
Abstract
It has become increasingly clear that inflammatory processes play a significant role in the pathophysiology of Alzheimer’s disease (AD). Neuroinflammation is characterized by the activation of astrocytes and microglia and the release of proinflammatory cytokines and chemokines. Vascular inflammation, mediated largely by the products of endothelial activation, is accompanied by the production and the release of a host of inflammatory factors which contribute to vascular, immune, and neuronal dysfunction. The complex interaction of these processes is still only imperfectly understood, yet as the mechanisms continue to be elucidated, targets for intervention are revealed. Although many of the studies to date on therapeutic or preventative strategies for AD have been narrowly focused on single target therapies, there is accumulating evidence to suggest that the most successful treatment strategy will likely incorporate a sequential, multifactorial approach, addressing direct neuronal support, general cardiovascular health, and interruption of deleterious inflammatory pathways.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Despite the best efforts of clinicians and researchers, Alzheimer’s disease (AD) remains the most common form of senile dementia (Wirths et al. 2004). Clinically, the disease presents as a severe impairment in memory, as well as other cognitive functions, which invariably cause a decline in an individual’s ability to function in society. Often, it is preceded by a prodromal state commonly referred to as mild cognitive impairment (MCI) characterized by milder cognitive deficits. With an aging population in developed countries, including the United States, it becomes increasingly imperative that pharmacological agents capable of modifying the course of the disease be developed. This is especially evident in light of the fact that there are currently over 5.3 million Americans afflicted with this disease, which has also become the seventh leading cause of death in this country (Galluzzi et al. 2010).
The major pathological hallmarks of AD include the presence of abnormal proteinaceous deposits known as senile plaques and neurofibrillary tangles (NFTs), along with extensive neuronal loss in specific cortical and subcortical regions such as the nucleus basalis of Meynert and the hippocampus (Tang 2009). Senile plaques are composed primarily of the protein fragment β-amyloid (Aβ), and are generally thought to be formed extracellularly (Plant et al. 2003), although there is also evidence from murine models which suggests that the process of oligomerization and subsequent deposition begins in intracellular compartments (Takahashi et al. 2002). NFTs are composed of the microtubule-associated protein (MAP) called tau. Tau becomes hyperphosphorylated during the disease process of AD, which results in its own aggregation as well as the disruption of various other cellular processes, including, for example, the function of other MAPs, which aid in the maintenance of the structure of axons and dendrites (Ballatore et al. 2007).
Additionally, cholinergic abnormalities such as the loss of presynaptic cholinergic markers have been described in the postmortem histological analyses of AD brains. This occurs along with the loss of acetylcholine (ACh) receptors in the putamen and the nucleus basalis of Meynert (Kihara and Shimohama 2004). Importantly, most of the pharmacological agents that have been approved to date for the treatment of AD do so by inhibiting the function of acetylcholinesterase (AChE) (Sabbagh and Cummings 2011).
Although amyloid plaques and neurofibrillary tangles are considered the major clinical pathological markers in AD (Hermann et al. 1991; Tang 2009), Alois Alzheimer himself was the first to describe the involvement of glial cells (perivascular gliosis) in AD in 1899 (Alzheimer 1991). Since then, much research has been devoted to elucidating the mechanisms whereby this progressive and highly prevalent (Lindesay et al. 2010) neurodegenerative disease wreaks destruction upon our brains, resulting in severe morbidity and mortality. Although we do not yet fully understand the disease process, and have few approved therapies—none of which is anything near curative—we are continuing to accumulate observations about the myriad biological processes that are associated with AD pathogenesis.
The picture that is emerging is very complex. Rather than a simple set of local phenomena directly injurious to the neurons whose demise is the most obvious symptom of the disease, the development and progression of AD involves physiological aberrations and reactions which are numerous, simultaneous, widespread, and interdependent. Part of this progression clearly involves a chronic inflammatory process involving neurons, astrocytes, and microglia as the former are the primary producers of Aβ, while the latter two constitute the immunocompetent cells of the central nervous system (CNS) and show a robust inflammatory response to Aβ (Agostinho et al. 2010). But as will be described below, inflammatory chemokines and cytokines, reactive oxygen species (ROS), and inflammatory enzymes produced by cells in the CNS as well as components of the cardiovascular, endocrine, and complement systems all have a role to play in the inflammatory pathways that coincide with the pathogenesis of AD (Heneka and O’Banion 2007; Lee et al. 2009; Carlsson 2010; McGeer and McGeer 2003).
Neurons and Aβ-induced inflammation
Far from being passive spectators of the inflammatory process that is initiated in the course of AD pathology, neurons are active participants. Despite the fact that glial cells such as astrocytes and microglia have traditionally been thought of as the primary mediators of immunity and thus inflammation in the CNS, neurons are capable of both producing and responding to molecules involved in the pathways of inflammation. In addition, many of these pathways have downstream impacts on the regulation of normal cellular processes in neurons.
Inflammation and Aβ
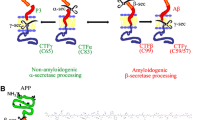
Aβ, derived from the membrane spanning amyloid precursor protein (APP), is one of the most studied molecules involved in the pathogenesis of AD. Functional Aβ 1–40 (Plant et al. 2003) or pathological Aβ 1–42 (Abramov et al. 2004) fragments are produced by intramembrane cleavage of APP by a gamma secretase encoded by the presenilin gene (Mundy 1994), as well as cleavage at an extracellular site by a beta secretase, β-site APP cleaving enzyme 1 (BACE-1) (Selkoe 2001). Alternatively, cleavage by an alpha secretase such as disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) does not result in the production of Aβ (Galimberti and Scarpini 2011). The primary (although by no means only) producers of Aβ in the CNS are neuronal cells. In general, the mechanisms by which toxic Aβ interacts with the progression of AD are not clearly understood. However, there is well-established evidence that it exerts numerous effects on inflammatory processes associated with AD.
It has been reported in many studies that inflammatory markers co-localize with the pathological hallmarks of AD, including, but not necessarily limited to, Aβ plaques (Rogers and Shen 2000) which form extracellularly from accumulations of Aβ 1–42 fragments along with other amyloid species. Many of these inflammatory markers, when expressed in the periphery have been shown to be highly cytotoxic, and it has also been shown that non-specific systemic inflammation can quicken the onset of the clinical manifestations of AD, possibly by further activation of already primed microglia (Perry et al. 2003). Aβ plaques, which are fibrillar in nature, exhibit dense populations of microglia at their core, as well as activated astrocytes at their periphery (Akiyama et al. 2000). This is in fact true not only for human patients with firm clinical indicators of the disease prior to postmortem histological examination of brain tissue, but also for those considered to be in a less mature disease state (i.e., MCI) (Lue et al. 1996; Eikelenboom and van Gool 2004). In addition, it has been shown in in vivo models of AD that Aβ-induced inflammation occurs early in the pathology of the disease, and can be blocked by the application of an inhibitor of glial activity (Craft et al. 2006).
In several studies, it has been shown that Aβ is capable of activating many different inflammatory and apoptotic pathways. For instance, in vitro studies have shown that fibrillar forms of Aβ are capable of activating the NACHT/LRR/PYD domains-containing protein 3 (NALP3) inflammasome via a caspase-1 dependent mechanism. This leads to the production of IL-1β and IL-18, which in turn stimulate microglia to produce further inflammatory cytokines (Halle et al. 2008). In another in vitro study, primary murine microglial cultures showed increased IL-6 and tumor necrosis factor-α (TNFα) production in response to exposure to aggregated Aβ (Walter et al. 2007). Further, it has been shown in positron emission tomography studies of AD patients that there is an increased activation of microglia in the region of senile plaques (Lue et al. 1996; Edison et al. 2008).
Channel hypothesis
Another possible pathway by which Aβ may induce inflammation has been suggested by the proponents of the “channel hypothesis”. Simply stated, it has been suggested that Aβ oligomers are capable of forming membrane spanning ion-permeable channels that exhibit poor specificity (Wirths et al. 2004; Kawahara et al. 2011). Studies have suggested that these channels are capable of inducing aberrant Ca2+ currents across the membranes of both neurons and glial cells (Kawahara et al. 2011; Abramov et al. 2004). It has also been shown that in hippocampal astrocytes co-cultured with neurons incubated with Aβ, these currents elicited the activation of a transient acidification of the extracellular media concurrent with the production of ROS (Abramov et al. 2004). As it has been suggested that acidosis may increase amyloidogenic processing of APP (Brewer 1997) and immunoreactivity of Aβ (Bell and Zlokovic 2009), the formation of Aβ channels may create a self-sustaining cycle that could potentially drive further inflammation.
Intracellular domain of amyloid precursor protein
Besides the extracellular cleavage products of APP, recent evidence also suggests that the intracellular domain of the protein (APP intracellular domain, AICD) may play an important role in AD as well. Specifically, a cleavage product of AICD, C31, has been implicated in the mediation of several deleterious processes by acting upon gene regulation in the cells in which it is released (Park et al. 2009). AICD has also been shown to impact the proliferation of hippocampal progenitor cells (HPCs) (Ghosal et al. 2010), a cell population that is associated with the processes of learning and memory (van Praag et al. 2002; Shors et al. 2001; Snyder et al. 2005; Wang et al. 2010; Klempin and Kempermann 2007). Specifically, it was observed that a mouse model induced to overproduce AICD exhibited a lower rate of production of daughter cells from the HPC population concurrent with an upregulation of several pro-inflammatory cytokines. When this model was treated with ibuprofen, the rate of cell proliferation in the HPC population fell more in line with that observed in wild-type (WT) controls (Park et al. 2009).
Toll-like receptors
One important mediator of Aβ-induced inflammation is the family of Toll-like receptors (TLRs), a class of pattern recognition receptors recently identified as having important roles in the innate immune system (Landreth and Reed-Geaghan 2009). Many TLRs, including TLR1, 2, 4, 5, 6, and 7 show increased expression as a function of age in mouse models (Liu et al. 2005; Walter et al. 2007), as one would expect of a receptor type capable of interacting with the pathogenic mediators of AD.
The primary subtypes of TLRs implicated in AD-associated inflammation include TLR2 and TLR4. Both subtypes have been implicated not only in mediating inflammation induced by Aβ, but also with aiding in the clearance of Aβ from the extracellular space. Specifically, blockage of TLR2 in vivo was shown to reduce the inflammatory response to Aβ, and when a TLR2−/− murine model was crossed with an APP/PS1 (presenilin-1) mutant, Aβ deposition was increased coinciding with a concomitant decrease in cognitive function. This effect was reversed by bone marrow gene therapy for TLR2 in this model (Triantafilou et al. 2004). Further, another study found that primary cultures of microglia from TLR2 knockout (KO) mice were incapable of increasing the expression of TNFα, IL-1β, IL-6, and inducible nitric oxide synthase (iNOS) following exposure to fibrillar forms of Aβ 1–42 (Udan et al. 2008).
Aβ has also been found to activate microglia and astrocytes via TLR4 as well as its co-receptor, cluster of differentiation 14 (CD14). The effects of Aβ on this receptor complex is similar to its effect on TLR2, in that KO models of TLR4 or CD14 exhibited decreased inflammatory markers in response to Aβ, and the ability of microglia in such models to clear Aβ is significantly impaired (Reed-Geaghan et al. 2009; Walter et al. 2007; Tahara et al. 2006). Additionally, cultured microglia with intact TLR4 and CD14 genotypes produce neurotoxic factors in response to incubation with Aβ 1–42 and also directly kill neurons which had sustained damage from Aβ in a CD14, contact dependent manner (Combs et al. 2001; Bate et al. 2004, 2006). Further, TLR4 was shown to be upregulated in in vitro neuronal populations in response to Aβ and 4-hydroxynonenal (4-HNE), a lipid peroxidation product. Within this population, a c-Jun N-terminal kinase (JNK) dependent, caspase-3 mediated apoptosis was observed, presumably in response to Aβ challenge. In the same study, TLR4 KO cell cultures were shown to be spared from this fate despite Aβ exposure. While it has been shown that TLR4 can be upregulated in neuronal populations exposed to Aβ both in vivo and in vitro, cortical neurons of end-stage AD patients exhibit reduced TLR4 expression. This may be a result of increased cell death in neurons that upregulate TLR4 earlier in the disease process (Tang et al. 2007, 2008). Finally, human carriers of the asp299gly allele, associated with decreased activity of TLR4 (Arbour et al. 2000) exhibit significant protection from the late onset AD (Minoretti et al. 2006).
Advanced glycation end products and their receptors
Another receptor/ligand axis that has been shown to have interactions with the production of Aβ as well as inflammatory response in AD is that of advanced glycation end products (AGEs) and their receptors (RAGEs) (Yan et al. 1996). RAGE expression is increased in neuronal and astroglial cell populations in AD patients, which correlates with increased levels of production of ROS by these cells. Further, Aβ is capable of binding to RAGE, and as such is a possible mediator of this effect (Yan et al. 1996). Aβ also activates RAGEs at the surface of microglia, which in turn stimulates these cells to produce proinflammatory cytokines and chemotactic factors, an effect that is abrogated by blocking the interaction between Aβ and RAGE (Yan et al. 1996, 2009). Another intriguing experimental observation is that activation of RAGEs by in vivo treatment with AGEs in mouse models caused a nuclear factor-κB (NF-κB) dependent upregulation of BACE-1 (Guglielmotto et al. 2010). This finding makes sense in light of evidence suggesting that there are NF-κB binding sites in the promoter regions of BACE-1, as well as presenilin and APP. The genetic sequence coding for RAGE also contains a binding site for NF-κB (Li and Schmidt 1997). Taken together, these findings suggest a NF-κB mediated positive feedback mechanism in which Aβ and RAGE each stimulate further production of the other.
Other intracellular signaling pathways
Aβ has also been shown to interact extensively with many intracellular signaling pathways within neurons. For instance, glycogen synthase kinase 3 (GSK3), and specifically its beta isoform (GSK3β), has been shown to increase amyloidogenic processing of APP (Phiel et al. 2003). At the same time, Aβ has been reported to be capable of activating GSK3β by as yet unidentified mechanisms (Takashima et al. 1993), establishing another means by which Aβ could possibly upregulate its own production. In addition to this effect, GSK3 has been shown to be capable of indirectly increasing NF-κB, signal transducer and activator of transcription (STAT)3/5, and mitogen-activated protein kinase kinase kinase 11 (MLK3), all of which can increase the production of inflammation mediating cytokines, such as IL-6, TNFα, and monocyte chemotactic protein-1 (MCP-1) (Beurel et al. 2010; Schmitz et al. 2004). Murine models induced to overexpress GSK3β exhibited increased microglial and astrocyte activation as well as neuronal structural abnormalities concomitant with deficits in spatial memory. This effect was rescued in a conditional KO model when the overexpression of GSK3β ceased (Lucas et al. 2001; Hernandez et al. 2002; Engel et al. 2006; Mines et al. 2011).
Additionally, an inflammatory milieu has been shown to be conducive to the hyperphosphorylation of the microtubule-associated protein, tau. This leads to the formation of intracellular insoluble plaques known as NFTs, a hallmark of AD pathology (Ballatore et al. 2007). The hyper-phosphorylation of tau is mediated by the activation of its kinases, and an inflammatory microenvironment is thought to promote the activation of these molecules (Ballatore et al. 2007; Iqbal et al. 2005). Further, GSK3 is one of the primary mediators of phosphorylation of the tau protein (Avila et al. 2010). Thus, GSK3 and its isoforms are capable of generating both the inflammatory effects observed in AD, as well as several pathological hallmarks of the disease.
Physiological role of Aβ
Strong evidence has been presented that Aβ is involved in the inflammatory and various other pathological mechanisms of AD. However, much research has been done in recent years suggesting a physiological role for Aβ in memory formation and retention (Morley et al. 2010; Puzzo et al. 2011; Barbagallo et al. 2010). Several studies have demonstrated that when otherwise healthy animal models were induced to produce Aβ at abnormally low levels, significant deficits in long-term potentiation (LTP), spatial memory, and synaptic plasticity were observed (Morley et al. 2010; Puzzo et al. 2011). Furthermore, ectopically introduced Aβ 1–42 was found to be sufficient to rescue the deficits in LTP only when it was allowed to oligomerize prior to application (Morley et al. 2010). Thus, Aβ clearly has a complex interaction with the cells of the CNS, and more work is required to characterize precisely what impact different forms and concentrations of this molecule have on cognitive function.
Role of microglia and astrocytes
Astrocytes and microglia are each a subset of the larger classification of glial cells which serve many supporting functions in the CNS. Astrocytes in a non-reactive state are involved in ion homeostasis, regulation of metabolic function and synaptic levels of glutamate, production of anti-oxidant species, maintenance of the blood-brain barrier (BBB), and other functions (Wang and Bordey 2008; Benarroch 2005; Shih et al. 2006; Attwell and Laughlin 2001; Abbott et al. 1992). Microglia are the resident immunocompetent cells of the CNS. When not actively engaged in an inflammatory response, they play the role of quiescent sentinels, constantly extending and contracting processes that probe the surrounding extracellular space as well as cellular neighbors for signs of both exogenously and endogenously derived danger signals (Banati et al. 1993).
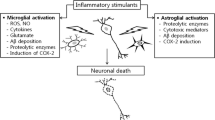
In terms of neuroinflammation in AD, microglia and astrocytes play a critical role. In situations which call for inflammatory response (i.e., pathogenic infiltration) astrocytes and microglia take on an activated phenotype and begin proliferating (Akiyama et al. 2000; McGeer and McGeer 2003). Neuroinflammation is mediated, at least in part, by arrays of cytokines which are released by reactive astrocytes and activated microglia (Tan et al. 1999; Heneka and O’Banion 2007). For example, levels of the potent pro-inflammatory cytokines interleukin (IL)-1β and IL-6 are elevated in the cerebrospinal fluid of AD patients (Griffin et al. 1989; Blum-Degen et al. 1995; Huell et al. 1995), while the ratio of IL-10, a cytokine with anti-inflammatory properties, to the pro-inflammatory IL-1β is decreased (Remarque et al. 2001).
During the progression of the AD disease process, it has been observed that there is a steady increase in the number of morphologically identifiable reactive astrocytes (Sastre et al. 2008). These are recruited by activated microglia and enhance the inflammatory response to extracellular Aβ deposits which colocalize with activated microglia. Local acute phase neuroinflammatory response mechanisms mediated by enzymes such as COX-2 and nitric oxide synthase (NOS) have been demonstrated to be major contributors to neuronal dysfunction and cell death (McAdam et al. 1999; Montine et al. 1999; Brown and Bal-Price 2003; Edison et al. 2008).
Microglia
The activation of microglia can occur in response to the formation of amyloid plaques. At the same time, APP and amyloid peptides can act as potent glial activators (Barger and Harmon 1997; Schubert et al. 2000). Increased microglial activation is an event that colocalizes with the areas of heavy Aβ concentration in the CNS of AD patients (Edison et al. 2008). Microglial activation is an early event in AD pathology (Craft et al. 2006; Vehmas et al. 2003) and progresses in line with the increase of Aβ deposition in the brains of AD afflicted individuals (Vehmas et al. 2003).
Despite the clear connection that microglial distribution in the CNS exhibits with the pathological markers of AD, there has been no small amount of disagreement over the role that these cells actually play at their sites of activation. For example, depletion of microglia in animal models can cause an increase in Aβ load (Majumdar et al. 2007), suggesting that they play a role in the clearance of Aβ by phagocytosis. At the same time, as the primary resident immune cells of the CNS, they are also capable of producing many pro-inflammatory factors that can potentially be neurotoxic in nature (Kreutzberg 1996).
Macrophages are thought to be one of the primary mediators of Aβ removal from the extracellular space in the brain as they have been shown capable in in vitro assays of internalizing Aβ via various receptor-mediated mechanisms (Berthiaume et al. 1995). The deposition of Aβ may reflect an overabundance of amyloidogenic processing of APP, overwhelming the ability of microglia to effectively clear this substance (Paresce et al. 1997). Microglial activation by pro-inflammatory cytokines such as macrophage colony stimulating factor (MCSF) and IL-6 can increase their ability to degrade Aβ (Majumdar et al. 2007). Alternatively, it has been suggested that CNS resident microglia may be capable of phagocytosing Aβ, but that their ability to degrade the peptide may be lacking due to a relatively high lysosomal pH. As such, the purported role of CNS-derived microglia as recruiters of peripheral macrophages to the sites of Aβ accumulation may be their primary mechanism of Aβ clearance (Majumdar et al. 2007; Simard and Rivest 2004).
Besides their role as clearers of Aβ, microglia are also capable of performing various other functions with potentially neuroprotective effects. For instance, a study has shown that microglia are capable of acting as sinks for glutamate during circumstances that might otherwise lead to N-methyl-d-aspartic acid (NMDA) receptor-linked Ca2+-mediated neurotoxicity (Persson et al. 2005). Additionally, microglia also release factors such as brain derived neurotrophic factor and thrombospondins (TSPs) capable of stimulating neurogenesis and synaptogenesis in the mature CNS (Kettenmann et al. 2011). Further, it has been suggested that microglia themselves may serve as potential precursors for other CNS cells, including neurons (Yokoyama et al. 2004). Microglia are also thought to function as effective removers of synapses of damaged neurons (Cullheim and Thams 2007).
While Aβ clearance by microglia can play a beneficial role in the pathogenesis of AD, this is not the only effect exerted by microglia in this process. As immune-associated cells, microglia are also important producers of pro-inflammatory factors including cytokines, chemokines, and reactive oxygen/nitrogen species (ROS/RNS) (Nathan et al. 2005; Block et al. 2007; Ard et al. 1996). One such pro-inflammatory cytokine, TNFα, has been shown to decrease the phagocytic function of CNS immune cells in vivo (Koenigsknecht-Talboo and Landreth 2005). This demonstrates one means by which inflammation limits the efficacy of microglia in curtailing the production of AD markers.
Astrocytes
Recent studies have demonstrated the prominence of astrogliosis—an increase in the number of astrocytes in response to neuronal distress—in AD brains. This is observed mainly in the regions surrounding amyloid plaques with processes of activated astrocytes participating in the formation of neuritic plaques (Nagele et al. 2003; Rodriguez et al. 2009). However, there is evidence suggesting that this activation is more widespread (Vehmas et al. 2003). A recent hypothesis posits that progression of astrocytosis per se may play a role in the observed cognitive deficits of aging populations beyond that which is attributed to those pathways that are more traditionally associated with the pathology of AD. It has been demonstrated that reactive glia are major contributors to ongoing neurodegeneration. Recent studies also suggest that neuroinflammation plays an important role in the early stage of AD pathology (Maccioni et al. 2009; Simpson et al. 2010).
There are several mechanisms by which astroglia are thought to interact with inflammation in AD. One idea that has recently received attention is that astroglia can exert neurotoxic effects simply by taking part in an inflammatory response. During this period, the normal role of astroglia in their support function is likely compromised (Fuller et al. 2010). Several studies have suggested that compromised glucose metabolism in neurons may be a contributing factor in AD, and that this effect is observable early in the disease process in both human and animal models (Freemantle et al. 2006; Drzezga et al. 2003; Mielke et al. 1998; Alexander et al. 2002; Small et al. 2000). As the ratio of astroglia to neurons in the CNS is quite high, it has been suggested that this deficit in glucose metabolism must be due at least in part to the loss of astroglial glucose uptake (Alexander et al. 2002). Additionally, a dearth of both glutamate transporter (GLT)-1, which functions in astrocytes to regulate synaptic glutamate concentration, and glutathione, which is an antioxidant produced by astrocytes, is a characteristic of AD (Li et al. 1997; Calabrese et al. 2006). In aggregate, this evidence suggests that one of the most deleterious effects of astroglial inflammatory activation is the neglect of their typical role as supporters of neuronal function.
It has been suggested that microglial activation precedes astroglial activation in AD (Zhang et al. 2009). At the same time, it has also been suggested that the precipitating event in astrocytic activation is the formation of fibrillar Aβ (Paris et al. 2002b; Hensley et al. 1998). A large body of literature has implicated various other possible mechanisms that may cause astrogliosis, including ischemia (Wakasa et al. 2009), infiltration by infectious agents (Okamoto et al. 2005), and other chemical exposure (El-Fawal and O’Callaghan 2008). Regardless of the causative event, the effects of astrocytic activation on neuroinflammation in AD have been well documented. Generally speaking, these fall into two categories: production of cytokines and chemokines and the production of free radical species.
With regard to the first of these, as activated astroglia are immunocompetent cells, it is not surprising that they are capable of producing cytokines and chemokines. Specifically, it has been reported that in co-cultures of neurons with astroglia, there is an increased production of IL-1β, IL-6, TNFα, and interferon-γ (IFNγ) which coincides with the introduction of Aβ 1–42 to the cells’ growth media (Jana and Pahan 2010). A more recent study has shown that IL-13, IL-17, and IFNγ induced protein-10 (IP-10) production were increased in a similar disease model (Garwood et al. 2011). This pattern of cytokine and chemokine production suggests a mechanism for reciprocal activation as, for example, IL-17 has been shown capable of inducing further IL-6 production in astrocytes (Anisman 2009). Additionally, IL-6 production is increased during the early stages of the appearance of major deposits of Aβ, suggesting a connection between IL-6 and Aβ (Huell et al. 1995).
As to the production of free radicals, astrocytes have been shown to be capable of producing both RNS and ROS (Hashioka et al. 2009), production of which results in oxidative stress in the physiological processes of cells, with downstream effects such as damage to lipids, proteins, and DNA. This may in turn result in necrosis or apoptosis (Simonian and Coyle 1996). Nitric oxide (NO), produced by activation of iNOS in particular, is a signaling molecule in this pathway with multiple effects. For example, it has been suggested that astroglial-derived NO can exert such varied effects as neuronal energy depletion (Bolanos et al. 1997), deleterious interactions with protein thiol groups and iron–protein complexes (Chen et al. 2001), and upregulation of inducible as well as constitutive cyclooxygenases (COX-2 and COX-1, respectively) (Calabrese et al. 2007). Oxidative stress induced by the production of ROS has been implicated in the early stages of the development of AD (Mondragon-Rodriguez et al. 2010). Therefore, the inhibition of the production of these radical species has received considerable attention in recent years as a potential avenue for intervention in the progression of the disease.
Under normal conditions in the brain, COX-2 is not produced by astroglia (Yermakova and O’Banion 2001). On the other hand, COX-2 activity is increased in astrocytes associated with the fibrillar Aβ deposits in a double transgenic murine model of AD (Matsuoka et al. 2001). COX production is particularly insidious with regard to its ability to mediate inflammation as its enzymatic activity produces both prostaglandins from arachidonic acid as well as oxidative species (Smith et al. 1996). The production of each of these molecules has been implicated in the enhanced production of inflammatory cytokines such as IL-6 in CNS cells (Fiebich et al. 2001). Furthermore, COX-2 production in astrocytes can be induced by IL-1β (O’Banion et al. 1996). In another link to AD pathology, it has been shown that APP production is induced in microglia and astrocytes by the activation of prostaglandin E2 (PGE2) receptors (Lee et al. 1999; Pooler et al. 2004). Despite these findings, it has also been reported that while COX-2 production is increased in the initial phases of AD, its expression is actually downregulated as the pathological hallmarks of the disease develop (Hoozemans et al. 2008; Krause and Muller 2010).
Cardiovascular inflammatory processes
Neurovascular interactions in AD
Recently, it has become clear that vascular dysfunction likely plays an important role in the pathogenesis of AD. Vascular dementia is a form of dementia which has a clinical presentation similar to that of AD, but which has a pathogenesis involving specific insult to the cerebrovasculature. It is well known that vascular dementia and AD share risk factors, and one-third of patients with a clinical and pathological diagnosis of AD have some degree of vascular pathology (Gearing et al. 1995; Ince et al. 2000) and vice versa (Sadowski et al. 2004). Furthermore, cardiovascular disease risk factors such as diabetes and hypertension have been well established as risk factors for AD. Among AD patients with similar amounts of plaques and tangles, only those with both AD and cardiovascular disease had significant dementia (Snowdon et al. 1997).
It has even been postulated that AD is primarily a vascular disorder with neurodegenerative consequences, with the initial pathogenic insult arising from cerebral hypoperfusion, and with impairment of NO bioactivity contributing to the progression of the disease (Hollenberg 2006). Indeed, reduced cerebral blood flow (Iadecola et al. 1999) and a reduction in vasoreactivity (Vicenzini et al. 2007) have been found in AD patients. Chronic hypoperfusion can result in oxidative stress, leading in turn to vascular endothelial permeability (Aliyev et al. 2004) and neuronal death associated with mitochondrial failure (Aliev et al. 2003). Hypoperfusion also results in decreased pH, which contributes to inflammatory processes in AD by increasing Aβ immunoreactivity (Bell and Zlokovic 2009).
Blood–neuron barriers in AD
Changes in the BBB make up a pronounced element of AD pathology, and the functional integrity of the BBB in AD has been called into question. It has been found that serum amyloid P component, a protein synthesized in the periphery which can serve to stabilize amyloid plaques, is associated with senile plaques and neurofibrillary tangles in postmortem AD brains even though it is not synthesized in the brain. This observation has lead to the suggestion that the BBB may be compromised in AD (Kalaria and Grahovac 1990). Indeed, both Aβ and extracellular tau have been posited to influence inflammatory responses in AD by contributing to vascular leakiness (Kovac et al. 2009).
In a healthy state, the BBB comprises endothelial cells joined by tight junctions, the basal lamina, and surrounding pericytes and astrocytic foot processes. Movement across this barrier of substances which cannot freely pass through the phospholipid cell membrane is tightly regulated by endothelial and astrocytic transport mechanisms. This protects the brain from harmful substances and oxidative damage. Importantly, the BBB regulates the entry of soluble Aβ, which is also produced in the periphery, into the CNS, via RAGEs, and the clearance of Aβ out of the CNS into the bloodstream, mediated by LDL-receptor related protein-1 (LRP-1) (Deane et al. 2003, 2004; Bell et al. 2007; Ji et al. 2001; Shibata et al. 2000; Zlokovic et al. 2000; Bading et al. 2002; Mackic et al. 1998; Tanzi et al. 2004). LRP-1 is downregulated in AD patients, with regional downregulation patterns consistent with the patterns of increased Aβ deposit density (Shibata et al. 2000). ATP binding cassette transport protein P-glycoprotein (P-gp) is also involved in Aβ export from the brain. P-gp is found to be downregulated in the vessel walls where there is accumulated Aβ. Unaffected capillaries have high P-gp expression, and it has been suggested that Aβ itself plays a role in downregulating expression of P-gp in the neurovasculature, thus exacerbating its effects (Brenn et al. 2011).
In addition to the BBB, the blood–cerebrospinal fluid barrier is another means by which neurons are protected from circulating factors. There is some evidence that this barrier undergoes damage early in the disease process, preceding Aβ and tau pathologies, although the functional significance of this with regard to passage of inflammatory factors is not yet fully known (Chalbot et al. 2011).
Vascular/endothelial involvement in AD
Diffusible mediators of inflammation
Endothelial cells are capable of producing a host of inflammatory, neurotoxic, and neuroprotective chemicals that interact with the progression AD. For example, while being more widely recognized for its role in the coagulation cascade, thrombin can also act as an inflammatory and neurotoxic protein that is produced by endothelial cells (Luo and Grammas 2010). Thrombin can induce expression of the protein endothelin-1 (ET-1), which has neuroprotective as well as vasoactive properties. On the other hand, ET-1 may play a deleterious role, as it can also interact synergistically with Aβ to cause increased vasoconstriction via activation of an inflammatory pathway (Paris et al. 2002a). Thrombin also induces the transcription factor hypoxia-inducible factor-1α (HIF-1α) which is involved in the regulation of pro-inflammatory gene expression. Thrombin, ET-1, and HIF-1α are all elevated in AD (Luo and Grammas 2010; Grammas et al. 2006).
Matrix metalloproteinases (MMPs) are endoproteases which cleave proteins in the extracellular matrix and participate in the regulation of growth factors, adhesion molecules, and receptors. One means by which MMPs may affect the progression of AD is through their ability to regulate the activity of nerve growth factor (NGF). Specifically, MMP-9 has been shown to be involved in the conversion of the mature form of NGF (mNGF) to a prefunctional form (proNGF). NGF-dependent cholinergic neurons in the nucleus basalis are differentially lost during the progression of AD. Further, MMP-9 activity is increased in the frontal and parietal cortical tissue harvested from humans afflicted with both AD and MCI relative to controls. The degree of MMP-9 upregulation correlated with the degree of mental impairment observed in the test subjects, which in turn supports the theory that it is a dysregulation of trophic factors that leads to the demise of cholinergic neurons in the nucleus basalis (Bruno et al. 2009). On the other hand, it has also been shown that MMP-2 and MMP-9 are released from AD-derived microvessels, but their activity may be suppressed as increased levels of thrombin associated with AD induce elevated levels of tissue inhibitor of matrix metalloproteinases-1 (TIMP-1) (Thirumangalakudi et al. 2006). Ultimately, while correlational evidence suggests an interaction, the effects that MMPs may have on the progression of AD, as well as the time course with which they occur, have not been fully elucidated.
In addition, microvessels in AD patients also express the angiogenic substances angiopoietin-2 and vascular endothelial growth factor (VEGF) (Thirumangalakudi et al. 2006), and the integrin αVβ3, a marker of angiogenesis, is also elevated in many brain regions in AD (Desai et al. 2009), which might account for the increased levels of capillary density observed in AD patients (Fioravanzo et al. 2010). Brain capillary endothelial cells have also been shown to proliferate in response to NGF, express NGF receptors, and secrete NGF after inflammation (Moser et al. 2004).
Nitric oxide
Nitric oxide plays a key role in maintaining normal vascular function and structure. Originally discovered by Robert Furchgott and named endothelial-derived relaxation factor, it is known to mediate the relaxation of vascular smooth muscle both in the brain and throughout the body, ensuring proper perfusion of tissues (Furchgott 1999). NO is produced enzymatically by NOS which is produced by cell types in many different systems. These employ different isoforms of the enzyme depending on the local function for which it is required. Neuronal NOS (nNOS) is the isoform expressed in the brain, and is believed to be used for neurotransmission. Inducible NOS (iNOS) is produced by astroglial cells and neurons in response to inflammatory conditions. Endothelial NOS (eNOS) influences vasodilation, and thus the state of cerebral perfusion (Hollenberg 2006).
There is some controversy regarding the role of NO in AD, and it likely plays both protective and detrimental roles. For example, NO is known to be involved in normal neurotransmission and is believed to play an important role in memory function as a retrograde transmitter in LTP (Kuiper et al. 2000). On the other hand, NO is a free radical which can cause both nitrosative and oxidative stress, lipid peroxidation, DNA damage, and impairment of mitochondrial function (Contestabile et al. 2006) and may play a role in the initiation of apoptosis (Warner et al. 2004), neuroinflammation, and neurodegeneration (Calabrese et al. 2000). Both chronic cerebral hypoperfusion and oxidative stress have been implicated in the pathogenesis of AD (Calabrese et al. 2000). As such, regulation of the expression and activation of the NOS isoforms presents itself as an attractive potential avenue of pharmacological modification.
Vascular amyloid deposits
In addition to its role in neuronal function and pathology, Aβ is also a physiological component of plasma (Altman and Rutledge 2010). It is transported across the blood–brain barrier into the plasma, and in vitro studies have shown that Aβ can be produced and released by activated platelets exposed to collagen, arachidonic acid, or thrombin in a process involving protein kinase C (PKC) and, under some conditions, COX (Skovronsky et al. 2001). Plasma Aβ 1–42 is elevated in AD patients prior to the development of symptoms (Mayeux et al. 1999, 2003). In addition, in AD, Aβ is also produced by cerebrovascular endothelial cells, which exhibit α-, β-, and γ-secretase like activity, and which may contribute to amyloidogenic deposits which have been found associated with the cerebral vasculature of AD patients (Davies et al. 1998). Indeed, more than 80 % of autopsied AD patients show some amount of cerebral amyloid angiopathy (Ellis et al. 1996), which is associated with an increased frequency of hemorrhage or ischemic lesions. These deposits contain predominantly the Aβ 1–42 isoform (Roher et al. 1993) and can have deleterious effects, compounding pathogenesis in AD as the distortion and occlusion of the capillaries leads to reduction in cerebral blood flow, further hindering the clearance of Aβ from the brain (Iadecola 2004).
Homocysteine
Elevated plasma levels of the amino acid homocysteine (Hcy) have been associated with an increased risk of both AD and vascular dementia (Seshadri 2006; Gallucci et al. 2004), and elevated Hcy levels in the brain can result from deficiencies in vitamins B6 and B12 (Weir and Molloy 2000). Hcy interferes with the activity of eNOS, induces iNOS, and produces ROS via a nicotinamide adenine dinucleotide phosphate (NADPH) oxidase dependent pathway, and can upregulate NF-κB and TNFα in endothelial cells (Faraci 2003). By increasing the expression of vascular cell-adhesion molecule-1 (VCAM-1) while simultaneously decreasing expression of glucose transporter-1 (GLUT-1) and eNOS, Hcy has been shown to induce endothelial dysfunction in rat brain (Lee et al. 2004). Neurotoxicity, abnormalities in brain chemistry, and cerebrovascular dysfunction were all observed in a murine model produced by dietary addition of Hcy, and these effects could be abolished by pharmacological modification (Troen 2005). However, the neurotoxic mechanisms may be more direct as well, as Hcy has been shown to kill cerebrocortical neurons via an NMDA dependent mechanism (Lipton et al. 1997).
Drug targets
Drugs currently in use
There are only five drugs which are currently approved for use in the treatment of AD. Of these, donepezil (Aricept®), galantamine (Reminyl®), rivastigmine (Exelon®), and tacrine (Cognex®) all belong to the class of drugs called acetylcholinesterase inhibitors (AchEI). The fifth drug, memantine (Namenda®), is a non-competitive antagonist at the NMDA glutamate receptor.
The AChEI have been proven to be effective in slowing the progression of dementia in AD (Sabbagh and Cummings 2011). All four drugs in this class are primarily prescribed for the treatment of mild to moderate stages of the disease, although donepezil has also been approved for use in severe AD as well. Although efficacy appears to be similar for drugs in this class, they differ in their mechanism of inhibition, selectivity, and pharmacokinetics (Schneider 2000), and differences in side effects may influence the choice of drug (Qaseem et al. 2008). Notably, tacrine, which was the first drug approved for clinical use, is now only rarely used due to a relatively high risk of hepatotoxicity (Watkins et al. 1994).
AD patients have reduced ACh synthesis and impaired cortical cholinergic function (Whitehouse et al. 1982). This is consistent with animal studies showing that Aβ reduces ACh production and release (Kar et al. 2004; Bales et al. 2006). The AChEI act by inhibiting the enzyme AChE which is anchored in the synaptic cleft at cholinergic synapses. AChE cleaves ACh into choline and acetate, thus limiting the ability of this neurotransmitter to bind to and activate receptors (Schneider 2000). By inhibiting this enzyme, the AChEI prolong the activity of the remaining endogenously released ACh, compensating at least partially for the cholinergic deficit.
Although it is not clearly understood how a cholinergic deficit contributes to AD pathophysiology, multiple mechanisms are likely involved. For example, activation of M1 muscarinic ACh receptors stimulates α-secretase cleavage of APP, reducing the production of toxic Aβ fragments (Nitsch et al. 1992), so a reduction in ACh would be expected to increase Aβ production. Furthermore, by the activation of α4 and α7 nicotinic ACh receptors, both donepezil and galantamine, but not tacrine have been shown to be protective against glutamate-induced neurotoxicity, via a mechanism involving the phosphatidylinositol 3-kinase-Akt/protein kinase B pathway (Takatori 2006). In addition to their effect at cholinergic synapses, the clinical efficacy of the AChEI likely results from other pharmacologic mechanisms as well, which may include effects on inflammatory processes both through α7 nicotinic ACh receptors peripherally (Wang et al. 2003), and directly, as donepezil has been shown to suppress iNOS gene expression, inhibit the production of NO, TNFα, and IL-1β, and inhibit inflammatory NF-κB signaling in microglial cell cultures (Hwang et al. 2010) as well as suppressing IL-1β and COX 2 production, and reducing neuroinflammation in vivo in a tau mutant mouse model of AD (Yoshiyama et al. 2010).
The NMDA receptor antagonist memantine is considered to be neuroprotective, and is currently approved by FDA for moderate to severe AD patients. It has been shown to produce a notable improvement in clinical deterioration in AD patients compared to placebo (Reisberg et al. 2003), and has been reported to have fewer side effects compared to AChEI (McShane et al. 2006). Furthermore, memantine can be used in combination with existing AChEIs (Tariot et al. 2004).
Glutamate is the principal excitatory amino acid neurotransmitter in cortical and hippocampal neurons, and NMDA receptors are important for many physiologically relevant processes, including learning and memory (Orrego and Villanueva 1993; Danysz and Parsons 1998). However, since part of the current passing through open NMDA receptor-mediated channels is carried by Ca2+, excessive stimulation of NMDA receptors can lead to Ca2+-mediated toxicity, which is detrimental in AD (Dong et al. 2009) and other neurodegenerative diseases (Dong et al. 2009; Levine et al. 1999; Klapstein and Colmers 1997; Klapstein and Levine 2005). Memantine exerts a neuroprotective effect by binding within and physically blocking the ion channel which is integral to the NMDA receptor, thereby preventing the entry of Ca2+.
As with the AChEI, however, memantine also has pharmacological actions in addition to those described above, which interfere with inflammatory processes. It has been shown to reduce inflammatory microglial activation and increase the release of neurotrophic factors by astroglia (Wu et al. 2009), both of which would be beneficial in AD.
Drugs in trial
Because of the role of inflammation in the pathogenesis of AD, several drug targets with anti-inflammatory or anti-oxidative properties have been investigated. An extensive review of recent studies and clinical trials involving disease-modifying treatments for AD has recently been published (Galimberti and Scarpini 2011); therefore, we shall briefly discuss some of those agents which have been postulated to be beneficial in the treatment of AD through interaction or interference with inflammatory processes.
By acting as agonists at the ligand-activated nuclear receptor peroxisome proliferator activated receptor-γ (PPARγ), non-steroidal anti-inflammatory drugs (NSAIDs), thiazolidinediones, and prostaglandin J2 all initiate neuroprotective mechanisms. Combs et al. (2000) have found that PPARγ agonists inhibit the secretion of proinflammatory products by microglia, as well as the production of IL-6 and TNFα in response to Aβ stimulation, and the differentiation of activated macrophages from monocytes. Indeed, some epidemiological studies correlate the use of NSAIDs with reduced risk of developing AD within 2–3 years if taken prior to age 65 (Hayden et al. 2007), but others show increased risk in older populations (Breitner et al. 2009). Disappointingly, there is no evidence that NSAIDs have any effect in changing disease progression, and their use may actually be detrimental (Willard et al. 2000; Moore and O’Banion 2002; Martin et al. 2008; Wolfson et al. 2002). Several compounds with anti-oxidative properties, such as vitamin E, alpha lipoic acid and resveratrol are also being investigated for effectiveness in slowing the progression of AD, but there has been no conclusive evidence of benefit (Granzotto and Zatta 2011; Isaac et al. 2008; Cho et al. 2010).
Ginkgo biloba has historically been used to improve cognition, and is known to increase cerebral blood flow, reduce blood viscosity, modify neurotransmitter systems, and scavenge free radicals. While negative side effects have not been shown, consistent beneficial evidence for the use of Ginkgo biloba in AD patients is lacking (Birks and Grimley Evans 2009).
Folic acid and vitamin B12 deficiency have been linked to elevated Hcy levels, which are associated with elevated AD risk, as described above. As such, it has been proposed that increasing levels of these vitamins might be of benefit in reducing risk or symptoms of AD. In fact, there is some evidence that folate and B12 supplementation can improve cognition in healthy older people with high levels of Hcy, but this effect has not been found in AD patients (Malouf and Grimley Evans 2008).
There is some evidence that cannabinoids may be a good target for AD drug development, as they may regulate excessive glutamate production and neuroinflammation. Although there is no current evidence of benefit to AD patients in using cannabinoids, extensive studies have not been done, and further research is warranted (Krishnan et al. 2009).
Colostrinin, a polypeptide with immunomodulatory properties derived from ovine colostrum (Blach-Olszewska and Leszek 2007), has been shown in animal models in vivo to improve cognitive performance, and in cell-based assays to reduce Aβ aggregation and neurotoxicity. Although clinical trials in AD patients were initially encouraging, showing modest improvements in mild AD (Leszek et al. 1999), this effect did not appear to be sustained with therapy continued more than 15 weeks (Bilikiewicz and Gaus 2004).
Another promising drug is Huperzine A, a chemical with AChEI activity, derived from the Chinese herb Huperzia serrata. Huperzine A has been shown to be protective against ischemia, hydrogen peroxide, glutamate, and Aβ-mediated toxicity. Clinical trials have shown some evidence of improvements in cognition, activities of daily living, and behavioral disturbances (Li et al. 2008), and in vitro studies have indicated that Huperzine, when added to cells exposed to Aβ, increased cell survival and activity of glutathione peroxidase and catalase, and produced decreased levels of malondialdehyde (Xiao et al. 2000).
Curcumin is a component of the spice turmeric and has been proven to contain antioxidant, anti-inflammatory, and cholesterol-lowering properties (Ringman et al. 2005). In animal studies, it was shown to reduce Aβ and amyloid levels (Yang et al. 2005). Clinical trials have not proven the direct effect of curcumin on AD progression, however, this could be due to time limitations or the experimental design (Baum et al. 2008), and further studies are warranted.
Summary
Inflammatory processes are extensively involved in the pathophysiological processes of AD. These processes provide numerous targets for intervention with therapeutic or preventative agents, but they must be approached with caution as many of the inflammatory processes are obviously involved in pathogenesis of the disease, yet others contribute to compensatory defense mechanisms and some play dual roles. Several compounds which interfere with inflammatory mechanisms have made their way to clinical trial, and many more have shown promise of therapeutic potential in preclinical studies. Of these compounds, few have shown unequivocal success, especially at later stages of the disease when neuronal death may render cerebral dysfunction irreversible. Yet hope remains. Many trials report modest but positive results. As we gather more of the pieces to the puzzle of AD, we may find that testing such drugs at different stages of the disease or in synergistic combinations may lead to significant, clinically relevant treatment strategies.
Abbreviations
- ACh:
-
Acetylcholine
- AChE:
-
Acetylcholinesterase
- AChEI:
-
Acetylcholinesterase inhibitors
- AGE:
-
Advanced glycation end product
- AD:
-
Alzheimer’s disease
- APP:
-
Amyloid precursor protein
- AICD:
-
Amyloid precursor protein intracellular domain
- BACE-1:
-
β-Site APP cleaving enzyme 1
- Aβ:
-
β-Amyloid
- BBB:
-
Blood–brain barrier
- CNS:
-
Central nervous system
- CD14:
-
Cluster of differentiation 14
- COX:
-
Cyclooxygenase
- eNOS:
-
Endothelial nitric oxide synthase
- ET-1:
-
Endothelin-1
- GLUT-1:
-
Glucose tansporter-1
- GLT-1:
-
Glutamate transporter-1
- GSK:
-
Glycogen synthase kinase
- HPC:
-
Hippocampal progenitor cell
- Hcy:
-
Homocysteine
- 4-HNE:
-
4-Hydroxynonenal
- HIF-1α:
-
Hypoxia-inducible factor-1α
- iNOS:
-
Inducible nitric oxide synthase
- IFNγ:
-
Interferon-γ
- IP-10:
-
Interferon-γ induced protein-10
- IL:
-
Interleukin
- JNK:
-
c-Jun N-terminal kinase
- KO:
-
Knockout
- LRP-1:
-
LDL-receptor related protein-1
- LTP:
-
Long-term potentiation
- LDL:
-
Low density lipoprotein
- MCSF:
-
Macrophage colony stimulating factor
- MMP:
-
Matrix metalloproteinase
- MAP:
-
Microtubule-associated protein
- MCI:
-
Mild cognitive impairment
- MLK-3:
-
Mitogen-activated protein kinase kinase kinase 11
- MCP-1:
-
Monocyte chemotactic protein-1
- NFT:
-
Neurofibrillary tangle
- NMDA:
-
N-methyl-d-aspartic acid
- NALP3:
-
NACHT/LRR/PYD domains-containing protein 3
- NGF:
-
Nerve growth factor
- nNOS:
-
Neuronal nitric oxide synthase
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- NSAID:
-
Non-steroidal anti-inflammatory drug
- NF-κB:
-
Nuclear factor-κB
- P-gp:
-
P-glycoprotein
- PPARγ:
-
Peroxisome proliferator activated receptor-γ
- PS-1:
-
Presenilin-1
- PGE2:
-
Prostaglandin E2
- PKC:
-
Protein kinase C
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- RAGE:
-
Receptor for advanced glycation end product
- STAT:
-
Signal transducer and activator of transcription
- TSP:
-
Thrombospondin
- TIMP-1:
-
Tissue inhibitor of MMP
- TLR:
-
Toll-like receptor
- TNFα:
-
Tumor necrosis factor-α
- VCAM-1:
-
Vascular cell-adhesion molecule
- VEGF:
-
Vascular endothelial growth factor
- WT:
-
Wild type
References
Abbott NJ, Revest PA, Romero IA (1992) Astrocyte-endothelial interaction: physiology and pathology. Neuropathol Appl Neurobiol 18(5):424–433
Abramov AY, Canevari L, Duchen MR (2004) Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim Biophys Acta 1742(1–3):81–87
Agostinho P, Cunha RA, Oliveira C (2010) Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr Pharm Des 16(25):2766–2778
Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K (2000) Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis Assoc Disord 14(Suppl 1):S47–S53
Alexander CL, Fitzgerald UF, Barnett SC (2002) Identification of growth factors that promote long-term proliferation of olfactory ensheathing cells and modulate their antigenic phenotype. Glia 37(4):349–364
Aliev G, Smith MA, Obrenovich ME, de la Torre JC, Perry G (2003) Role of vascular hypoperfusion-induced oxidative stress and mitochondria failure in the pathogenesis of Alzheimer disease. Neurotox Res 5(7):491–504
Aliyev A, Seyidova D, Rzayev N, Obrenovich ME, Lamb BT, Chen SG, Smith MA, Perry G, de la Torre JC, Aliev G (2004) Is nitric oxide a key target in the pathogenesis of brain lesions during the development of Alzheimer’s disease? Neurol Res 26(5):547–553
Altman R, Rutledge JC (2010) The vascular contribution to Alzheimer’s disease. Clin Sci (Lond) 119(10):407–421
Alzheimer A (1991) A contribution concerning the pathological anatomy of mental disturbances in old age, 1899. Alzheimer Dis Assoc Disord 5(2):69–70
Anisman H (2009) Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. J Psychiatry Neurosci 34(1):4–20
Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA (2000) TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet 25(2):187–191
Ard MD, Cole GM, Wei J, Mehrle AP, Fratkin JD (1996) Scavenging of Alzheimer’s amyloid beta-protein by microglia in culture. J Neurosci Res 43(2):190–202
Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21(10):1133–1145
Avila J, Wandosell F, Hernandez F (2010) Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev Neurother 10(5):703–710
Bading JR, Yamada S, Mackic JB, Kirkman L, Miller C, Calero M, Ghiso J, Frangione B, Zlokovic BV (2002) Brain clearance of Alzheimer’s amyloid-beta40 in the squirrel monkey: a SPECT study in a primate model of cerebral amyloid angiopathy. J Drug Target 10(4):359–368
Bales KR, Tzavara ET, Wu S, Wade MR, Bymaster FP, Paul SM, Nomikos GG (2006) Cholinergic dysfunction in a mouse model of Alzheimer disease is reversed by an anti-A beta antibody. J Clin Invest 116(3):825–832
Ballatore C, Lee VM, Trojanowski JQ (2007) Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci 8(9):663–672
Banati RB, Gehrmann J, Schubert P, Kreutzberg GW (1993) Cytotoxicity of microglia. Glia 7(1):111–118
Barbagallo AP, Weldon R, Tamayev R, Zhou D, Giliberto L, Foreman O, D’Adamio L (2010) Tyr(682) in the intracellular domain of APP regulates amyloidogenic APP processing in vivo. PLoS One 5(11):e15503
Barger SW, Harmon AD (1997) Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 388(6645):878–881
Bate C, Veerhuis R, Eikelenboom P, Williams A (2004) Microglia kill amyloid-beta1-42 damaged neurons by a CD14-dependent process. Neuroreport 15(9):1427–1430
Bate C, Kempster S, Williams A (2006) Prostaglandin D2 mediates neuronal damage by amyloid-beta or prions which activates microglial cells. Neuropharmacology 50(2):229–237
Baum L, Lam CW, Cheung SK, Kwok T, Lui V, Tsoh J, Lam L, Leung V, Hui E, Ng C, Woo J, Chiu HF, Goggins WB, Zee BC, Cheng KF, Fong CY, Wong A, Mok H, Chow MS, Ho PC, Ip SP, Ho CS, Yu XW, Lai CY, Chan MH, Szeto S, Chan IH, Mok V (2008) Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J Clin Psychopharmacol 28(1):110–113
Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 118(1):103–113
Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV (2007) Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 27(5):909–918
Benarroch EE (2005) Neuron-astrocyte interactions: partnership for normal function and disease in the central nervous system. Mayo Clin Proc 80(10):1326–1338
Berthiaume EP, Medina C, Swanson JA (1995) Molecular size-fractionation during endocytosis in macrophages. J Cell Biol 129(4):989–998
Beurel E, Michalek SM, Jope RS (2010) Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol 31(1):24–31
Bilikiewicz A, Gaus W (2004) Colostrinin (a naturally occurring, proline-rich, polypeptide mixture) in the treatment of Alzheimer’s disease. J Alzheimers Dis 6(1):17–26
Birks J, Grimley Evans J (2009) Ginkgo biloba for cognitive impairment and dementia. Cochrane Database Syst Rev (1):CD003120
Blach-Olszewska Z, Leszek J (2007) Mechanisms of over-activated innate immune system regulation in autoimmune and neurodegenerative disorders. Neuropsychiatr Dis Treat 3(3):365–372
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8(1):57–69
Blum-Degen D, Muller T, Kuhn W, Gerlach M, Przuntek H, Riederer P (1995) Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett 202(1–2):17–20
Bolanos JP, Almeida A, Stewart V, Peuchen S, Land JM, Clark JB, Heales SJ (1997) Nitric oxide-mediated mitochondrial damage in the brain: mechanisms and implications for neurodegenerative diseases. J Neurochem 68(6):2227–2240
Breitner JC, Haneuse SJ, Walker R, Dublin S, Crane PK, Gray SL, Larson EB (2009) Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology 72(22):1899–1905
Brenn A, Grube M, Peters M, Fischer A, Jedlitschky G, Kroemer HK, Warzok RW, Vogelgesang S (2011) Beta-amyloid downregulates MDR1-P-glycoprotein (Abcb1) expression at the blood-brain barrier in mice. Int J Alzheimers Dis 2011:690121
Brewer GJ (1997) Effects of acidosis on the distribution of processing of the beta-amyloid precursor protein in cultured hippocampal neurons. Mol Chem Neuropathol 31(2):171–186
Brown GC, Bal-Price A (2003) Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol 27(3):325–355
Bruno MA, Mufson EJ, Wuu J, Cuello AC (2009) Increased matrix metalloproteinase 9 activity in mild cognitive impairment. J Neuropathol Exp Neurol 68(12):1309–1318
Calabrese V, Bates TE, Stella AM (2000) NO synthase and NO-dependent signal pathways in brain aging and neurodegenerative disorders: the role of oxidant/antioxidant balance. Neurochem Res 25(9–10):1315–1341
Calabrese V, Sultana R, Scapagnini G, Guagliano E, Sapienza M, Bella R, Kanski J, Pennisi G, Mancuso C, Stella AM, Butterfield DA (2006) Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer’s disease. Antioxid Redox Signal 8(11–12):1975–1986
Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM (2007) Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 8(10):766–775
Carlsson CM (2010) Type 2 diabetes mellitus, dyslipidemia, and Alzheimer’s disease. J Alzheimers Dis 20(3):711–722
Chalbot S, Zetterberg H, Blennow K, Fladby T, Andreasen N, Grundke-Iqbal I, Iqbal K (2011) Blood-cerebrospinal fluid barrier permeability in Alzheimer’s disease. J Alzheimers Dis 25(3):505–515
Chen Y, Vartiainen NE, Ying W, Chan PH, Koistinaho J, Swanson RA (2001) Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanism. J Neurochem 77(6):1601–1610
Cho JY, Um HS, Kang EB, Cho IH, Kim CH, Cho JS, Hwang DY (2010) The combination of exercise training and alpha-lipoic acid treatment has therapeutic effects on the pathogenic phenotypes of Alzheimer’s disease in NSE/APPsw-transgenic mice. Int J Mol Med 25(3):337–346
Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE (2000) Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci 20(2):558–567
Combs CK, Karlo JC, Kao SC, Landreth GE (2001) beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci 21(4):1179–1188
Contestabile A, Fila T, Bartesaghi R, Contestabile A, Ciani E (2006) Choline acetyltransferase activity at different ages in brain of Ts65Dn mice, an animal model for Down’s syndrome and related neurodegenerative diseases. J Neurochem 97(2):515–526
Craft JM, Watterson DM, Van Eldik LJ (2006) Human amyloid beta-induced neuroinflammation is an early event in neurodegeneration. Glia 53(5):484–490
Cullheim S, Thams S (2007) The microglial networks of the brain and their role in neuronal network plasticity after lesion. Brain Res Rev 55(1):89–96
Danysz W, Parsons CG (1998) Glycine and N-methyl-d-aspartate receptors: physiological significance and possible therapeutic applications. Pharmacol Rev 50(4):597–664
Davies TA, Billingslea AM, Long HJ, Tibbles H, Wells JM, Eisenhauer PB, Smith SJ, Cribbs DH, Fine RE, Simons ER (1998) Brain endothelial cell enzymes cleave platelet-retained amyloid precursor protein. J Lab Clin Med 132(4):341–350
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B (2003) RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 9(7):907–913
Deane R, Wu Z, Zlokovic BV (2004) RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 35(11 Suppl 1):2628–2631
Desai BS, Schneider JA, Li JL, Carvey PM, Hendey B (2009) Evidence of angiogenic vessels in Alzheimer’s disease. J Neural Transm 116(5):587–597
Dong XX, Wang Y, Qin ZH (2009) Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 30(4):379–387
Drzezga A, Lautenschlager N, Siebner H, Riemenschneider M, Willoch F, Minoshima S, Schwaiger M, Kurz A (2003) Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer’s disease: a PET follow-up study. Eur J Nucl Med Mol Imaging 30(8):1104–1113
Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, Hammers A, Tai YF, Fox N, Kennedy A, Rossor M, Brooks DJ (2008) Microglia, amyloid, and cognition in Alzheimer’s disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis 32(3):412–419
Eikelenboom P, van Gool WA (2004) Neuroinflammatory perspectives on the two faces of Alzheimer’s disease. J Neural Transm 111(3):281–294
El-Fawal HA, O’Callaghan JP (2008) Autoantibodies to neurotypic and gliotypic proteins as biomarkers of neurotoxicity: assessment of trimethyltin (TMT). Neurotoxicology 29(1):109–115
Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A (1996) Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology 46(6):1592–1596
Engel T, Hernandez F, Avila J, Lucas JJ (2006) Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci 26(19):5083–5090
Faraci FM (2003) Hyperhomocysteinemia: a million ways to lose control. Arterioscler Thromb Vasc Biol 23(3):371–373
Fiebich BL, Schleicher S, Spleiss O, Czygan M, Hull M (2001) Mechanisms of prostaglandin E2-induced interleukin-6 release in astrocytes: possible involvement of EP4-like receptors, p38 mitogen-activated protein kinase and protein kinase C. J Neurochem 79(5):950–958
Fioravanzo L, Venturini M, Liddo RD, Marchi F, Grandi C, Parnigotto PP, Folin M (2010) Involvement of rat hippocampal astrocytes in beta-amyloid-induced angiogenesis and neuroinflammation. Curr Alzheimer Res 7(7):591–601
Freemantle E, Vandal M, Tremblay-Mercier J, Tremblay S, Blachere JC, Begin ME, Brenna JT, Windust A, Cunnane SC (2006) Omega-3 fatty acids, energy substrates, and brain function during aging. Prostaglandins Leukot Essent Fatty Acids 75(3):213–220
Fuller S, Steele M, Munch G (2010) Activated astroglia during chronic inflammation in Alzheimer’s disease—do they neglect their neurosupportive roles? Mutat Res 690(1–2):40–49
Furchgott RF (1999) Endothelium-derived relaxing factor: discovery, early studies, and identification as nitric oxide. Biosci Rep 19(4):235–251
Galimberti D, Scarpini E (2011) Disease-modifying treatments for Alzheimer’s disease. Ther Adv Neurol Disord 4(4):203–216
Gallucci M, Zanardo A, De Valentin L, Vianello A (2004) Homocysteine in Alzheimer disease and vascular dementia. Arch Gerontol Geriatr Suppl 9:195–200
Galluzzi KE, Appelt DM, Balin BJ (2010) Modern care for patients with Alzheimer disease: rationale for early intervention. J Am Osteopath Assoc 110(9 Suppl 8):S37–S42
Garwood CJ, Pooler AM, Atherton J, Hanger DP, Noble W (2011) Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis 2:e167
Gearing M, Mirra SS, Hedreen JC, Sumi SM, Hansen LA, Heyman A (1995) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part X. Neuropathology confirmation of the clinical diagnosis of Alzheimer’s disease. Neurology 45(3 Pt 1):461–466
Ghosal K, Stathopoulos A, Pimplikar SW (2010) APP intracellular domain impairs adult neurogenesis in transgenic mice by inducing neuroinflammation. PLoS One 5(7):e11866
Grammas P, Samany PG, Thirumangalakudi L (2006) Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: implications for disease pathogenesis. J Alzheimers Dis 9(1):51–58
Granzotto A, Zatta P (2011) Resveratrol acts not through anti-aggregative pathways but mainly via its scavenging properties against Abeta and Abeta-metal complexes toxicity. PLoS One 6(6):e21565
Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL 3rd, Araoz C (1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 86(19):7611–7615
Guglielmotto M, Aragno M, Tamagno E, Vercellinatto I, Visentin S, Medana C, Catalano MG, Smith MA, Perry G, Danni O, Boccuzzi G, Tabaton M (2010) AGEs/RAGE complex upregulates BACE1 via NF-kappaB pathway activation. Neurobiol Aging. doi:10.1016/j.neurobiolaging.2010.05.026
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9(8):857–865
Hashioka S, Klegeris A, Schwab C, McGeer PL (2009) Interferon-gamma-dependent cytotoxic activation of human astrocytes and astrocytoma cells. Neurobiol Aging 30(12):1924–1935
Hayden KM, Zandi PP, Khachaturian AS, Szekely CA, Fotuhi M, Norton MC, Tschanz JT, Pieper CF, Corcoran C, Lyketsos CG, Breitner JC, Welsh-Bohmer KA (2007) Does NSAID use modify cognitive trajectories in the elderly? The Cache County study. Neurology 69(3):275–282
Heneka MT, O’Banion MK (2007) Inflammatory processes in Alzheimer’s disease. J Neuroimmunol 184(1–2):69–91
Hensley K, Maidt ML, Yu Z, Sang H, Markesbery WR, Floyd RA (1998) Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J Neurosci 18(20):8126–8132
Hermann C, Stern RG, Losonzcy MF, Jaff S, Davidson M (1991) Diagnostic and pharmacological approaches in Alzheimer’s disease. Drugs Aging 1(2):144–162
Hernandez F, Borrell J, Guaza C, Avila J, Lucas JJ (2002) Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J Neurochem 83(6):1529–1533
Hollenberg NK (2006) Organ systems dependent on nitric oxide and the potential for nitric oxide-targeted therapies in related diseases. J Clin Hypertens (Greenwich) 8(12 Suppl 4):63–73
Hoozemans JJ, Rozemuller JM, van Haastert ES, Veerhuis R, Eikelenboom P (2008) Cyclooxygenase-1 and -2 in the different stages of Alzheimer’s disease pathology. Curr Pharm Des 14(14):1419–1427
Huell M, Strauss S, Volk B, Berger M, Bauer J (1995) Interleukin-6 is present in early stages of plaque formation and is restricted to the brains of Alzheimer’s disease patients. Acta Neuropathol 89(6):544–551
Hwang J, Hwang H, Lee HW, Suk K (2010) Microglia signaling as a target of donepezil. Neuropharmacology 58(7):1122–1129
Iadecola C (2004) Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci 5(5):347–360
Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA (1999) SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci 2(2):157–161
Ince P, Xuereb J, MacKenzie J, Rossi M, Lowe J, Morris HJ, Matthews F, Brayne C, Esiri MM (2000) Neuropathology of a community sample of elderly demented and nondemented people. Brain Pathol 10:591–593. doi:10.1111/j.1750-3639.2000.tb00310.x
Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I (2005) Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 1739(2–3):198–210
Isaac MG, Quinn R, Tabet N (2008) Vitamin E for Alzheimer’s disease and mild cognitive impairment. Cochrane Database Syst Rev (3):CD002854
Jana A, Pahan K (2010) Fibrillar amyloid-beta-activated human astroglia kill primary human neurons via neutral sphingomyelinase: implications for Alzheimer’s disease. J Neurosci 30(38):12676–12689
Ji Y, Permanne B, Sigurdsson EM, Holtzman DM, Wisniewski T (2001) Amyloid beta40/42 clearance across the blood-brain barrier following intra-ventricular injections in wild-type, apoE knock-out and human apoE3 or E4 expressing transgenic mice. J Alzheimers Dis 3(1):23–30
Kalaria RN, Grahovac I (1990) Serum amyloid P immunoreactivity in hippocampal tangles, plaques and vessels: implications for leakage across the blood-brain barrier in Alzheimer’s disease. Brain Res 516(2):349–353
Kar S, Slowikowski SP, Westaway D, Mount HT (2004) Interactions between beta-amyloid and central cholinergic neurons: implications for Alzheimer’s disease. J Psychiatry Neurosci 29(6):427–441
Kawahara M, Ohtsuka I, Yokoyama S, Kato-Negishi M, Sadakane Y (2011) Membrane incorporation, channel formation, and disruption of calcium homeostasis by Alzheimer’s beta-amyloid protein. Int J Alzheimers Dis 2011:304583
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91(2):461–553
Kihara T, Shimohama S (2004) Alzheimer’s disease and acetylcholine receptors. Acta Neurobiol Exp (Wars) 64(1):99–105
Klapstein GJ, Colmers WF (1997) Neuropeptide Y suppresses epileptiform activity in rat hippocampus in vitro. J Neurophysiol 78(3):1651–1661
Klapstein GJ, Levine MS (2005) Age-dependent biphasic changes in ischemic sensitivity in the striatum of Huntington’s disease R6/2 transgenic mice. J Neurophysiol 93(2):758–765
Klempin F, Kempermann G (2007) Adult hippocampal neurogenesis and aging. Eur Arch Psychiatry Clin Neurosci 257(5):271–280
Koenigsknecht-Talboo J, Landreth GE (2005) Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci 25(36):8240–8249
Kovac A, Zilkova M, Deli MA, Zilka N, Novak M (2009) Human truncated tau is using a different mechanism from amyloid-beta to damage the blood-brain barrier. J Alzheimers Dis 18(4):897–906
Krause DL, Muller N (2010) Neuroinflammation, microglia and implications for anti-inflammatory treatment in Alzheimer’s disease. Int J Alzheimers Dis 2010:732806. doi:10.4061/2010/732806
Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19(8):312–318
Krishnan S, Cairns R, Howard R (2009) Cannabinoids for the treatment of dementia. Cochrane Database Syst Rev (2):CD007204
Kuiper MA, Teerlink T, Visser JJ, Bergmans PL, Scheltens P, Wolters EC (2000) l-glutamate, l-arginine and l-citrulline levels in cerebrospinal fluid of Parkinson’s disease, multiple system atrophy, and Alzheimer’s disease patients. J Neural Transm 107(2):183–189
Landreth GE, Reed-Geaghan EG (2009) Toll-like receptors in Alzheimer’s disease. Curr Top Microbiol Immunol 336:137–153
Lee RK, Knapp S, Wurtman RJ (1999) Prostaglandin E2 stimulates amyloid precursor protein gene expression: inhibition by immunosuppressants. J Neurosci 19(3):940–947
Lee H, Kim HJ, Kim JM, Chang N (2004) Effects of dietary folic acid supplementation on cerebrovascular endothelial dysfunction in rats with induced hyperhomocysteinemia. Brain Res 996(2):139–147
Lee KS, Chung JH, Choi TK, Suh SY, Oh BH, Hong CH (2009) Peripheral cytokines and chemokines in Alzheimer’s disease. Dement Geriatr Cogn Disord 28(4):281–287
Leszek J, Inglot AD, Janusz M, Lisowski J, Krukowska K, Georgiades JA (1999) Colostrinin: a proline-rich polypeptide (PRP) complex isolated from ovine colostrum for treatment of Alzheimer’s disease. A double-blind, placebo-controlled study. Arch Immunol Ther Exp (Warsz) 47(6):377–385
Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, Vargas ME, Jokel ES, Carpenter EM, Zanjani H, Hurst RS, Efstratiadis A, Zeitlin S, Chesselet MF (1999) Enhanced sensitivity to N-methyl-d-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J Neurosci Res 58(4):515–532
Li J, Schmidt AM (1997) Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem 272(26):16498–16506
Li S, Mallory M, Alford M, Tanaka S, Masliah E (1997) Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol 56(8):901–911
Li J, Wu HM, Zhou RL, Liu GJ, Dong BR (2008) Huperzine A for Alzheimer’s disease. Cochrane Database Syst Rev (2):CD005592
Lindesay J, Bullock R, Daniels H, Emre M, Forstl H, Frolich L, Gabryelewicz T, Martinez-Lage P, Monsch AU, Tsolaki M, van Laar T (2010) Turning principles into practice in Alzheimer’s disease. Int J Clin Pract 64(9):1198–1209
Lipton SA, Kim WK, Choi YB, Kumar S, D’Emilia DM, Rayudu PV, Arnelle DR, Stamler JS (1997) Neurotoxicity associated with dual actions of homocysteine at the N-methyl-d-aspartate receptor. Proc Natl Acad Sci USA 94(11):5923–5928
Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, Heine H, Penke B, Neumann H, Fassbender K (2005) LPS receptor (CD14): a receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain 128(Pt 8):1778–1789
Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J (2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J 20(1–2):27–39
Lue LF, Brachova L, Civin WH, Rogers J (1996) Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of Alzheimer’s disease neurodegeneration. J Neuropathol Exp Neurol 55(10):1083–1088
Luo J, Grammas P (2010) Endothelin-1 is elevated in Alzheimer’s disease brain microvessels and is neuroprotective. J Alzheimers Dis 21(3):887–896
Maccioni RB, Rojo LE, Fernandez JA, Kuljis RO (2009) The role of neuroimmunomodulation in Alzheimer’s disease. Ann N Y Acad Sci 1153:240–246
Mackic JB, Weiss MH, Miao W, Kirkman E, Ghiso J, Calero M, Bading J, Frangione B, Zlokovic BV (1998) Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer’s amyloid beta peptide in aged squirrel monkey with cerebral amyloid angiopathy. J Neurochem 70(1):210–215
Majumdar A, Cruz D, Asamoah N, Buxbaum A, Sohar I, Lobel P, Maxfield FR (2007) Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Mol Biol Cell 18(4):1490–1496
Malouf R, Grimley Evans J (2008) Folic acid with or without vitamin B12 for the prevention and treatment of healthy elderly and demented people. Cochrane Database Syst Rev (4):CD004514
Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JC, Craft S, Evans D, Green R, Mullan M (2008) Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol 65(7):896–905
Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, Olschowka JA, Fonseca MI, O’Banion MK, Tenner AJ, Lemere CA, Duff K (2001) Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer’s disease. Am J Pathol 158(4):1345–1354
Mayeux R, Tang MX, Jacobs DM, Manly J, Bell K, Merchant C, Small SA, Stern Y, Wisniewski HM, Mehta PD (1999) Plasma amyloid beta-peptide 1–42 and incipient Alzheimer’s disease. Ann Neurol 46(3):412–416
Mayeux R, Honig LS, Tang MX, Manly J, Stern Y, Schupf N, Mehta PD (2003) Plasma A[beta]40 and A[beta]42 and Alzheimer’s disease: relation to age, mortality, and risk. Neurology 61(9):1185–1190
McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA (1999) Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci USA 96(1):272–277
McGeer EG, McGeer PL (2003) Inflammatory processes in Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry 27(5):741–749
McShane R, Areosa Sastre A, Minakaran N (2006) Memantine for dementia. Cochrane Database Syst Rev (2):CD003154
Mielke R, Kessler J, Szelies B, Herholz K, Wienhard K, Heiss WD (1998) Normal and pathological aging—findings of positron-emission-tomography. J Neural Transm 105(8–9):821–837
Mines MA, Beurel E, Jope RS (2011) Regulation of cell survival mechanisms in Alzheimer’s disease by glycogen synthase kinase-3. Int J Alzheimers Dis 2011:861072. doi:10.4061/2011/861072
Minoretti P, Gazzaruso C, Vito CD, Emanuele E, Bianchi M, Coen E, Reino M, Geroldi D (2006) Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on susceptibility to late-onset Alzheimer’s disease. Neurosci Lett 391(3):147–149
Mondragon-Rodriguez S, Basurto-Islas G, Lee HG, Perry G, Zhu X, Castellani RJ, Smith MA (2010) Causes versus effects: the increasing complexities of Alzheimer’s disease pathogenesis. Expert Rev Neurother 10(5):683–691
Montine TJ, Sidell KR, Crews BC, Markesbery WR, Marnett LJ, Roberts LJ 2nd, Morrow JD (1999) Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology 53(7):1495–1498
Moore AH, O’Banion MK (2002) Neuroinflammation and anti-inflammatory therapy for Alzheimer’s disease. Adv Drug Deliv Rev 54(12):1627–1656
Morley JE, Farr SA, Banks WA, Johnson SN, Yamada KA, Xu L (2010) A physiological role for amyloid-beta protein: enhancement of learning and memory. J Alzheimers Dis 19(2):441–449
Moser KV, Reindl M, Blasig I, Humpel C (2004) Brain capillary endothelial cells proliferate in response to NGF, express NGF receptors and secrete NGF after inflammation. Brain Res 1017(1–2):53–60
Mundy DI (1994) Identification of the multicatalytic enzyme as a possible gamma-secretase for the amyloid precursor protein. Biochem Biophys Res Commun 204(1):333–341
Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang HY (2003) Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res 971(2):197–209
Nathan C, Calingasan N, Nezezon J, Ding A, Lucia MS, La Perle K, Fuortes M, Lin M, Ehrt S, Kwon NS, Chen J, Vodovotz Y, Kipiani K, Beal MF (2005) Protection from Alzheimer’s-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J Exp Med 202(9):1163–1169
Nitsch RM, Slack BE, Wurtman RJ, Growdon JH (1992) Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science 258(5080):304–307
O’Banion MK, Miller JC, Chang JW, Kaplan MD, Coleman PD (1996) Interleukin-1 beta induces prostaglandin G/H synthase-2 (cyclooxygenase-2) in primary murine astrocyte cultures. J Neurochem 66(6):2532–2540
Okamoto M, Wang X, Baba M (2005) HIV-1-infected macrophages induce astrogliosis by SDF-1alpha and matrix metalloproteinases. Biochem Biophys Res Commun 336(4):1214–1220
Orrego F, Villanueva S (1993) The chemical nature of the main central excitatory transmitter: a critical appraisal based upon release studies and synaptic vesicle localization. Neuroscience 56(3):539–555
Paresce DM, Chung H, Maxfield FR (1997) Slow degradation of aggregates of the Alzheimer’s disease amyloid beta-protein by microglial cells. J Biol Chem 272(46):29390–29397
Paris D, Townsend KP, Humphrey J, Obregon DF, Yokota K, Mullan M (2002a) Statins inhibit A beta-neurotoxicity in vitro and A beta-induced vasoconstriction and inflammation in rat aortae. Atherosclerosis 161(2):293–299
Paris D, Townsend KP, Obregon DF, Humphrey J, Mullan M (2002b) Pro-inflammatory effect of freshly solubilized beta-amyloid peptides in the brain. Prostaglandins Other Lipid Mediat 70(1–2):1–12
Park SA, Shaked GM, Bredesen DE, Koo EH (2009) Mechanism of cytotoxicity mediated by the C31 fragment of the amyloid precursor protein. Biochem Biophys Res Commun 388(2):450–455
Perry VH, Newman TA, Cunningham C (2003) The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci 4(2):103–112
Persson M, Brantefjord M, Hansson E, Ronnback L (2005) Lipopolysaccharide increases microglial GLT-1 expression and glutamate uptake capacity in vitro by a mechanism dependent on TNF-alpha. Glia 51(2):111–120
Phiel CJ, Wilson CA, Lee VM, Klein PS (2003) GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423(6938):435–439
Plant LD, Boyle JP, Smith IF, Peers C, Pearson HA (2003) The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J Neurosci 23(13):5531–5535
Pooler AM, Arjona AA, Lee RK, Wurtman RJ (2004) Prostaglandin E2 regulates amyloid precursor protein expression via the EP2 receptor in cultured rat microglia. Neurosci Lett 362(2):127–130
Puzzo D, Privitera L, Fa M, Staniszewski A, Hashimoto G, Aziz F, Sakurai M, Ribe EM, Troy CM, Mercken M, Jung SS, Palmeri A, Arancio O (2011) Endogenous amyloid-beta is necessary for hippocampal synaptic plasticity and memory. Ann Neurol 69(5):819–830
Qaseem A, Snow V, Cross JT Jr, Forciea MA, Hopkins R Jr, Shekelle P, Adelman A, Mehr D, Schellhase K, Campos-Outcalt D, Santaguida P, Owens DK (2008) Current pharmacologic treatment of dementia: a clinical practice guideline from the American College of Physicians and the American Academy of Family Physicians. Ann Intern Med 148(5):370–378
Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE (2009) CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci 29(38):11982–11992
Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ (2003) Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 348(14):1333–1341
Remarque EJ, Bollen EL, Weverling-Rijnsburger AW, Laterveer JC, Blauw GJ, Westendorp RG (2001) Patients with Alzheimer’s disease display a pro-inflammatory phenotype. Exp Gerontol 36(1):171–176
Ringman JM, Frautschy SA, Cole GM, Masterman DL, Cummings JL (2005) A potential role of the curry spice curcumin in Alzheimer’s disease. Curr Alzheimer Res 2(2):131–136
Rodriguez JJ, Olabarria M, Chvatal A, Verkhratsky A (2009) Astroglia in dementia and Alzheimer’s disease. Cell Death Differ 16(3):378–385
Rogers J, Shen Y (2000) A perspective on inflammation in Alzheimer’s disease. Ann N Y Acad Sci 924:132–135
Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, Ball MJ (1993) beta-Amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci USA 90(22):10836–10840
Sabbagh M, Cummings J (2011) Progressive cholinergic decline in Alzheimer’s disease: consideration for treatment with donepezil 23 mg in patients with moderate to severe symptomatology. BMC Neurol 11:21
Sadowski M, Pankiewicz J, Scholtzova H, Li YS, Quartermain D, Duff K, Wisniewski T (2004) Links between the pathology of Alzheimer’s disease and vascular dementia. Neurochem Res 29(6):1257–1266
Sastre M, Walter J, Gentleman SM (2008) Interactions between APP secretases and inflammatory mediators. J Neuroinflammation 5:25
Schmitz ML, Mattioli I, Buss H, Kracht M (2004) NF-kappaB: a multifaceted transcription factor regulated at several levels. Chembiochem 5(10):1348–1358
Schneider LS (2000) A critical review of cholinesterase inhibitors as a treatment modality in Alzheimer’s disease. Dialogues Clin Neurosci 2(2):111–128
Schubert P, Morino T, Miyazaki H, Ogata T, Nakamura Y, Marchini C, Ferroni S (2000) Cascading glia reactions: a common pathomechanism and its differentiated control by cyclic nucleotide signaling. Ann N Y Acad Sci 903:24–33
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81(2):741–766
Seshadri S (2006) Elevated plasma homocysteine levels: risk factor or risk marker for the development of dementia and Alzheimer’s disease? J Alzheimers Dis 9(4):393–398
Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV (2000) Clearance of Alzheimer’s amyloid-beta(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 106(12):1489–1499
Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH (2006) Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci 26(41):10514–10523
Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E (2001) Neurogenesis in the adult is involved in the formation of trace memories. Nature 410(6826):372–376
Simard AR, Rivest S (2004) Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J 18(9):998–1000
Simonian NA, Coyle JT (1996) Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol 36:83–106
Simpson JE, Ince PG, Haynes LJ, Theaker R, Gelsthorpe C, Baxter L, Forster G, Lace GL, Shaw PJ, Matthews FE, Savva GM, Brayne C, Wharton SB (2010) Population variation in oxidative stress and astrocyte DNA damage in relation to Alzheimer-type pathology in the ageing brain. Neuropathol Appl Neurobiol 36(1):25–40
Skovronsky DM, Lee VM, Pratico D (2001) Amyloid precursor protein and amyloid beta peptide in human platelets. Role of cyclooxygenase and protein kinase C. J Biol Chem 276(20):17036–17043
Small GW, Ercoli LM, Silverman DH, Huang SC, Komo S, Bookheimer SY, Lavretsky H, Miller K, Siddarth P, Rasgon NL, Mazziotta JC, Saxena S, Wu HM, Mega MS, Cummings JL, Saunders AM, Pericak-Vance MA, Roses AD, Barrio JR, Phelps ME (2000) Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc Natl Acad Sci USA 97(11):6037–6042
Smith WL, Garavito RM, DeWitt DL (1996) Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem 271(52):33157–33160
Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR (1997) Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 277(10):813–817
Snyder JS, Hong NS, McDonald RJ, Wojtowicz JM (2005) A role for adult neurogenesis in spatial long-term memory. Neuroscience 130(4):843–852
Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K (2006) Role of toll-like receptor signalling in Abeta uptake and clearance. Brain 129(Pt 11):3006–3019
Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK (2002) Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 161(5):1869–1879
Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K (1993) Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc Natl Acad Sci USA 90(16):7789–7793
Takatori Y (2006) Mechanisms of neuroprotective effects of therapeutic acetylcholinesterase inhibitors used in treatment of Alzheimer’s disease. Yakugaku Zasshi 126(8):607–616
Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M (1999) Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science 286(5448):2352–2355
Tang BL (2009) Neuronal protein trafficking associated with Alzheimer disease: from APP and BACE1 to glutamate receptors. Cell Adh Migr 3(1):118–128
Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP (2007) Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci USA 104(34):13798–13803
Tang SC, Lathia JD, Selvaraj PK, Jo DG, Mughal MR, Cheng A, Siler DA, Markesbery WR, Arumugam TV, Mattson MP (2008) Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp Neurol 213(1):114–121
Tanzi RE, Moir RD, Wagner SL (2004) Clearance of Alzheimer’s Abeta peptide: the many roads to perdition. Neuron 43(5):605–608
Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I (2004) Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA 291(3):317–324
Thirumangalakudi L, Samany PG, Owoso A, Wiskar B, Grammas P (2006) Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J Alzheimers Dis 10(1):111–118
Triantafilou M, Morath S, Mackie A, Hartung T, Triantafilou K (2004) Lateral diffusion of Toll-like receptors reveals that they are transiently confined within lipid rafts on the plasma membrane. J Cell Sci 117(Pt 17):4007–4014
Troen AM (2005) The central nervous system in animal models of hyperhomocysteinemia. Prog Neuropsychopharmacol Biol Psychiatry 29(7):1140–1151
Udan ML, Ajit D, Crouse NR, Nichols MR (2008) Toll-like receptors 2 and 4 mediate Abeta(1–42) activation of the innate immune response in a human monocytic cell line. J Neurochem 104(2):524–533
van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH (2002) Functional neurogenesis in the adult hippocampus. Nature 415(6875):1030–1034
Vehmas AK, Kawas CH, Stewart WF, Troncoso JC (2003) Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol Aging 24(2):321–331
Vicenzini E, Ricciardi MC, Altieri M, Puccinelli F, Bonaffini N, Di Piero V, Lenzi GL (2007) Cerebrovascular reactivity in degenerative and vascular dementia: a transcranial Doppler study. Eur Neurol 58(2):84–89
Wakasa S, Shiiya N, Tachibana T, Ooka T, Matsui Y (2009) A semiquantitative analysis of reactive astrogliosis demonstrates its correlation with the number of intact motor neurons after transient spinal cord ischemia. J Thorac Cardiovasc Surg 137(4):983–990
Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W, Fassbender K (2007) Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem 20(6):947–956
Wang DD, Bordey A (2008) The astrocyte odyssey. Prog Neurobiol 86(4):342–367
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421(6921):384–388
Wang JM, Singh C, Liu L, Irwin RW, Chen S, Chung EJ, Thompson RF, Brinton RD (2010) Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 107(14):6498–6503
Warner DS, Sheng H, Batinic-Haberle I (2004) Oxidants, antioxidants and the ischemic brain. J Exp Biol 207(Pt 18):3221–3231
Watkins PB, Zimmerman HJ, Knapp MJ, Gracon SI, Lewis KW (1994) Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 271(13):992–998
Weir DG, Molloy AM (2000) Microvascular disease and dementia in the elderly: are they related to hyperhomocysteinemia? Am J Clin Nutr 71(4):859–860
Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR (1982) Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science 215(4537):1237–1239
Willard LB, Hauss-Wegrzyniak B, Danysz W, Wenk GL (2000) The cytotoxicity of chronic neuroinflammation upon basal forebrain cholinergic neurons of rats can be attenuated by glutamatergic antagonism or cyclooxygenase-2 inhibition. Exp Brain Res 134(1):58–65
Wirths O, Multhaup G, Bayer TA (2004) A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide—the first step of a fatal cascade. J Neurochem 91(3):513–520
Wolfson C, Perrault A, Moride Y, Esdaile JM, Abenhaim L, Momoli F (2002) A case-control analysis of nonsteroidal anti-inflammatory drugs and Alzheimer’s disease: are they protective? Neuroepidemiology 21(2):81–86
Wu HM, Tzeng NS, Qian L, Wei SJ, Hu X, Chen SH, Rawls SM, Flood P, Hong JS, Lu RB (2009) Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology 34(10):2344–2357
Xiao XQ, Wang R, Tang XC (2000) Huperzine A and tacrine attenuate beta-amyloid peptide-induced oxidative injury. J Neurosci Res 61(5):564–569
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382(6593):685–691
Yan SF, Yan SD, Ramasamy R, Schmidt AM (2009) Tempering the wrath of RAGE: an emerging therapeutic strategy against diabetic complications, neurodegeneration, and inflammation. Ann Med 41(6):408–422
Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM (2005) Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 280(7):5892–5901
Yermakova AV, O’Banion MK (2001) Downregulation of neuronal cyclooxygenase-2 expression in end stage Alzheimer’s disease. Neurobiol Aging 22(6):823–836
Yokoyama A, Yang L, Itoh S, Mori K, Tanaka J (2004) Microglia, a potential source of neurons, astrocytes, and oligodendrocytes. Glia 45(1):96–104
Yoshiyama Y, Kojima A, Ishikawa C, Arai K (2010) Anti-inflammatory action of donepezil ameliorates tau pathology, synaptic loss, and neurodegeneration in a tauopathy mouse model. J Alzheimers Dis 22(1):295–306
Zhang D, Hu X, Qian L, Wilson B, Lee C, Flood P, Langenbach R, Hong JS (2009) Prostaglandin E2 released from activated microglia enhances astrocyte proliferation in vitro. Toxicol Appl Pharmacol 238(1):64–70
Zlokovic BV, Yamada S, Holtzman D, Ghiso J, Frangione B (2000) Clearance of amyloid beta-peptide from brain: transport or metabolism? Nat Med 6(7):718–719
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Broussard, G.J., Mytar, J., Li, Rc. et al. The role of inflammatory processes in Alzheimer’s disease. Inflammopharmacol 20, 109–126 (2012). https://doi.org/10.1007/s10787-012-0130-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-012-0130-z
Keywords
- Alzheimer’s disease
- Inflammation
- Neuron
- Neuronal
- Astrocyte
- Microglia
- Cardiovascular
- Microvessels
- Vascular
- Endothelial
- Cytokine
- Chemokine
- Immune
- Neurodegenerative
- Aβ
- Amyloid
- β-amyloid
- Pathogenesis
- Pathology
- Reactive oxygen species
- Reactive nitrogen species
- Interleukin
- Blood–brain barrier
- Cyclooxygenase
- Inflammatory mediators
- Activated endothelium