactions to maintain homeostasis of the internal environment. There are many efficient antioxidant defense systems in the body, including the antioxidant defense enzymes. Once the antioxidant system is compromised, the elimination ability of free radicals becomes impaired. Excess oxygen free radicals can connect with proteins and cross-link with DNA. Thus, all resulting abnormal reactions will severely interfere with normal metabolisms, induce cell apoptosis and neovascularization in the retina, and ultimately promote the development of DR [14, 15].
Cells contain different types of antioxidant defense mechanisms, including an enzymatic antioxidant system and a non-enzymatic antioxidant system. NF-E2-related factor 2 (Nrf2)/Kelch-like ECH-associated protein 1 (Keap1)/antioxidant response element (ARE) signaling pathway has been considered one of the major pathways associated with the pathological process of DR [16]. Nrf2 activation can prevent DR progression and its complications, such as nephropathy, neuropathy, retinopathy, and metabolic syndrome [17]. As a redox-regulating transcription factor, Nrf2 plays a protective role in oxidative stress reaction. Under normal conditions, Nrf2 exists in the cytosol and closely binds with its inhibitor, Keap1. When cellular oxidative stress occurs, Nrf2 dissociates from Keap1, transfers to the nucleus, and binds with ARE to promote its downstream antioxidant gene expression, such as glutamate cysteine ligase (GCL), glutathione peroxidase (GPX), and hemeoxygenase-1 [18]. The GCL catalytic subunit (GCLC) was demonstrated to be the rate-limiting active component responsible for the biosynthesis of the antioxidant factor, glutathione (GSH), which is mainly regulated by Nrf2. In the retinas of diabetic rats, although Nrf2 expression and its binding with Keap1 were increased in the cytosol, GSH synthesis and DNA binding with Nrf2 in the nuclei were declined [19, 20]. Nrf2 has been confirmed to be significantly expressed in Müller cells of mouse and human retinas, and Nrf2-deficient diabetic mice showed blood–retina barrier dysfunction, increased levels of superoxide, and decreased GSH at the early stage [21].
Probucol, a well-known effective anti-atherosclerosis drug, displays potent anti-oxidative and anti-inflammatory effects. Zhou et al. [22] found that probucol exerted a protective activity in diabetic nephropathy (DN) by decreasing the level of nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox2) and inhibiting ROS generation. The redox enzyme, p66Shc, was recently discovered to be a biomarker for kidney damage and is increased in DN patients. Yang et al. [23], however, demonstrated that probucol can suppress the expression of p66Shc. Endothelial progenitor cells (EPCs), which possess a critical function in atherosclerosis prevention, are remarkably decreased in hyperlipidemia ambience. In addition, probucol could protect EPCs from oxidized low-density lipoprotein by reducing ROS production [24]. Spinal cord injury (SCI) causes accumulation of inflammatory factors and eventually influences nerve recovery. In a recent study, Zhou et al. [25] showed that activation of the Nrf2/ARE signaling pathway by probucol could promote functional recovery by reducing inflammatory responses and inhibiting neuronal apoptosis after SCI. Through our previous studies, we revealed that probucol could reduce oxidative stress and improve vision function in patients with DR; however, the underlying mechanisms are yet to be clearly illustrated [26, 27]. Based on the aforementioned findings, we explored the effects and possible protective mechanisms of probucol on Müller cells cultured in high glucose conditions.
Materials and methods
Primary human retinal Müller cell culture
All donated human eyeballs were provided by the AIER Eye Bank of Changsha (Changsha, China). Donors and their legally authorized representatives agreed to the donation of their eyeballs after death for scientific research. Consent forms were also signed. All experimental approaches were approved by the Ethics Committee of AIER Eye Group. Eyeballs without any contamination and impairment could be included in this research.
Retinas were carefully dissociated from human eyes within 6 h after death and washed thrice with phosphate-buffer saline (PBS). The retinas were digested with trypsin–EDTA (Gibco, Grand Island, USA) for 5 min and centrifuged at 800 rmp/min for 5 min. All cells were seeded on a six-well plate containing 2 mL of complete DMEM [10% fetal bovine serum (FBS) (Gibco) and 1% penicillin–streptomycin (Gibco)]. Primary cells were cultured in an incubator (37 °C, 5% CO2). Medium change was performed every 2–3 days.
The adherent primary Müller cells were digested with 0.25% trypsin–EDTA (Gibco) for 2 min, centrifuged at 800 rmp/min for 5 min, and passaged at a 1:2 ratio. Primary cells were routinely identified by the typical morphology expected as well as the glutamine synthetase (GS) antibody. Subsequent studies were performed with the third-generation cells.
Morphological characteristic of primary Müller cells by H&E staining
Müller cells on a glass coverslip were fixed with 95% ethyl alcohol, rinsed twice with PBS, stained with hematoxylin reagent (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) for 2 min, and finally treated with eosin (Nanjing Jiancheng Bioengineering Institute) for 1 min. The cellular morphological characteristics were observed and captured with an inverted optical microscope (Zeiss, Germany).
Immunofluorescence staining
Primary Müller cells on a coverslip were preliminarily fixed with 4% paraformaldehyde for 15 min, rinsed with PBS, and then permeated using 0.1% Triton-X100 for 20 min. After washing with PBS, the cells were blocked with 5% bull serum albumin (BSA) (Beijing Solarbio Technology, Beijing, China) at 37 °C for 1 h and, respectively, reacted overnight with GS (1:100; Santa Cruz, Dallas, Texas, USA), Keap1 (1:100; Abcam, Cambridge, UK), Nrf2 (1:500; Abcam, Cambridge, UK), and GCLC antibody (1:100; Abcam, Cambridge, UK) at 4 °C. PBS was employed as negative control. After two rounds of washing with PBS, the cells were reacted with CY3 (1:100; Molecular Probes, Eugene, Oregon, USA) and FITC (1:500; Molecular Probes, Eugene, Oregon, USA) secondary antibodies, away from light, for 1 h. Finally, the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (1:4000; Beijing Solarbio Technology) for 5 min. Images were observed and captured by a fluorescence microscope (Olympus, Japan).
Groups
The third-generation Müller cells were randomly divided into the following three groups: normoglycemia group (NG; 5.5 mmol/L), hyperglycemia group (HG; 30 mmol/L), and hyperglycemia group (30 mmol/L) with probucol (HGPB; Food and Drug Inspection Institute of China, Beijing, China) (10 μmol/L). To exclude serum interference, all cells were washed thrice with basal DMED. The following studies were performed after 24 h of intervention.
Intracellular reactive oxygen species assay
2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA; Sigma-Aldrich, USA) was used to detect ROS activity in cells. DCFH-DA, a non-fluorescent probe, can freely permeate cells and be hydrolyzed for DCFH production. When oxidative stress occurs, DCFH can be oxidized to DCF, displaying fluorescence. The fluorescence value indicates the level of intracellular ROS. To eliminate the effect of probucol, cells were rinsed thrice with DMEM. The cells from each group were incubated with DCFH-DA probe for 20 min in the dark at 37 °C. Intracellular fluorescence images were observed and captured by a fluorescence microscope (Olympus, Japan) at an excitation wavelength of 488 nm and emission wavelength of 525 nm. The quantitative values of the fluorescence were evaluated with a multi-functional microplate reader (Synerg™).
Cell proliferation assay
Cell proliferation was assayed with a CCK-8 kit (Shanghai BestBio, Shanghai, China). The solution color is directly proportional to the speed of cell proliferation, and generally, the darker the solution, the stronger is the proliferative activity. All assay processes were performed under manufacturer instruction. Optical density (OD) values were measured with a multi-functional microplate reader (Synerg™) at 450 nm.
Detection of apoptosis by flow cytometry
Double staining was used for flow cytometric detection of cell apoptosis. Annexin V-FITC, a specific biomarker of apoptosis, can specifically bind to membrane phospholipid phosphatidylserine (PS). Propidium iodide (PI) is a nucleic acid dye that can only permeate inactive cells. Applying a combination of those two stains (Nanjing Vazyme Biotech, Nanjing, China) allows the identification of valid cells, apoptotic cells, and necrotic cells. Pre-treated Müller cells were washed twice with cold PBS, resuspended in 500 μL binding buffer (Nanjing Vazyme Biotech) at a concentration of 1 × 106 cells/mL, reacted with Annexin V-FITC (5 μL) and PI (5 μL) by gentle eddying, and incubated for 15 min at 25 °C. The stained cells were detected within 1 h by flow cytometry (Beckman Coulter, USA) with the Summit Software, version 5.0 (Beckman Coulter, USA). A total of 10,000 cell counts were recorded for each sample.
Statistical analyses
Each group of experiments was performed independently more than three times. Statistical analysis was performed with SPSS software (Chicago, IL, USA) version 18. All data are expressed as mean ± standard deviation (M ± SD) and were statistically analyzed by Student’s t test for paired groups or one-way analysis of variance for multiple groups. A P < 0.05 indicated statistical significance.
Results
Morphology of isolated primary cells
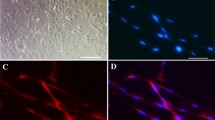
In the primary cell culture, cells were observed to gradually adhere to the plate 3 days after seeding. After approximately 2 weeks, approximately 80% of the 6-well plate was covered with cells. Cells morphology reflected characteristics of typical Müller cells with regular elongation, rich plasma, and large nuclei (Fig. 1).

Morphology of the isolated primary cells presented as typical Müller cells. a Third-generation isolated primary cells (4×) showed stable morphology, and purity of cells was above 95%. b H&E staining (40×) indicated that most of the isolated primary cells were fused together with a rich plasma and large nuclei. Some Müller cells contained more than one nucleus
Identification of Müller cells by GS immunofluorescence staining
In the retina tissue, GS is only expressed in Müller cells. Therefore, the isolated primary cells were further identified by GS immunofluorescence stain and the nuclei were visualized with DAPI stain (blue). Figure 2 shows that almost all of the cytoplasm was positively stained by GS (red, Fig. 2a, d) compared to the nuclei (blue, Fig. 2b, e). These results indicate that the isolated primary cells were indeed Müller cells and were thus used for subsequent experiments.

Identification of Müller cells by GS immunofluorescence staining. a GS expression in Müller cells (10×). b The nuclei of the corresponding cells were stained with DAPI (10×). c The merged image of GS and DAPI. d GS expression in Müller cells (40×). e The nuclei of the corresponding cells were stained with DAPI (40×). f The merged image of GS and DAPI
Probucol prevented the hyperglycemia-induced decrease in Müller cells proliferation
CCK-8 was utilized to detect cellular proliferation. As shown in Fig. 3, OD value of the HG was lower than that of the NG (0.58 ± 0.11 compared to 0.98 ± 0.23, P < 0.01). However, probucol treatment (HGPB) significantly prevented hyperglycemia–induced decrease in cell proliferation (0.73 ± 0.11 compared to 0.58 ± 0.11, P < 0.01). These data indicate that probucol acted as a protective drug to promote the proliferation of Müller cells in high glucose condition.

Probucol prevented the hyperglycemia-induced decrease in Müller cell proliferation. Cell proliferation of Müller cells in the normoglycemia group (NG), hyperglycemia group (HG), and hyperglycemia with 10 μM probucol group (HGPB) was assayed with a CCK-8 kit. Values represent mean ± SD. #P < 0.01 compared to HG, n = 48; and P < 0.01 compared to HGPB, n = 48
Probucol attenuated hyperglycemia-induced apoptosis of Müller cells
Annexin V-FITC/PI staining was used to evaluate cellular apoptosis by flow cytometry. As shown in Fig. 4, the cellular apoptotic rate of the HG was higher than that of the NG (7.70 ± 0.44% compared to 2.79 ± 0.52%, P < 0.001). Nevertheless, probucol treatment (HGPB) successfully attenuated hyperglycemia-induced apoptosis (HG) (4.00 ± 0.95% compared to 7.70 ± 0.44%, P < 0.01). These data demonstrate that in a high glucose environment, probucol can inhibit apoptosis of Müller cells.

Probucol attenuated the hyperglycemia-induced apoptosis of Müller cells Cellular apoptotic rate in the normoglycemia group (NG), hyperglycemia group (HG), and hyperglycemia with 10 μmol/L probucol group (HGPB) was detected by flow cytometry with Annexin V-FITC/PI staining. Summary data are presented as mean ± SD. #P < 0.001 compared to HG, n = 3; and P < 0.01 compared to HGPB, n = 3
Probucol prohibited hyperglycemia-induced ROS generation in Müller cells
To further explore the mechanism of hyperglycemia-induced cellular proliferation and apoptosis, and the protective activity of probucol, cellular ROS in Müller cells were detected. After incubation with the DCFH-DA probe, the intensity of DCF fluorescence, which was directly proportional to intracellular ROS level, was analyzed by a fluorescence microplate reader. Fluorescence intensity of the HG (Fig. 5b, d) was stronger than that of the NG (Fig. 5a, d; 55.17 ± 14.07 compared to 20.89 ± 5.14, P < 0.01). Similar to the above tests, this hyperglycemia-induced ROS generation was significantly inhibited by probucol treatment (HGPB) (Fig. 5c, d; 26.28 ± 4.73 compared to 55.17 ± 14.07, P < 0.01). These data suggest that high glucose could induce the generation of ROS in Müller cells, and probucol could act as an effective anti-oxidative drug to inhibit hyperglycemia-induced ROS production in these cells. The cellular protective activity of probucol may also occur by prohibiting ROS generation.

Probucol prohibited hyperglycemia-induced ROS generation in Müller cells Intracellular ROS in Müller cells of the normoglycemia group (NG), hyperglycemia group (HG), and hyperglycemia with 10 μmol/L probucol group (HGPB) were assayed using a DCFH-DA probe. a ROS in NG group (40×); b ROS in HG group (40×); and c ROS in HGPB group (40×). d Each column represents mean ± SD. #P < 0.01 compared to HG, n = 18; and P < 0.01 compared to HGPB, n = 18
Expression of Nrf2, Keap1, and GCLC in Müller cells
Immunofluorescence staining was used to detect the protein expression of anti-oxidative factors, such as Nrf2, Keap1, and GCLC in Müller cells. The GCLC (Fig. 6a), Nrf2 (Fig. 6e), and Keap1 (Fig. 6f) proteins were clearly found to be abundantly expressed in the cytoplasm of Müller cells.

Expression of Nrf2, Keap1, and GCLC in Müller cells a GCLC expression in Müller cells (40 ×). b GS expression in Müller cells (40×). c The nuclei of the corresponding cells were stained with DAPI. d The merged image of GCLC and GS. e Nrf2 expression in Müller cells (40×). f Keap1 expression in Müller cells (40×). g The nuclei of the corresponding cells were stained with DAPI (40 ×). h The merged image of Nrf2 and Keap1
Discussion
Diabetes induces many morphological and functional alterations in retinas, including retinal microvascular dysfunction (a decrease in retinal blood flow, blood–retinal barrier dysfunction, death of pericytes and capillary endothelial cells), inflammatory response, and dysfunction of retinal neurons and glial cells [1]. In a high glucose environment, Müller cells that span the entire neuroretina generate ROS in the mitochondrion, causing oxidative stress. Under normal circumstances, Müller cells can transport extracellular glutamate into the cytosol followed by transformation to glutamine. Oxidative stress can, however, injure glutamate transportation to induce mitochondrion dysfunction in Müller cells [28]. In addition, Müller cell-derived VEGF contributes to retinal inflammation, retinal neovascularization, and vascular leakage. Therefore, it is of great significance to examine the alterations in Müller cells in the process of DR [29].
In this study, glucose treatment was employed to simulate DR. In the presence of high glucose, we observed an increase in intracellular ROS compared to the levels observed under normal glucose conditions. This was accompanied by a decrease in the proliferative activity and an increase in the apoptotic rate of Müller cells in the high glucose group. Liemburg-Apers et al. [30] found that high glucose stimulated the production of intracellular ROS and this increase contributed to the absorption of glucose by cells. Thus, an interaction between glucose and ROS can ultimately result in cell death. Recent studies proved that high glucose induced the apoptosis of Müller cells in miceretina [31, 32]. In addition, Fu et al. [33] observed that streptozotocin(STZ) injection gradually decreased retinal Müller cells in mice, with an evident reduction in the density of Müller cells at 10 months after development of STZ-induced diabetes. These results are consistent with the data obtained in the present study.
In the probucol-treated groups, we found that probucol promoted hyperglycemia-induced cellular proliferation and inhibited cellular apoptosis with a reduction in ROS generation in Müller cells. Interestingly, Zhang et al. [24] found that probucol had a protective effect on EPCs from oxidized low-density lipoprotein. This occurred through the restraining of both extracellular and intracellular ROS formation and an increase in the level of superoxide dismutase (SOD) in serum. Sheng et al. [34] revealed that probucol could inhibit hydrogen peroxide-induced apoptosis of vascular muscle cells. In diabetes, the activation of matrix metalloproteinase-9 (MMP-9) injured mitochondrial basement membranes and damaged retinal mitochondrion. In vitreous samples from patients with proliferative diabetic retinopathy (PDR), the level of MMP-9 was significantly higher than that in non-diabetic patients. This increase in MMP-9 promoted the progress of DR by accelerating VEGF production, which facilitated retinal neovascularization. Notably, probucol could suppress MMP-9 expression in apoE knockout mice and human umbilical vein endothelial cells (HUVECs) [7, 35,36,37]. These underlying mechanisms may contribute to the antioxidant activity of probucol.
In our study, Nrf2, Keap1, and GCLC were found to be expressed in Müller cells. Keap1 is an inhibitory factor of Nrf2 that binds tightly in the cytosol. When oxidative stress occurs, Nrf2 separates from Keap1, translocates to the nucleus, and binds with ARE to promote the transcription of downstream genes, including GCL (GCLC as the critical subunit of GCL). This indicates that Keap1/Nrf2/ARE antioxidant signaling pathway may exist in Müller cells. Nrf2-deficient diabetic mice showed blood–retinal barrier dysfunction with increased superoxide and decreased GSH at the early stage [21]. Hyperglycemia altered the epigenetic modifications at the Keap1 promoter, facilitated its binding with the transcription factor specificity protein 1 (SP1), and upregulated its expression. This increase in Keap1 suppressed the transfer of Nrf2 to the nucleus, impairing its transcriptional activity and promoting the development of DR [38]. In rat brain models with permanent middle cerebral artery occlusion, probucol exhibited neuroprotection in rat brains by upregulating Nrf2 [39]. Probucol also prevented the progress of renal disease by inhibiting SP1 expression in the DN rat model [40]. Through our previous studies, probucol was found to possess the potential to reduce oxidative stress and improve vision function in patients with DR [26, 27]. In brief, it is likely that probucol may protect Müller cells by activating the Nrf2/ARE signaling pathway. However, the underlying mechanism of this process requires further research.
References
Stitt AW, Curtis TM, Chen M et al (2016) The progress in understanding and treatment of diabetic retinopathy. Prog Retin Eye Res 51(3):156–186
Biedermann B, Bringmann A, Franze K et al (2004) GABA(A) receptors in Müller glial cells of the human retina. Glia 46(3):302–310
Krugel K, Wurm A, Pannicke T et al (2011) Involvement of oxidative stress and mitochondrial dysfunction in the osmotic swelling of retinal glial cells from diabetic rats. Exp Eye Res 92(1):87–93
Wang J, Xu X, Elliott MH et al (2010) Müller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes 59(9):2297–2305
Zhong Y, Li J, Chen Y et al (2012) Activation of endoplasmic reticulum stress by hyperglycemia is essential for Müller cell-derived inflammatory cytokine production in diabetes. Diabetes 61(2):492–504
Bai Y, Ma JX, Guo J et al (2009) Müller cell-derived VEGF is a significant contributor to retinal neovascularization. J Pathol 219(4):446–454
Abu El-Asrar AM, Siddiquei MM, Nawaz MI et al (2016) Coexpression of heparanase activity, cathepsin L, tissue factor, tissue factor pathway inhibitor, and MMP-9 in proliferative diabetic retinopathy. Mol Vis 22:424–435
Durning SP, Preston-Hurlburt P, Clark PR et al (2016) The receptor for advanced glycation endproducts drives T cell survival and inflammation in type 1 diabetes mellitus. J Immunol 197(8):3076–3085
Kline CL, Schrufer TL, Jefferson LS et al (2006) Glucosamine-induced phosphorylation of the alpha-subunit of eukaryotic initiation factor 2 is mediated by the protein kinase R-like endoplasmic-reticulum associated kinase. Int J Biochem Cell Biol 38(5–6):1004–1014
Kowluru RA, Mishra M (2015) Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim Biophys Acta 1852(11):2474–2483
Lorenzi M (2007) The polyol pathway as a mechanism for diabetic retinopathy: attractive, elusive, and resilient. Exp Diabetes Res 2007:61038
Tarr JM, Kaul K, Chopra M et al (2013) Pathophysiology of diabetic retinopathy. ISRN Ophthalmol 2013:343560
Wautier MP, Tessier FJ, Wautier JL (2014) Advanced glycation end products: a risk factor for human health. Ann Pharm Fr 72(6):400–408
Kowluru RA, Kowluru A, Mishra M et al (2015) Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog Retin Eye Res 48:40–61
Li C, Miao X, Li F et al (2017) Oxidative stress-related mechanisms and antioxidant therapy in diabetic retinopathy. Oxid Med Cell Longev 2017:9702820
Cao Y, Li X, Wang CJ et al (2015) Role of NF-E2-related factor 2 in neuroprotective effect of l-carnitine against high glucose-induced oxidative stress in the retinal ganglion cells. Biomed Pharmacother 69:345–348
Jimenez-Osorio AS, Gonzalez-Reyes S, Pedraza-Chaverri J (2015) Natural Nrf2 activators in diabetes. Clin Chim Acta 448:182–192
Chen B, Lu Y, Chen Y et al (2015) The role of Nrf2 in oxidative stress-induced endothelial injuries. J Endocrinol 225(3):R83–R99
Zhong Q, Mishra M, Kowluru RA (2013) Transcription factor Nrf2-mediated antioxidant defense system in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci 54(6):3941–3948
Satoh T, Mckercher SR, Lipton SA (2013) Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic Biol Med 65:645–657
Xu Z, Wei Y, Gong J et al (2014) NRF2 plays a protective role in diabetic retinopathy in mice. Diabetologia 57(1):204–213
Zhou G, Wang Y, He P et al (2013) Probucol inhibited Nox2 expression and attenuated podocyte injury in type 2 diabetic nephropathy of db/db mice. Biol Pharm Bull 36(12):1883–1890
Yang S, Zhao L, Han Y et al (2017) Probucol ameliorates renal injury in diabetic nephropathy by inhibiting the expression of the redox enzyme p66Shc. Redox Biol 13:482–497
Zhang Q, Chen L, Si Z et al (2016) Probucol protects endothelial progenitor cells against oxidized low-density lipoprotein via suppression of reactive oxygen species formation in vivo. Cell Physiol Biochem 39(1):89–101
Zhou Z, Liu C, Chen S et al (2017) Activation of the Nrf2/ARE signaling pathway by probucol contributes to inhibiting inflammation and neuronal apoptosis after spinal cord injury. Oncotarget 8(32):52078–52093
Chen Z-P, Liu J-H, Jiang D-Y et al (2011) Observation of oxidative stress condition and macular edema in patients of early diabetic retinopathy treated by probucol. Zhong Guo Yi Shi Za Zhi 13(2):154–157
Chen Z-P, Tang S-B, Wang Q-C et al (2013) Clinical study of probucol in the treatment of hyperlipidemia diabetic macular edema. Zhong Hua Yan Di Bing Za Zhi 29(5):490–494
Toft-Kehler AK, Skytt DM, Svare A et al (2017) Mitochondrial function in Müller cells—Does it matter? Mitochondrion 36:43–51
Wang JJ, Zhu M, Le YZ (2015) Functions of Müller cell-derived vascular endothelial growth factor in diabetic retinopathy. World J Diabetes 6(5):726–733
Liemburg-Apers DC, Willems PH, Koopman WJ et al (2015) Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch Toxicol 89(8):1209–1226
Han N, Yu L, Song Z et al (2015) Agmatine protects Müller cells from high-concentration glucose-induced cell damage via N-methyl-d-aspartic acid receptor inhibition. Mol Med Rep 12(1):1098–1106
Xi X, Gao L, Hatala DA et al (2005) Chronically elevated glucose-induced apoptosis is mediated by inactivation of Akt in cultured Müller cells. Biochem Biophys Res Commun 326(3):548–553
Fu S, Dong S, Zhu M et al (2015) Müller glia are a major cellular source of survival signals for retinal neurons in diabetes. Diabetes 64(10):3554–3563
Sheng L, Jiao B, Shao L et al (2013) Probucol inhibits hydrogen peroxide to induce apoptosis of vascular smooth muscle cells. Mol Med Rep 7(4):1185–1190
Kowluru RA, Shan Y (2017) Role of oxidative stress in epigenetic modification of MMP-9 promoter in the development of diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol 255(5):955–962
Wang YY, Li H, Wang XH et al (2016) Probucol inhibits MMP-9 expression through regulating miR-497 in HUVECs and apoE knockout mice. Thromb Res 140:51–58
Katagiri M, Shoji J, Inada N et al (2018) Evaluation of vitreous levels of advanced glycation end products and angiogenic factors as biomarkers for severity of diabetic retinopathy. Int Ophthalmol 38(2):607–615
Mishra M, Zhong Q, Kowluru RA (2014) Epigenetic modifications of Keap1 regulate its interaction with the protective factor Nrf2 in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci 55(11):7256–7265
Du Y, Zhang X, Ji H et al (2012) Probucol and atorvastatin in combination protect rat brains in MCAO model: upregulating Peroxiredoxin2, Foxo3a and Nrf2 expression. Neurosci Lett 509(2):110–115
Duan SB, Liu GL, Wang YH et al (2012) Epithelial-to-mesenchymal transdifferentiation of renal tubular epithelial cell mediated by oxidative stress and intervention effect of probucol in diabetic nephropathy rats. Ren Fail 34(10):1244–1251
Acknowledgements
We thank the staff at the Research Institute of AIER Eye for their assistance in this research and Professor Chang-Luo for his help in writing this paper.
Funding
This study was supported by the Natural Science Foundation Project of Hunan Province (No. 2018JJ2001), China, and the Research Fund Project of AIER Eye Hospital Group (No. AF1601D5), China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhou, X., Ai, S., Chen, Z. et al. Probucol promotes high glucose-induced proliferation and inhibits apoptosis by reducing reactive oxygen species generation in Müller cells. Int Ophthalmol 39, 2833–2842 (2019). https://doi.org/10.1007/s10792-019-01130-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10792-019-01130-8