
Abstract
Alzheimer´s disease (AD) stands out as the most common chronic neurodegenerative disorder. AD is characterized by progressive cognitive decline and memory loss, with neurodegeneration as its primary pathological feature. The role of neuroinflammation in the disease course has become a focus of intense research. While microglia, the brain’s resident macrophages, have been pivotal to study central immune inflammation, recent evidence underscores the contributions of other cellular entities to the neuroinflammatory process. In this article, we review the inflammatory role of microglia and astrocytes, focusing on their interactions with AD’s core pathologies, amyloid beta deposition, and tau tangle formation. Additionally, we also discuss how different modes of regulated cell death in AD may impact the chronic neuroinflammatory environment. This review aims to highlight the evolving landscape of neuroinflammatory research in AD and underscores the importance of considering multiple cellular contributors when developing new therapeutic strategies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Alzheimer’s disease and neurodegeneration
Aging is a global phenomenon, with projections indicating that by 2050, 16% of the world's population will be aged 65 or older, compared to 9% in 2019 [235]. This demographic shift poses a significant challenge, as aging is a major risk factor for dementia, imposing an estimated economic cost exceeding 2.8 trillion dollars. Alzheimer's disease (AD) is the most prevalent form of dementia affecting 50 million people at present. AD cases are expected to double every 20 years [26, 163]
AD patients experience progressive cognitive decline, notably in episodic memory and executive functions, leading to interference with their daily activities. This cognitive impairment manifests as short-term memory deterioration, visuospatial processing issues, executive problems, and expressive speech difficulties. Together, this results in a growing burden on patients, caregivers, and families, impacting patients and relatives independence [118]. Despite extensive efforts, current treatments for AD are mainly symptomatic with limited efficacy, stressing the urgent development of therapies capable of halting the pathogenic process [110, 146]. Alois Alzheimer first reported AD in 1906 based on the examination of a single patient's brain, identifying key neuropathological features including deposition of so-called senile plaques composed of extracellular amyloid-β (Aβ) peptide aggregates and the formation of intraneuronal neurofibrillary tangles (NFTs) consisting mainly of hyperphosphorylated tau protein [5, 6]. He also depicted and described a massive glial reaction; however, it took several decades until the role of immune processes, such as microglial and astroglial reactivity, have become a major focus of research [92].
The predominant hypothesis for AD pathogenesis is the amyloid cascade hypothesis, proposed by Hardy and Higgins (1992) and supported by Selkoe and Hardy (2002) and others subsequently. Mutations in the amyloid precursor protein (APP), Presenilin-1 (PSEN1), and Presenilin-2 (PSEN2) genes, associated with early onset familial AD (EOAD), cause AD in individuals as young as their 30s and 40s [88, 89]. EOAD mutations in APP and PS1/2 alter Aβ biogenesis, resulting in an altered formation of amyloidogenic Aβ peptides [43, 52, 91, 152, 172, 240, 249]. In EOAD, symptoms follow the development of Aβ pathology by up to a decade, marked by the aggregation and deposition of Aβ peptides [13, 89, 104, 105]. This hypothesis suggests that accumulating Aβ peptides initiate a cascade of pathological events involving neuroinflammation, synaptic loss, tau pathology, and neuronal loss, leading to progressive symptoms, cerebral atrophy, and ultimately death.
Neurodegenerative diseases (NDDs), including AD, can be distinctively identified by characteristics such as brain region susceptibility or protein aggregate composition [70]. Interestingly, they also share important commonalities like pathological protein aggregation, synaptic and neuronal network dysfunction, aberrant proteostasis, cytoskeletal abnormalities, altered energy metabolism, DNA and RNA defects, inflammation, and neuronal cell death [257]. These commonalities do not arise from a single dominant molecular pathway. Instead, the neurodegenerative process seems to result from the synergistic action of various interrelated mechanisms including abnormal protein dynamics, increased production of reactive oxygen species (ROS), defective autophagy, mitochondrial dysfunctions, impairment of axonal transport, and, notably, neuroinflammation [106].
Characterized by the activation of microglia and astrocytes, neuroinflammation has garnered attention in recent years due to the early presence in the disease course, even before histopathological and pathological features of degeneration can be detected, representing an attractive therapeutic approach [92, 93, 257]. This immune activation was detected in close proximity to areas of neuronal damage and degeneration, leading to the obvious yet simplistic conclusion that it was a consequence of neuronal death. Now, a growing body of evidence challenge this assumption, demonstrating that neuroinflammation may also play an active role in the disease. (1) In many NDDs, signs of neuroinflammation are often detected prior to the appearance of neurodegenerative biomarkers and clinical symptoms [63], suggesting a driving role for disease pathogenesis rather than a pure bystander reaction [124]. (2) This view is further supported by the correlation between inflammatory markers, such as soluble TREM2 (sTREM2), YKL-40 or glial fibrillary acidic protein (GFAP), and disease severity [46, 47, 97]. (3) Genome-wide association studies (GWAS) and next-generation sequencing approaches have identified over 80 independent genetic loci modulating the risk of AD. Notably, many of these risk loci are located in genes that encode for proteins which form integral parts of microglia key signaling pathways including immunoreceptors (TREM2, SPI1, CD33) [250], agonistic ligands (IL34 and APOE) or effector mechanisms (ABI3 and EPHA1), suggesting a connection of inflammation to disease pathogenesis [16, 120, 168]. (4) Furthermore, preclinical in vivo models of extracellular Aβ accumulation or intraneuronal formation of NFTs show widespread innate immune activation that correlates with neuropathology. In keeping with this, inflammation-targeting genetic modifications greatly impact disease outcome in rodent models of neurodegenerative disease in general [21, 261] and AD in particular [94, 258]. (5) Persistent activation of CNS glial cells, such as microglia and astrocytes, results in elevated levels of inflammatory cytokines (IL-1β, TNFα, and IL-6) for prolonged periods of time. This, in turn, prompts the upregulation of matrix metalloprotease 9 (MMP9) in brain endothelial cells (BECs). MMP9 targets components of the endothelial basal layer and tight junctions (TJs), thereby possibly affecting blood–brain barrier (BBB) integrity [18, 262]. (6) Neurons exposed to microglia-derived pro-inflammatory cytokines, such as IL-1β, IL-2, IL-6, or TNFα, show enhanced spine loss and reduced hippocampal long-term potentiation (LTP) [48, 161]. (7) Epidemiological data suggest that individuals with chronic inflammatory conditions are at a higher risk of developing neurodegenerative diseases [72, 199, 227]. Taken together, current evidence strongly supports the idea that neuroinflammation represents an influential player able to contribute to the progression of pathological processes.
The intricate interplay between neuroinflammation and neurodegeneration emerges as a future target for therapeutics. Interrupting the mutual interaction between both components may be exceedingly complex; but offer the opportunity to develop effective therapies. This undertaking, however, requires a deep understanding of all the implicated players in the brain and the periphery. In this review, we aim to highlight the inflammatory contribution to AD neurodegenerative process throughout the lens of the different major cells types involved, including microglia, astrocytes, and neurons, but also others, such as T cells, oligodendrocytes, pericytes, and border-associated macrophages.
The neuroinflammatory disease component
Inflammation is an evolutionarily conserved process involving the activation of both immune and non-immune cells. This response protects the host from bacteria, viruses, toxins, and infections by eliminating pathogens and facilitating tissue repair and recovery [30]. Neuroinflammation refers to the inflammatory process in the CNS in response to injury, infection, mental illness, ROS and RNS, redox iron, and different oligomers of τ- and β-amyloid [75]. Despite the negative connotations, inflammation per se is neither harmful nor maladaptive, but a survival response aimed to reinstate the cellular homeostasis upon an inflammatory challenge. This response usually includes four different components: inflammatory triggers, sensors, inflammatory mediators, and the affected tissues [153]. In classical NDDs, such as AD, Parkinson’s disease (PD), or amyotrophic lateral sclerosis (ALS), the neuroinflammatory process translates into increased reactivity of glia cells, like astrocytes and microglia, together with the elevated production and release of pro-inflammatory molecules.
Besides Aβ plaques and intracellular NFTs, neuroinflammation has been identified as the third core feature in AD pathogenesis [92, 129] that may serve a link between the pathologies [78, 225, 268]. The old prevailing belief of “immune privilege” excluded the capacity of brain cells to initiate an immune response [36, 207]. Evidence demonstrated that the brain indeed is not privileged, but rather specialized [28, 138]. By now, multiple lines of research have established the presence of reactive microglia in AD brains as well as its unequivocal interaction with amyloid plaques and NFTs, both AD pathological hallmarks. Likewise, biomarker studies have widely demonstrated that this reactive phenotype results in increased levels of immune mediators, such as cytokines, chemokines, inflammasomes, and ROS [27, 37]. Latter publications have not only validated the profound immune alterations under AD conditions but have also confirmed the implication of other cellular entities, such as astrocytes, oligodendrocytes, and even neurons, in the inflammatory equation [84, 179]. While in the early phase of the pathogenic process, tissue repair and control of the inflammatory response is attempted through the release of anti-inflammatory interleukins like IL-10, IL-4, IL-13, and transforming growth factor β, later stages are characterized by chronic inflammation in which microglia switches to an activated phenotype, releasing ROS, NO, and producing pro-inflammatory cytokines, including IL-1β, IL-6, IL-12, IL-23, and TNF-α [40].
Naturally, evolution has provided “protein quality control systems” to maintain the proteostasis. Mechanisms, such as proteasome degradation, autophagy or the unfolded protein response, and specialized proteins such as chaperones or heat shock proteins (HSPs), work together serving that purpose. In AD, Aβ dyshomeostasis may arise from an imbalance between Aβ neuronal production and extracellular clearance of Aβ [89, 202]. As this imbalance persists over time, it leads to the accumulation of Aβ deposits, marking a critical transition from a state of balanced proteostasis to one of pathological aggregation. These toxic Aβ aggregates are detected as danger or pathogen associated molecular patterns (DAMPs/PAMPs) by a group of receptors collectively known as pattern recognition receptors (PRRs) [23]. PRRs comprise a number of different surface receptors which can sense bacterial or viral products, neurodegenerative proteins, DNA, and neuronal debris [276]. Aβ activates microglia through multiple receptors including the receptor for advanced glycation end products (RAGE), nucleotide-binding oligomerization domain-like receptors (NLRs), or Toll-like receptors (TLRs) [125]. Microglial Aβ phagocytosis can take place via CD36, involving the formation of a TLR2–TLR6 heterodimer and subsequent NFKB activation [221]. Additionally, it can occur through CD14, which acts as a coreceptor for TLR4, TLR6, TLR9, α6β1 integrin, and SCARAq [66, 136, 247]. PRRs ligation triggers different inflammatory signaling cascades leading to the production and release of inflammatory mediators, such as complement factors, cytokines IL-1β, IL-6, IL-18, and TNF-α, chemokines such as C–C and C-X-C motif chemokine ligand 1 (CCL1, CXCL1), CCL5, small-molecule messengers, prostaglandins, nitric oxide (NO), and ROS [57, 92, 95].
NFTs, AD’s other core pathology, are made of hyperphosphorylated tau protein [4, 101]. Under normal physiological conditions, tau plays important roles in a wide range of biological processes spanning from control of microtubule dynamics and stability, to glucose metabolism, and extending to phenomena such as hibernation [8, 58, 145]. Tau protein undergoes multiple posttranslational modifications (PTMs), among them phosphorylation, glycation, ubiquitination, or truncation [265], which critically contribute to tau proteostasis, dysfunction, and aggregation. In AD, multiple protein kinases (PKs) are known to phosphorylate tau at nearly 40 AD-relevant epitopes. Glycogen synthase kinase (GSK-3β) and cyclin-dependent kinase (CDK5) are the two most studied kinases involved in tau hyperphosphorylation, while PP2A is the main phosphatase related with tau dephosphorylation [12]. When hyperphosphorylated, tau reduces its affinity for microtubules, detaches, and aggregates in the neuronal cytoplasm and extracellular spaces, later propagating through the brain in a prion-like manner [160]. Accumulation of toxic tau forms is considered to lead to impairment of intraneuronal processes, specifically protein degradation, energy metabolism, membrane integrity, intracellular transport, and signal transmission [165]. Pathological tau not only disrupts microtubule stability but also instigates an immune response. Interestingly, NLRP3 inflammasome, one of the most important immune pathways in microglia, can also be activated by tau, and indeed, elevated levels can be found in brain tissue and CSF of tauopathy cases [107]. In line, NLRP3 loss of function leads to reduced tau hyperphosphorylation and aggregation by regulating tau kinases and phosphatases [102].
Reactive microglia is observed in the vicinity of NFTs [56] and tau internalization by microglia has been demonstrated in vitro and in vivo [22], but the underlying mechanisms are still elusive [273]. Likewise, microglia implication in tau pathology and propagation has been extensively demonstrated in multiple tau-transgenic mouse lines. Virginia Lee’s group observed that treatment of P301S Tg mice with FK506 immunosuppressant inhibited microglial activation and attenuated tau pathology [266]. Also, in another study, microglia depletion through two independent approaches (PLX3397&CSF1R) in two independent tauopathy models significantly suppressed the propagation of tau measured by AT8 + [9]. The activation of microglia and the subsequent propagation of tau pathology could potentially be driven by the joint action of several agents like pro-inflammatory cytokines (IL-1β) [117], transcription factors (NF-κB) [248], and signaling pathways (CX3CL1/CX3CR1) [19]. Taken together, these studies demonstrate that tau can serve as DAMP, activating microglia, and triggering and immune response.
Here, we offer a comprehensive review of the inflammatory processes that contribute to the neurodegenerative progression in AD from a cellular perspective. Our focus centers on the principal cellular contributors, notably microglia, astrocytes, and neurons, while also examining the intricate interactions among them. Specifically, we delve into the inflammatory response of microglia and astrocytes within the context of AD's core pathologies, amyloid, and tau pathology. Additionally, we explore the inflammatory contribution of neurons, considering the various modes of programmed cell death, as well as other cells types, including T cells, oligodendrocytes, pericytes, and border-associated macrophages. This cellular-centric approach aims to provide deeper insights into the complex interplay between neuroinflammation and the neurodegenerative process in AD.
Microglia
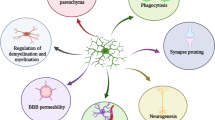
Microglia are the resident macrophages and represent the major component of the innate immune system of the CNS. They are unique in their origin, deriving from embryonic yolk sac precursors, similar to perivascular, meningeal, and retinal macrophages [79, 180]. Before the formation of the BBB, myeloid progenitor cells migrate to the neuroepithelium, proliferate, and spread throughout the CNS, where they differentiate into microglia [254]. They account for 0.5–16.6% [156] of the total number of cells in the human brain and slowly renew at a yearly median rate of 28% [184]. Microglia are highly dynamic cells whose states depend on their location and neighboring entities, translating into epigenomic, transcriptomic, proteomic, metabolomic, and functional changes. The combination of single-cell multiomics technologies and protein expression analysis has allowed the identification of multiple of these states such as disease-associated microglia (DAMs), interferon response microglia (IRM), antigen-presenting response (HLA), or ribosomal microglia (RM), among others [142, 179, 245]. Importantly, evidence suggest that these states are fluid and highly context dependent [223]. Microglia play an essential role maintaining brain homeostasis. They provide neurotrophic factors, scale, and prune synapses, thereby contributing to neuronal plasticity. Further, they engulf and degrade excess metabolic byproducts and damaged tissues, promote development and myelination of oligodendrocytes, and perform fundamental housekeeping functions [32]. These functions are tightly regulated by genes encoding chemokine and chemoattractant receptors, phagocytosis, and synaptic pruning and remodeling.
In basal physiological conditions, homeostatic microglia are highly motile by nature, with multiple ramifications that constantly extend and retract, enabling active surveillance of their immediate microenvironment [166]. In AD, Aβ accumulation induces a sustained microglia activation, leading to a continuous release of inflammatory elements that impair their phagocytic and degrative capacities. This aggravates Aβ accumulation, promoting tau propagation, and leading to neuronal death in vivo and in vitro [173, 220], ultimately advancing the progression of the disease [98, 154]. This central role of microglia in AD pathogenesis has been extensively demonstrated from different angles. Genome-wide association studies (GWAS) have shown that mutations in key microglia genes, such as TREM2, dramatically increase AD risk [222]. Furthermore, Aβ clearance seems to be impaired not only in late but early onset forms of AD [149, 178], and its regulation and effectiveness may depend on factors such as age or stage of the disease [122]. In line, mouse models of Aβ deposition have repeatedly shown impaired microglia phagocytic capacity [98, 201, 255]. Finally, in vitro experiments have robustly demonstrated that exposure of microglia cultures to Aβ leads to the production of neurotoxic ROS and RNS, NLRP3 (NOD-, LRR-, and pyrin domain-containing protein 3) inflammasome activation [69], and production of pro-inflammatory cytokines, such as pro-IL-1β, IL-6, or TNF-α [134, 140, 246].
Ineffective microglia clearance of Aβ leads to the sustained release of the pro-inflammatory mediators mentioned above. Over time, this inflammatory activation turns into a chronic, deleterious process. This unbridled microglia activity not only creates a pro-neurodegenerative environment but also promotes the initial Aβ pathology. Different Aβ species can trigger microglia activation through a wide variety of cell receptors, such as CD40, CD36/TLR4/6, CD33, TREM2, and RAGE [61, 221]. CD40 and CD36/TLR4/6 activate the nuclear factor-κB (NFκB) transcription factor, resulting in the expression of auto-inhibited NLRP3, as well as the release of ROS/NOS [17, 204]. Internalized Aβ damages mitochondria and lysosomes, causing the release of ROS and Cathepsin B (CatB). CatB in turn serves as the triggering signal for NLRP3 inflammasome formation [41]. After NLRP3 activation, the adaptor protein known as apoptosis-associated speck-like protein containing a CARD (ASC) is mobilized to assemble ASC helical fibrils specks. ASC recruits procaspase-1, followed by cleavage and activation of caspase-1 through induced proximity autocatalysis [139]. Active caspase-1 cleaves Gasdermin D (GSDMD) which releases its N-terminal domain (NT-GSDMD) to form membrane pores. Lytic cell death, or pyroptosis, occurs when the extent of pore formation exceeds the cell's capacity to repair its membrane [137]. Upon cell membrane rupture, intracellular content gets released including recently formed ASC specks as well as inflammatory cytokines such as IL-1β and IL-18 [183]. Released extracellular ASC specks not only propagate inflammation to surrounding cells [68] but bind Aβ acting like “seeds”, accelerating the ongoing amyloidogenic process [69, 239] (Fig. 1). In line, microglia depletion in experimental models of AD has shown numerous benefits including reduction of both intraneuronal amyloid as well as neuritic plaque deposition [215], neuronal loss, and mitigation of neuritic dystrophy [143, 216].

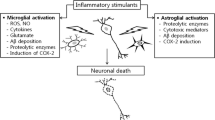
Microglia-mediated inflammation in AD. In the earliest stages of AD, the abnormal cleavage of APP by β- and γ-secretases leads to the formation of Aβ. Aβ monomers are inherently disordered and tend to aggregate into plaques, a process further promoted by genetic mutations in APP or PSEN1/PSEN2 genes. These Aβ aggregates activate microglia, which initially attempt to clear Aβ through phagocytosis and proteolysis. Upon failure, microglia become persistently activated, leading to Inflammasome activation and the release of pro-inflammatory components. This process culminates with the membrane rupture and release of the intracellular contents through pyroptosis. The release of ASC specks will further promote Aβ aggregation, closing the first inflammatory loop. Activated microglia in a chronic state release inflammatory cytokines along with detrimental substances like reactive ROS and NO, heightening the immune response and contributing to neuronal damage. Moreover, the release of DMAPs from dying neurons, including ATP, HMGB1, S100B, and DNA, exacerbates inflammation, creating a reinforcing loop. This inflammatory environment leads to the excessive phosphorylation of tau protein, its detachment from microtubules, and subsequent formation of tau tangles within neurons. Hence, inflammation acts as a pivotal intermediary linking Aβ aggregation to the accumulation of tau tangles in AD pathology. (Figure created using BioRender.com)
Interestingly, tau pathology is also amplified by these same Aβ-induced cytokines, with tau kinases like CDK5, GSK-3β, and p38-Mitogen-activated protein kinase (MAPK), increasing their activity, and, consequently, tau hyperphosphorylation [59]. Increased hyperphosphorylation is associated with microtubule disruption and tau polymerization into filaments and NFTs [4]. Moreover, tau monomers and oligomers also operate as DAMPs, resulting in NRLP3-ASC activation [102]. In parallel to this inflammatory cascade of events, microglia serve as a mobile carrier for the propagation of protein aggregates with several mechanisms proposed, including heparan sulfate proteoglycans (HSPGs)-mediated endocytosis, exosome fusion, receptor-mediated endocytosis, phagocytosis, and, more recently, tunneling nanotubes (TNTs) [38, 237]. Astrocytes are also indirectly affected by activated microglia. Through the release of IL-1α, TNF and complement component 1 (C1), subcomponent q (C1q) by microglia, astrocytes transition into the A1 astrocytic state where basic functions such promotion of neuronal survival, outgrowth, synaptogenesis, and phagocytosis get compromised, further contributing to the ongoing neurodegenerative process [132]. In a similar manner, the TNF-Signal Transducer and Activator of Transcription 3 (STAT3)-α1-antichymotrypsin (α1ACT) signaling axis can also induce Guanylate Binding Protein 2+ (GBP2+) astrocytic activation, leading to BBB dysfunction through increased levels of Serpina3n/α1ACT [112] (Fig. 1).
Age is the primary risk factor for most NDDs [100]. Microglia have been identified as remarkably long-lived cells [71]. Aging microglia can enter a senescent state displaying modified surveillance phenotype, reduced branching and motility, and persistent inflammatory response upon damage [49]. Senescence markers include an irreversible cell-cycle arrest induced by the activation of p53/p21 and p16INK4a/Retinoblastoma (Rb) pathways, and the formation of Senescence-associated heterochromatin foci (SAHF) [82]. Senescent-associated microglia (SAM) might substantially contribute to the AD neurodegenerative process through different mechanisms. First, an important characteristic of SAM is the Senescence-associated secretory phenotype (SASP) which involves the activation of cGAS–cGAMP–STING pathway and the successive release of type I interferons (IFN), MMPs, High mobility group box 1 (HMGB1) protein, and other pro-inflammatory mediators, including IL1β, IL-6, and IL-8 [126, 209]. Through a process called paracrine senescence, SASP can propagate to neighboring cells via NF-κB and IL-1 signaling, contributing to the inflammatory process [3]. Second, cell-cycle arrest impose substantial constrains on cell functionality like clearance of cellular debris. APP/PS1 mice have a twofold-to-fivefold decrease in expression of the Aβ-binding scavenger receptors scavenger receptor A (SRA), CD36, and RAGE, and the Aβ-degrading enzymes insulysin, neprilysin, and MMP9, compared with their littermate controls [98]. Moreover, characterization post-mortem AD brain tissue revealed senescent microglia associated with inflammatory activation and downregulated phagocytic capacity [65]. Finally, senolytic therapy consistently decreases neuroinflammation in AD, tau and aging mouse models [81, 169, 269]. Taken together, these data demonstrate the interplay of cellular senescence between inflammation and AD.
Astrocytes
Astrocytes are integral for neuronal survival and function of CNS and are involved in multiple fundamental processes, such as synaptic pruning and remodeling, blood flow regulation, neural metabolism, clearance of synaptic and neuronal debris, or circadian rhythms [238]. Interestingly, changes in the transcriptomic and functional characteristics of astrocytes occur in both the aging brain [171] and NDDs [85, 241]. In the past years, the role of astrocytes as key regulators of innate and adaptative immune responses has been demonstrated in multiple in vivo and in vitro studies [31, 44, 90, 253]. Reactive astrogliosis is a common feature in neurodegenerative disorders which encompasses morphological, transcriptional, biochemical, metabolic, and physiological changes as a result of a pathological insult. Typically, these alterations manifest as elevated levels of Glial Fibrillary Acidic Protein (GFAP) and vimentin, along with heightened production of pro-inflammatory cytokines, such as INF-γ, IL-1β, IL-6, and TNFα [24, 214]. Additionally, there is an upregulation in the expression of innate immune-related genes like Lipocalin 2 (Lcn2) and the protease inhibitor 1-antichymotrypsin (Serpina3n) [274].
Astroglial pathological changes can be broadly grouped into three categories: (i) astrodegeneration, involving astroglial atrophy and functional loss; (ii) the pathological remodeling of astrocytes; and (iii) reactive astrogliosis. The first two categories, representing non-reactive pathological transformations of astrocytes, can be collectively referred to as astrocytopathies, distinguishing them from reactive astrogliosis [242]. Other documented altered functions in reactive astrocytes include impaired phagocytosis, decreased glutamate uptake, loss of astrocyte foot processes accompanied by the loss of polarized localization of AQP4, and release of neurotoxic compounds [132]. From a phenotypic perspective, recent single-cell and single-nucleus RNAseq analysis in human brains and mouse models of chronic neurodegenerative pathologies have shown multiple stage dependent transcriptomes [195, 212]. Similar to microglia, astrocytes can adopt multiple intermediate states, undergoing morphological, molecular, and functional changes, thus moving beyond outdated classical polarization views [181]. In AD, astrocytes display an upregulation of monoaminoxidase-B, translating into increased levels of GABA and H2O2 [42]. The classical neurocentric view of AD attributed these changes to a nonspecific secondary response to the disease process [164]. An increasing body of evidence indicates that astrocytes, not solely due to the impairment of their inherent homeostatic functions, but also through active processes, might play a role in the advancement of AD pathology.
Several astrocytic functional processes like glutamate removal, extracellular potassium regulation, calcium signaling [133], and energetic metabolism are known to be impaired in AD [2]. Moreover, genetic studies also suggest a pivotal role of astroglia in AD, with several risk factor genes, such as Apolipoprotein E (APOE), Sortilin-related receptor 1 (SORL1), clusterin (ApoJ), and Fermitin family homolog 2 (FERMT2), primarily expressed by astrocytes. Post-mortem tissue studies in humans with mild cognitive impairment or preclinical AD have confirmed the presence of reactive astrocytes even before the formation of amyloid plaques [60, 189, 243], aspect that has been extensively demonstrated in animal models [7, 96, 229]. Reactive astrogliosis can be also visualized in later stages of the disease (assessed by the expression of GFAP and neurotrophic factor S100β) where astrocytes can be found in the plaques vicinity, with a marked upregulation of intermediate filament proteins, such as synemin, vimentin, nestin [64, 224], and adopting different pathological phenotypes [113]. In addition to exhibiting astroglial reactivity, AD also features atrophic astrocytes characterized by reduced volume and thinner processes. These atrophic astrocytes have been confirmed in post-mortem brains of AD patients [188], mouse models of AD [217], and induced pluripotent stem (iPS) cells derived from patients with both familial and sporadic forms of the disease [108]. Noteworthily, both phenomena seem to be spatial dependent, with cells surrounding the plaques undergoing gliosis but those distant from them becoming atrophied [187]. Furthermore, different signaling pathways have been implicated in AD astrogliosis among them the cyclic Adenosine monophosphate (cAMP) pathway [177], the Janus Kinase (JAK) /STAT3 pathway [86], the NF-κB pathway, the calcineurin/nuclear factor of activated T cells (CN/NFAT) pathway [1], and the MAPK pathways [196].
While robust data support the interaction of astrocytes with Aβ, there is no substantial evidence yet to confirm a significant role in Aβ production by astrocytes under physiological conditions [127, 272]. Reactive astrocytes seem to cluster around neuritic plaques forming a barrier and isolating them from the surrounding neuropil [103]. Whether the final outcome of this mechanism is detrimental or neuroprotective is still under debate. The creation of physical limits around the aggregate could limit its growth, reducing its neurotoxicity for neighboring cells [176]. Conversely, it is well documented that astrocytes can phagocytose Aβ [80, 228] and synapses [128]. Under normal physiological conditions, astrocytes uptake and transport Aβ from the brain into the perivascular space through the BBB. Aβ degradation occurs via neprylisin (NEP), insulin-degrading enzyme (IDE) and MMP-9 [10]. However, during AD, accumulation of Aβ forms resistant to degradation, such as N-terminally truncated forms [51], alongside heightened synaptic pruning activity [232], could potentially exacerbate the neurodegenerative process. Ablation of astrocytes in AD transgenic mice models aggravates amyloid pathology and leads to an increase in the expression of pro-inflammatory markers such as IL-6 and Il-1β [50, 111]. These data suggest a dual role of reactive astrocytes in AD. At first, they might play a significant role in Aβ uptake and degradation [11], as well as containing inflammation through glia-scar formation [213, 244]. As inflammation perpetuates, the sustained release of inflammatory mediators might hamper this capacity, leading to the formation of secondary deposits through the death of Aβ-loaded astrocytes [162]. Astrocytes can sense Aβ in the TLR/RAGE-dependent manner leading to morphological changes and increased levels of GFAP and S100β [162]. Aβ-reactive astrocytes provide neuroprotection through the release of neurotrophic factors, but they also participate in the inflammatory process via the afore stated signaling pathways with the release of ROS, NO, cytokines (e.g., IL-1β, IL-6, TNF-α, IFN-α, granulocyte–macrophage colony-stimulating factor), and chemokines (e.g., MCP-1, MIP1-α, CCL4, IL-8, IFN-γ-inducible protein-10) [135]. The synthesis of pro-inflammatory mediators contributes to Aβ pathology by disturbing APP processing equilibrium and boosting β- and γ-secretases activity by astrocytes [10]. In a similar manner, activated microglia can also activate astrocytes through the release of cytokines, such as IL-1α, IL-1β, IL-6, TNF-α, and C1q, stimulating again astrocytic β-amyloid production [132, 271] (Fig. 2).

Astrocytes-mediated inflammation in AD. Upon sensing Aβ plaques in a TLR/RAGE manner, astrocytes become activated and undergo significant changes in gene expression and signaling pathways. Activated astrocytes exhibit elevated levels of GFAP, S100β, and intermediate filament proteins, such as synemin, vimentin, and nestin, indicating their reactive state. Furthermore, there is an upregulation of pro-inflammatory cytokines, such as INF-γ, IL-1β, and IL-6, along with increased expression of Lcn2 and Serpina3n. Multiple signaling pathways have been implicated including cyclic Adenosine monophosphate (cAMP) pathway, the Janus Kinase (JAK)/STAT3 pathway, the NF-κB pathway, the calcineurin/nuclear factor of activated T cells (CN/NFAT) pathway, and the MAPK pathway. Additionally, microglia can further activate astrocytes by releasing neuroinflammatory contents, such as TNF-α, IL-1α, complement component 1q (C1q), IL-1β, and IL-6, leading to increased production of Aβ by astrocytes. In parallel, aggregated and hyperphosphorylated forms of tau are released by dying neurons. These forms could be uptake by another recipient neuron or by glial cells. The activation of astrocytes leads to the release of pro-inflammatory cytokines (IL1α, IL1β, IL6, and TNFα), chemokines (CCL2, CCL3, CCL4, and CXCL10), and nitric oxide. This pro-inflammatory milieu increases neurotoxicity, but also affects microglia, leading to the release of TNF-α, IL-1α, complement component 1q (C1q), IL-1β, further activating astrocytes. Additionally, the release of aggregated tau forms by astrocytes might also enhance the propagation of tau in a prion-like manner. (Figure created using BioRender.com)
The levels of tau in healthy astrocytes have been reported very low [175]; however, hyperphosphorylated tau has been observed in astrocytes in AD [185]. Astrocytes can internalize different forms of tau including monomers, tau preformed fibrils (PFFs), and aggregates [62, 147, 158]. Astrocytes may play as well a dual role during the disease course; while the uptake of tau from the extracellular environment might help mitigate the direct neurotoxic impacts of tau on neurons, the internalization and release of pathological tau species by astrocytes could potentially exert adverse effects on astrocytic function and propagate tau pathology [67]. Tau accumulation within astrocytes could also elicit an inflammatory response, further contributing to the neurodegenerative process. The available data regarding tau-mediated inflammation by astrocytes are limited. In one of the few reports available, Wang and Ye triggered an astrocytic inflammatory reaction using αV/β1 integrin by exposing them to tau monomers and PFFs. The administration of PFFs resulted in a robust elevation of numerous pro-inflammatory cytokines (IL1α, IL1β, IL6, and TNFα) and chemokines (CCL2, CCL3, CCL4, and CXCL10). Conversely, exposure to monomeric tau elicited a decreased inflammatory response [251]. In another paper, Ungerleider and colleagues exposed human astrocytes to tau monomers, PFFs, or a combination of both. All 3 treatments increased expression of IL-1β and IL-8 mRNA at 24 h. Also, a delayed increased in TNFα levels and Nitric oxide synthase 2 (NOS2), indicating an increase in oxidative stress following the initial inflammatory response [234]. Uptake of pathological tau by cells could potentially modify the secretion of cytokines by astrocytes, indicating a feedback loop where tau and inflammation mutually exacerbate the neurodegenerative process. The inflammatory state will also lead to a sustained astrogliosis, with trophic and metabolic support functions potentially comprised (Fig. 2). Tau-mediated astrocytic dysfunction has been demonstrated both in vivo and in vitro, resulting in a reduced neurosupportive capacity and increased neuronal loss [208]. Interestingly, this phenomenon seems to be an early event in the pathogenic process of AD that may serve as a bridge between Aβ pathology and early tau phosphorylation [15].
Microglia–astrocyte crosstalk has recently become a central focus in glial research. New evidence shows that signals originating from microglia and astrocytes are crucial in determining the functions and fates of each other [141, 192, 256]. In the AD context, neurons release self-antigens or aberrant protein forms that activate homeostatic microglia. Upon migration to the damaged site, microglia exhibit beneficial phagocytic activity by clearing toxic Aβ forms [210]; however, their involvement becomes deleterious when chronic activation occurs, leading to unresolved inflammation and acceleration of the neurodegenerative process. In this state, secretion of TNF-α, TNF-related apoptosis-inducing ligand (TRAIL), IL1α, and C1q by microglia is sufficient to induce A1 astrocytes with impaired support of synaptogenesis, reduced phagocytosis, and decreased expression of neurotrophic factors [29, 132]. Crosstalk between microglia and astrocytes is reciprocal, and therefore, microglia functions are also influenced by different cytokines and chemokines, such as IL-1β, IL-10, IL-15, TNF-α, nitric oxide, and CCL2 that are secreted by astrocytes [20] (Fig. 2). Data from animal models suggest that these functional alterations might significantly influence AD progression. Using an APP transgenic mouse model, Lian et al. showed that Aβ acts as an upstream activator of astroglial NF-κB, leading to the release of C3. The interaction of C3 with microglial C3a receptor leads to impaired Aβ phagocytosis [131]. In another report, McAlpine et al. utilized a 3D microfluidic triculture system comprising microglia derived from human iPS cells and the 5xFAD transgenic mouse model. Their study demonstrated that astrocyte-produced IL-3 plays a central role in directing microglial activity, leading to increased mobility and enhanced capability to cluster around and eliminate Aβ and tau aggregates [151]. Another important route of cross talk is the gut–brain axis. Through the metabolites of dietary tryptophan, commensal and pathogenic enteric bacteria may impact the production of Vascular Endothelial Growth Factor-β (VEGF-β) and transforming growth factor alpha (TGFα) by microglia, thereby regulating astrocyte pathogenic processes during inflammation and neurodegeneration [193]. Recently, several other routes of activation have been identified including complement protein C3 [259] and C8γ [114], human antimicrobial peptide LL-37 [39], extracellular vesicles [190], or TNTs [191].
Neurons
Neurodegeneration defines as an irreversible detrimental process for neurons that presents in NDDs, and to a lesser extent, during aging. Neurons themselves do not possess a pro-inflammatory machinery, and therefore, the classical unidirectional relation is that neuroinflammation acts upon neurons. However, neurons can also contribute to the inflammatory process through the different modes of programmed cell death [144]. Apoptosis is a form of regulated cell death (RCD) that can be initiated by various perturbations in both the intracellular [DNA damage, endoplasmic reticulum (ER) stress, ROS overload, replication stress, microtubular alterations, etc.] and extracellular microenvironments (through plasma membrane receptors)[73]. Among all RCD implicated in AD, apoptosis is the least detrimental with the production of anti-inflammatory factors, such as lactate, IL-10, and TGF-β, and with a cellular clearance without release of the intracellular components. However, accumulating evidence also suggest that the catalytic activity of caspases implicated in the apoptotic process may influence AD neuroinflammatory process. Caspase 3, for instance, has the capacity to cleave both APP and tau, producing additional cytotoxic APP fragments and disrupting synaptic communication, respectively [170, 174] (Fig. 3). Solid evidence of the presence of apoptosis is AD post-mortem tissue remains elusive [231], with few reports showing neurons with morphological features of apoptosis [218, 219]. Several reasons could explain this absence. First, apoptotic cells are usually degraded by microglia; hence, accumulation of them is only possible if microglia functions are severely impaired. Also, upregulation of anti-apoptotic B-cell lymphoma 2 (Bcl-2) family proteins and down-regulation of both the pro-apoptotic protein Bax and several pro-apoptotic BH3-only proteins, confer mature neurons natural resistant to apoptotic cell death [99]. Some lines of evidence indicate the presence of apoptotic markers in AD, but to what extend apoptosis contributes to neuron death in the disease course remains under debate [144].

Regulated cell death (RCD) & Inflammation. RCD modes (Apoptosis, Pyroptosis, and Necroptosis) and their relationship with inflammation in AD are outlined here, focusing on triggers, effectors, hallmarks, and inflammatory outcomes. Apoptosis, distinguished by its quiet nature and lack of inflammation, coordinates the removal of cells without releasing intracellular contents. It can be induced by various stimuli, culminating in Caspase-3 cleavage. Apoptotic death entails nuclear condensation and membrane blebbing, while also exerting an anti-inflammatory effect through the release of TGF-β, IL-10, and Lactate. Necroptosis occurs in the absence of Caspase-8 and after the activation of death receptors, such as TNFR, TRAIL, and FAS. This process involves RIPK1 autophosphorylation, RIPK3 activation, and subsequent MLKL phosphorylation, leading to oligomerization of phosphorylated MLKL at the plasma membrane, disrupting cell polarity, and ultimately resulting in lysis. Necroptosis incites inflammation by releasing intracellular contents as DAMPs together with pro-inflammatory cytokines. Pyroptosis is an inherently inflammatory RCD that induces cell lysis via membrane pore formation with two signals required for initiation. The first one primes the inflammasome, while the second activates intracellular pattern recognition receptors (e.g., NLRP3, NLRP1, and AIM2), forming inflammasome complexes. This cascade triggers Caspase-1 activation, cleaving substrates including pro-IL-1b, pro-1L-18, and GSDM. Active GSDM forms pores resulting in membrane integrity loss, cell lysis, and release of pro-inflammatory cytokines and intracellular DAMPs. (Figure created using BioRender.com)
Necroptosis, on the contrary, represents an inflammatory RCD characterized by the loss of membrane integrity and the release of intracellular content including pro-inflammatory cytokines, such as IL-1β, IL6, IL-33 or TNF-α, and DAMPs. Necroptosis is usually triggered by pro-inflammatory cytokines and death signaling ligands released by inflammatory cells like astrocytes and microglia. This process leads to the autophosphorylation of receptor-interacting protein kinase 1 (RIPK1) and the latter recruitment of RIPK3 to form a large molecular complex named necrosome. Activated RIPK3 then phosphorylates and activates the pseudokinase mixed lineage kinase domain-like protein (MLKL), which oligomerizes and translocates to the plasma membrane, disrupting its integrity and causing cell death [252] (Fig. 3). Different studies point toward a prominent role for necroptosis in AD. Caccamo and colleagues provided direct evidence for the activation of necroptosis in human AD brains, as well as in a mouse model of AD. Necroptosis levels positively correlated with Braak stages, and inversely correlated with brain weight and cognitive scores [33]. In another study, Salvadores et al. using post-mortem human AD brain tissue showed that Aβ oligomers correlates with the expression of key markers of necroptosis activation. Furthermore, pharmacological or genetic inhibition of necroptosis resulted in reduced neurodegeneration and memory impairment triggered by Aβ oligomers in mice [197]. Finally, in human post-mortem AD brains, Koper and colleagues demonstrated the presence of all three activated necrosome components when granulovacuolar degeneration (GVD) neuronal lesions were analyzed, suggesting an AD specific form of necroptosis [119]. Necroptosis machinery (RIPK1, RIPK3, and MLKL) promotes a robust and sustained pro-inflammatory response [45, 275]. Upon membrane rupture, released DAMPs might get recognized by nearby bystander glia (microglia and astrocytes) serving as pro-inflammatory triggers. However, in the context of AD, the situation might be even more detrimental, due to the release of pathological intraneuronal tau forms. These forms could not only act as DAMPs, but also have the potential to spread tau pathology within the local environment [25].
Pyroptosis represents an alternative type of inflammatory RCD characterized by DNA fragmentation, cellular swelling, and membrane disruption that is triggered by inflammasomes. Neuronal pyroptosis seems to be highly dependent on caspase-1 activation although recent reports have shown that other caspases, including caspase-3/4/5/6/8/9/11, may also cause pyroptosis [267]. The cleavage of GSDMD by caspase-1 results in the formation of membrane pores, leading to membrane rupture and subsequent release of cellular contents into the extracellular environment. Most pyroptosis-related AD research has focused on glial cells, but recent evidence suggests activation of the neuronal NLRP1 inflammasome may play an important role [264]. Increased levels of neuronal pyroptosis caused by NLRP1/caspase-1 activation were found in cultured cortical neurons exposed to Aβ. In the same study, NLRP1 or caspase-1 deficiency resulted in significantly lower levels of neuronal pyroptosis and reversed cognitive impairments [226]. The presence of other pyroptosis-related proteins has been also demonstrated in AD. In a recent paper analyzing human AD samples, Moonen and colleagues demonstrated the presence of cleaved GSDM in neurons. Interestingly, caspase-8 and non-canonical inflammasome protein caspase-4 were also detected, suggesting novel mechanisms for GSDMD cleavage [157]. Another set of experiments showed increased levels of GSDMD p30, NLRP3 protein, and cleaved caspase-1 following the incubation of mouse cortical neurons with Aβ1-42 [87]. The relation between phosphorylated tau (p-tau) and neuronal pyroptosis is barely explored. In one of the few reports available, hyperphosphorylated tau induced pyroptosis and release of IL-1β and IL-18 in PC12 cells treated with forskolin [130]. Additionally, increased concentrations of GSDMD, total tau (t-tau), and tau181p have been detected in the CSF of AD patients in comparison to both, the overall population and individuals with vascular dementia [205](Fig. 3).
Other cell types
While the involvement of the aforementioned cellular types in the neuroinflammatory phenomena of AD is well documented, increasing evidence is introducing new players into the field. The adaptive immune system is increasingly acknowledged for its role in the pathogenesis of AD. Integrity loss of the BBB in AD has been extensively investigated [123] and allows peripheral lymphocytes such as B and T cells, access to the brain parenchyma. This has been demonstrated in brains of transgenic AD mice, where the presence of mature B cells led to immunoglobulin deposits which were often colocalized with Aβ plaques and activated microglia. Moreover, loss of B cells was significantly reduced Aβ burden and behavioral impairments [115]. In line, individuals with mild cognitive impairment and AD exhibited elevated levels of inflammatory CD8 + CD45RA + T effector memory (TEMRA) cells in peripheral blood, along with their clonal expansion in the CSF [77]. Recently, these results have been confirmed using a novel a 3D human neuroimmune axis model [109].
Brain pericytes are key constituent cells of the neurovascular unit playing various functions including angiogenesis, vascular remodeling, regulation of microcirculation, and the formation and maintenance of the BBB itself. Pericyte loss and/or degeneration has also been reported in AD [155, 206], which results in BBB disruption. This allows the extravasation of toxic-blood derived products leading to immune response and secondary neuronal degenerative changes [14]. Interestingly, pericyte reduction has been shown to correlate not only with BBB dysfunction but also amyloid plaque load [203]. Brain pericytes can internalize and clear Aβ via an LRP1/APOE [35]. In AD, aberrant deposition of Aβ leads to progressive capillary constriction [167], decreasing oxygen and blood levels, leading to neuronal loss. Indeed, exposure to Aβ1–40/42 promoted membrane release of key pericyte proteins, including proteoglycan NG2 and platelet-derived growth factor receptor β (PDGFRβ) [200]. Perivascular cells are also involved in immune regulation [194]. Through the release of diverse immunomodulators, such as IL-1β, TNF-α, IFN-γ, or IL-6, pericytes can induce pro-inflammatory states in microglia, astrocytes, and endothelial cells [148]. Lipopolysaccharide immune activation have been shown in human and mice pericytes cultures, resulting in significant release of pro-inflammatory factors, such as L-1α, TNF-α, IL-3, IL-9, IL-10, IL-13, iNOS, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 [83, 121]. Finally, anti-inflammatory roles have also been proven in mouse models, with the release of CX3CL1 and IL-33, decreasing microglia activation and promoting their anti-inflammatory phenotype [34, 263].
While AD is often viewed primarily as a gray-matter disease, white-matter changes are also frequently reported [76, 150]. Remarkably, these lesions have also been shown in preclinical AD [53, 186], indicating a role in the disease etiology. In this regard, the group of Klaus-Armin Nave demonstrated in AD mice models, that myelin defects upstream amyloid deposition, recruit away plaque-corralling microglia, leading to a less efficient Aβ clearance. Additionally, RNA-seq analysis revealed a more inflammatory profile of microglia with upregulation of classical DAM signature genes, such as Clec7a, Gpnmb, APOE, Spp1, Axl, and Itgax [54]. Downstream amyloid deposition, the exact mechanisms leading to myelin loss in AD remain unknown but are likely to include cytotoxicity, RNA metabolism disruption, and neuroinflammation [74, 116, 260]. Additionally, independent lines of evidence suggest that oligodendrocytes might be active contributors in the disease development. Indeed, amyloidogenic-processing machinery is abundant in oligodendrocytes [198], generating detectable Aβ levels in vitro [211] and possible contributing to the Aβ burden. Aligned, another recent study showed that suppression of oligodendrocyte-derived Aβ rescued neuronal dysfunction in vivo [182]. Finally, both in AD patients and an AD mouse model, oligodendrocytes exhibited NLRP3-dependent GSDMD-associated inflammatory injury, concomitant with demyelination and axonal degeneration [270].
Recently, another population of tissue-resident macrophages was identified [159]. Border-associated macrophages (BAMs) are defined and resident macrophages in non-parenchymal tissues. They play critical roles in maintaining CNS homeostasis and differ phenotypically and functionally from microglial cells [55]. Also, several subgroups of BAMs have been identified based on their anatomical site: subdural/leptomeningeal macrophages (sdΜΦ), dural macrophages (dmΜΦ), stromal choroid plexus macrophages (cpΜΦ), choroid epiplexus macrophages (cpepiΜΦ), and perivascular macrophages (PVMs) [236]. Owing to their recent discovery, BAMs role in AD needs further research. In one recent study, Tg2576 mice perivascular and leptomeningeal compartments were repopulated through CD36−/− or CD36+/+ bone marrow transplantation. Deletion of CD36 in BAMs suppressed ROS production and improved neurovascular function, with complete rescue of cognitive function [233]. In another paper, immune responses induced by amyloid-targeting antibodies and CAA-induced microhemorrhages were analyzed using mouse models of AD. Anti-Aβ (3D6) immunotherapy lead to increased occurrences of microhemorrhages, altered cerebrovascular structure, and the formation of an antibody immune complexes with vascular amyloid deposits associated with perivascular macrophage [230].
Conclusion
Neuroinflammation is a conserved immune response aimed to protect neurons from a deleterious stimulus and resolve the homeostatic disbalance. In AD, chronic neuroinflammation emerges as a central driver for the neurodegenerative process due to the orchestrated interaction among multiple cell types including but not only, microglia, astrocytes, and neurons. Persistent ligand–receptor interactions in the CNS microenvironment overactivate cells for sustained periods of time causing chronic NDDs. The concept of microglia as the sole instigators of inflammation oversimplifies the complex interplay within the neuroinflammatory landscape. It is crucial to acknowledge and explore the contribution of other cellular entities such as astrocytes or neurons. Microglia and astrocytes, crucial components of the brain’s immune system, are activated by pathological forms of proteins such as Aβ and tau, initiating a cascade of inflammatory responses that self-sustain over the disease course. An over-extended inflammatory status then translates into increased cytokine and chemokine production, systemic stress, and, eventually, neural damage. In AD, neuronal damage sets off a subsequent harmful cycle, wherein multiple DAMPs, including toxic protein aggregates, and additional pro-inflammatory molecules are discharged into the extracellular space through the different RCD modes. This closes an iterative loop where neuroinflammation leads to neurodegeneration and neurodegeneration boost neuroinflammation. The complex interplay between cellular types makes the breaking point identification a formidable challenge. Therapies aimed to stop or halt this loop are urgently required.
Abbreviations
- ACT:
-
Antichymotrypsin
- AD:
-
Alzheimer disease
- Aβ:
-
Amyloid-β
- APOE:
-
Apolipoprotein E
- ApoJ/Clusterin:
-
Apolipoprotein J
- ALS:
-
Amyotrophic lateral sclerosis
- ASC:
-
Apoptosis-associated Speck-like protein containing a CARD
- APP:
-
Amyloid precursor protein
- BAMs:
-
Border-associated macrophages
- BBB:
-
Blood–brain barrier
- Bcl:
-
B-cell lymphoma
- BECs:
-
Brain endothelial cells
- C:
-
Complement component
- C1q:
-
Subcomponent q
- CCL:
-
C–C motif chemokine ligand
- CD40L:
-
CD40 ligand
- CDK:
-
Cyclin-dependent kinase
- CNS:
-
Central nervous system
- CN:
-
Calcineurin
- COX:
-
Cyclooxygenase
- DAMPs:
-
Damage-associated molecular patterns
- EOAD:
-
Early onset familial alzheimer disease
- FERMT:
-
Fermitin family homolog
- GFAP:
-
Glial fibrillary acidic protein
- GMF:
-
Glia maturation factor
- GBP:
-
Guanylate binding protein
- GSK:
-
Glycogen synthase kinase
- GWAS:
-
Genome-wide association studies
- HSPs:
-
Heat shock proteins
- HSPGs:
-
Heparan sulfate proteoglycans
- HMGB:
-
High mobility group box
- ICAM-1:
-
Intercellular adhesion molecule 1
- IDE:
-
Insulin-degrading enzyme
- IFN:
-
Interferon
- IL:
-
Interleukin
- iPS:
-
Induced pluripotent stem
- JAK:
-
Janus kinase
- MAPK:
-
Mitogen-activated protein kinase
- MMP9:
-
Matrix metalloprotease 9
- MLKL:
-
Mixed lineage kinase domain-like
- MMP:
-
Matrix metalloproteinase
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NDDs:
-
Neurodegenerative diseases
- NEP:
-
Neprylisin
- NFAT:
-
Nuclear factor of activated T cells
- NFκB:
-
Nuclear factor-κB
- NLRs:
-
Nucleotide-binding oligomerization domain-like receptors
- NFTs:
-
Neurofibrillary tangles
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- NOD:
-
Nucleotide-binding oligomerization domain
- PAR:
-
Protease-activated receptors
- PAMPs:
-
Pathogen-associated molecular patterns
- PD:
-
Parkinson’s disease
- PDGFRβ:
-
Platelet-derived growth factor receptor β
- PG:
-
Prostaglandin
- PKs:
-
Protein kinases
- PFFs:
-
Preformed fibrils
- PRRs:
-
Pattern recognition receptors
- PSEN:
-
Presenilin
- PVMs:
-
Perivascular macrophages
- RIPK:
-
Receptor-interactive protein kinases
- RAGE:
-
Receptor for advanced glycation end products
- RB:
-
Retinoblastoma
- ROS:
-
Reactive oxygen species
- RNS:
-
Reactive nitrogen species
- RCD:
-
Regulated cell death
- SAM:
-
Senescent-associated microglia
- SASP:
-
Senescence-associated secretory phenotype
- SAHF:
-
Senescence-associated heterochromatin foci
- SORL:
-
Sortilin-related receptor
- SP:
-
Substance P
- STAT:
-
Signal transducer and activator of transcription
- SRs:
-
Scavenger receptors
- TNTs:
-
Tunnelling nanotubes
- TNF:
-
Tumor necrosis factor
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- TREM:
-
Triggering receptor expressed on myeloid cells
- TGF:
-
Transforming growth factor
- TLRs:
-
Toll-like receptors
- VEGF:
-
Vascular endothelial growth factor
References
Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM et al (2009) Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci Off J Soc Neurosci 29:12957–12969. https://doi.org/10.1523/JNEUROSCI.1064-09.2009
Acosta C, Anderson HD, Anderson CM (2017) Astrocyte dysfunction in Alzheimer disease. J Neurosci Res 95:2430–2447. https://doi.org/10.1002/jnr.24075
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP et al (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15:978–990. https://doi.org/10.1038/ncb2784
Alonso AC, Grundke-Iqbal I, Iqbal K (1996) Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med 2:783–787. https://doi.org/10.1038/nm0796-783
Alzheimer A (1907) Uber eine eigenartige Erkrankung der Hirnrinde. Zentralbl Nervenh Psych 18:177–179
Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR (1995) An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde.” Clin Anat N Y N 8:429–431. https://doi.org/10.1002/ca.980080612
Andersen JV, Skotte NH, Christensen SK, Polli FS, Shabani M, Markussen KH et al (2021) Hippocampal disruptions of synaptic and astrocyte metabolism are primary events of early amyloid pathology in the 5xFAD mouse model of Alzheimer’s disease. Cell Death Dis 12:1–13. https://doi.org/10.1038/s41419-021-04237-y
Arendt T, Stieler J, Strijkstra AM, Hut RA, Rüdiger J, Van der Zee EA et al (2003) Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci Off J Soc Neurosci 23:6972–6981. https://doi.org/10.1523/JNEUROSCI.23-18-06972.2003
Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T et al (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18:1584–1593. https://doi.org/10.1038/nn.4132
Avila-Muñoz E, Arias C (2014) When astrocytes become harmful: functional and inflammatory responses that contribute to Alzheimer’s disease. Ageing Res Rev 18:29–40. https://doi.org/10.1016/j.arr.2014.07.004
Basak JM, Verghese PB, Yoon H, Kim J, Holtzman DM (2012) Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes. J Biol Chem 287:13959–13971. https://doi.org/10.1074/jbc.M111.288746
Basheer N, Smolek T, Hassan I, Liu F, Iqbal K, Zilka N et al (2023) Does modulation of tau hyperphosphorylation represent a reasonable therapeutic strategy for Alzheimer’s disease? From preclinical studies to the clinical trials. Mol Psychiatry 28:2197–2214. https://doi.org/10.1038/s41380-023-02113-z
Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC et al (2012) Clinical and biomarker changes in dominantly inherited alzheimer’s disease. N Engl J Med 367:795–804. https://doi.org/10.1056/NEJMoa1202753
Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R et al (2010) Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68:409–427
Bellaver B, Povala G, Ferreira PCL, Ferrari-Souza JP, Leffa DT, Lussier FZ et al (2023) Astrocyte reactivity influences amyloid-β effects on tau pathology in preclinical Alzheimer’s disease. Nat Med 29:1775–1781. https://doi.org/10.1038/s41591-023-02380-x
Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N et al (2022) New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet 54:412–436. https://doi.org/10.1038/s41588-022-01024-z
Berberich I, Shu GL, Clark EA (1994) Cross-linking CD40 on B cells rapidly activates nuclear factor-kappa B. J Immunol 153:4357–4366. https://doi.org/10.4049/jimmunol.153.10.4357
Bettcher BM, Tansey MG, Dorothée G, Heneka MT (2021) Peripheral and central immune system crosstalk in Alzheimer disease—a research prospectus. Nat Rev Neurol 17:689–701. https://doi.org/10.1038/s41582-021-00549-x
Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT (2010) Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68:19–31. https://doi.org/10.1016/j.neuron.2010.08.023
Bhusal A, Afridi R, Lee W-H, Suk K (2023) Bidirectional communication between microglia and astrocytes in neuroinflammation. Curr Neuropharmacol 21:2020–2029. https://doi.org/10.2174/1570159X21666221129121715
Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G et al (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312:1389–1392. https://doi.org/10.1126/science.1123511
Bolós M, Llorens-Martín M, Jurado-Arjona J, Hernández F, Rábano A, Avila J (2016) Direct evidence of internalization of tau by microglia in vitro and in vivo. J Alzheimers Dis JAD 50:77–87. https://doi.org/10.3233/JAD-150704
Borst K, Dumas AA, Prinz M (2021) Microglia: immune and non-immune functions. Immunity 54:2194–2208. https://doi.org/10.1016/j.immuni.2021.09.014
Bouvier DS, Jones EV, Quesseveur G, Davoli MA, Ferreira AT, Quirion R et al (2016) High resolution dissection of reactive glial nets in Alzheimer’s disease. Sci Rep 6:24544. https://doi.org/10.1038/srep24544
Brelstaff JH, Mason M, Katsinelos T, McEwan WA, Ghetti B, Tolkovsky AM et al (2021) Microglia become hypofunctional and release metalloproteases and tau seeds when phagocytosing live neurons with P301S tau aggregates. Sci Adv 7:eabg4980. https://doi.org/10.1126/sciadv.abg4980
Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM (2007) Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc 3:186–191. https://doi.org/10.1016/j.jalz.2007.04.381
Brosseron F, Krauthausen M, Kummer M, Heneka MT (2014) Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Mol Neurobiol 50:534–544. https://doi.org/10.1007/s12035-014-8657-1
Buckley MW, McGavern DB (2022) Immune dynamics in the CNS and its barriers during homeostasis and disease. Immunol Rev 306:58–75. https://doi.org/10.1111/imr.13066
Burgaletto C, Munafò A, Di Benedetto G, De Francisci C, Caraci F, Di Mauro R et al (2020) The immune system on the TRAIL of Alzheimer’s disease. J Neuroinflammation 17:298. https://doi.org/10.1186/s12974-020-01968-1
Burini RC, Anderson E, Durstine JL, Carson JA (2020) Inflammation, physical activity, and chronic disease: an evolutionary perspective. Sports Med Health Sci 2:1–6. https://doi.org/10.1016/j.smhs.2020.03.004
Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN et al (1999) Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23:297–308. https://doi.org/10.1016/S0896-6273(00)80781-3
Butovsky O, Weiner HL (2018) Microglial signatures and their role in health and disease. Nat Rev Neurosci 19:622–635. https://doi.org/10.1038/s41583-018-0057-5
Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS et al (2017) Necroptosis activation in Alzheimer’s disease. Nat Neurosci 20:1236–1246. https://doi.org/10.1038/nn.4608
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM et al (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9:917–924. https://doi.org/10.1038/nn1715
Casey CS, Atagi Y, Yamazaki Y, Shinohara M, Tachibana M, Fu Y et al (2015) Apolipoprotein E inhibits cerebrovascular pericyte mobility through a RhoA protein-mediated pathway*. J Biol Chem 290:14208–14217. https://doi.org/10.1074/jbc.M114.625251
Castellani G, Croese T, Peralta Ramos JM, Schwartz M (2023) Transforming the understanding of brain immunity. Science 380:eabo7649. https://doi.org/10.1126/science.abo7649
Chai YL, Lee JH, Chong JR, Ballard C, Francis PT, Kennedy BK et al (2023) Inflammatory panel cytokines are elevated in the neocortex of late-stage Alzheimer’s disease but not Lewy body dementias. J Neuroinflammation 20:111. https://doi.org/10.1186/s12974-023-02789-8
Chakraborty R, Nonaka T, Hasegawa M, Zurzolo C (2023) Tunnelling nanotubes between neuronal and microglial cells allow bi-directional transfer of α-Synuclein and mitochondria. Cell Death Dis 14:1–12. https://doi.org/10.1038/s41419-023-05835-8
Chen X, Deng S, Wang W, Castiglione S, Duan Z, Luo L et al (2022) Human antimicrobial peptide LL-37 contributes to Alzheimer’s disease progression. Mol Psychiatry 27:4790–4799. https://doi.org/10.1038/s41380-022-01790-6
Cherry JD, Olschowka JA, O’Banion MK (2014) Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation 11:98. https://doi.org/10.1186/1742-2094-11-98
Chevriaux A, Pilot T, Derangère V, Simonin H, Martine P, Chalmin F et al (2020) Cathepsin B is required for NLRP3 inflammasome activation in macrophages, through NLRP3 interaction. Front Cell Dev Biol. https://doi.org/10.3389/fcell.2020.00167
Chun H, Im H, Kang YJ, Kim Y, Shin JH, Won W et al (2020) Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer’s disease via H2O2- production. Nat Neurosci 23:1555–1566. https://doi.org/10.1038/s41593-020-00735-y
Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G et al (1997) Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat Med 3:67–72. https://doi.org/10.1038/nm0197-67
Colombo E, Farina C (2016) Astrocytes: key regulators of neuroinflammation. Trends Immunol 37:608–620. https://doi.org/10.1016/j.it.2016.06.006
Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Núñez G et al (2017) Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci 114:E961–E969. https://doi.org/10.1073/pnas.1613305114
Craig-Schapiro R, Perrin RJ, Roe CM, Xiong C, Carter D, Cairns NJ et al (2010) YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol Psychiatry 68:903–912. https://doi.org/10.1016/j.biopsych.2010.08.025
Crols R, Saerens J, Noppe M, Lowenthal A (1986) Increased GFAp levels in CSF as a marker of organicity in patients with Alzheimer’s disease and other types of irreversible chronic organic brain syndrome. J Neurol 233:157–160. https://doi.org/10.1007/BF00314423
Cunningham AJ, Murray CA, O’Neill LA, Lynch MA, O’Connor JJ (1996) Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett 203:17–20. https://doi.org/10.1016/0304-3940(95)12252-4
Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT (2011) Age-related alterations in the dynamic behavior of microglia. Aging Cell 10:263–276. https://doi.org/10.1111/j.1474-9726.2010.00660.x
Davis N, Mota BC, Stead L, Palmer EOC, Lombardero L, Rodríguez-Puertas R et al (2021) Pharmacological ablation of astrocytes reduces Aβ degradation and synaptic connectivity in an ex vivo model of Alzheimer’s disease. J Neuroinflammation 18:73. https://doi.org/10.1186/s12974-021-02117-y
De Kimpe L, van Haastert ES, Kaminari A, Zwart R, Rutjes H, Hoozemans JJM et al (2013) Intracellular accumulation of aggregated pyroglutamate amyloid beta: convergence of aging and Aβ pathology at the lysosome. Age Dordr Neth 35:673–687. https://doi.org/10.1007/s11357-012-9403-0
De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W et al (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391:387–390. https://doi.org/10.1038/34910
Dean DC III, Hurley SA, Kecskemeti SR, O’Grady JP, Canda C, Davenport-Sis NJ et al (2017) Association of amyloid pathology with myelin alteration in preclinical alzheimer disease. JAMA Neurol 74:41–49. https://doi.org/10.1001/jamaneurol.2016.3232
Depp C, Sun T, Sasmita AO, Spieth L, Berghoff SA, Nazarenko T et al (2023) Myelin dysfunction drives amyloid-β deposition in models of Alzheimer’s disease. Nature 618:349–357. https://doi.org/10.1038/s41586-023-06120-6
Dermitzakis I, Theotokis P, Evangelidis P, Delilampou E, Evangelidis N, Chatzisavvidou A et al (2023) CNS border-associated macrophages: ontogeny and potential implication in disease. Curr Issues Mol Biol 45:4285–4300. https://doi.org/10.3390/cimb45050272
DiPatre PL, Gelman BB (1997) Microglial cell activation in aging and Alzheimer disease: partial linkage with neurofibrillary tangle burden in the hippocampus. J Neuropathol Exp Neurol 56:143–149. https://doi.org/10.1097/00005072-199702000-00004
DiSabato DJ, Quan N, Godbout JP (2016) Neuroinflammation: the devil is in the details. J Neurochem 139(Suppl 2):136–153. https://doi.org/10.1111/jnc.13607
Dixit R, Ross JL, Goldman YE, Holzbaur ELF (2008) Differential regulation of dynein and kinesin motor proteins by tau. Science 319:1086–1089. https://doi.org/10.1126/science.1152993
Domingues C, da Cruz E, Silva OAB, Henriques AG (2017) Impact of cytokines and chemokines on alzheimer’s disease neuropathological hallmarks. Curr Alzheimer Res 14:870–882. https://doi.org/10.2174/1567205014666170317113606
Ekonomou A, Savva GM, Brayne C, Forster G, Francis PT, Johnson M et al (2015) Stage-specific changes in neurogenic and glial markers in Alzheimer’s disease. Biol Psychiatry 77:711–719. https://doi.org/10.1016/j.biopsych.2014.05.021
El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD (1996) Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature 382:716–719. https://doi.org/10.1038/382716a0
Eltom K, Mothes T, Libard S, Ingelsson M, Erlandsson A (2024) Astrocytic accumulation of tau fibrils isolated from Alzheimer’s disease brains induces inflammation, cell-to-cell propagation and neuronal impairment. Acta Neuropathol Commun 12:34. https://doi.org/10.1186/s40478-024-01745-8
Engelhart MJ, Geerlings MI, Meijer J, Kiliaan A, Ruitenberg A, van Swieten JC et al (2004) Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch Neurol 61:668–672. https://doi.org/10.1001/archneur.61.5.668
Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A et al (2021) Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci 24:312–325. https://doi.org/10.1038/s41593-020-00783-4
Fancy NN, Smith AM, Caramello A, Tsartsalis S, Davey K, Muirhead RCJ et al (2024) Characterisation of premature cell senescence in Alzheimer’s disease using single nuclear transcriptomics. Acta Neuropathol (Berl) 147:78. https://doi.org/10.1007/s00401-024-02727-9
Fassbender K, Walter S, Kühl S, Landmann R, Ishii K, Bertsch T et al (2004) The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J Off Publ Fed Am Soc Exp Biol 18:203–205. https://doi.org/10.1096/fj.03-0364fje
Fleeman RM, Proctor EA (2021) Astrocytic propagation of tau in the context of alzheimer’s disease. Front Cell Neurosci 15:645233. https://doi.org/10.3389/fncel.2021.645233
Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G et al (2014) ASC has extracellular and prionoid activities that propagate inflammation. Nat Immunol 15:727–737. https://doi.org/10.1038/ni.2913
Friker LL, Scheiblich H, Hochheiser IV, Brinkschulte R, Riedel D, Latz E et al (2020) β-Amyloid clustering around ASC fibrils boosts its toxicity in microglia. Cell Rep 30:3743-3754.e6. https://doi.org/10.1016/j.celrep.2020.02.025
Fu H, Hardy J, Duff KE (2018) Selective vulnerability in neurodegenerative diseases. Nat Neurosci 21:1350–1358. https://doi.org/10.1038/s41593-018-0221-2
Füger P, Hefendehl JK, Veeraraghavalu K, Wendeln A-C, Schlosser C, Obermüller U et al (2017) Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat Neurosci 20:1371–1376. https://doi.org/10.1038/nn.4631
Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C et al (2019) Chronic inflammation in the etiology of disease across the life span. Nat Med 25:1822–1832. https://doi.org/10.1038/s41591-019-0675-0
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P et al (2018) Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ 25:486–541. https://doi.org/10.1038/s41418-017-0012-4
Gaminde-Blasco A, Senovilla-Ganzo R, Balantzategi U, García-Moreno F, Matute C, Baleriola J et al (2024) Amyloid-β increases MBP and MOBP translation in oligodendrocytes through dysregulation of hnRNP A2 dependent RNA dynamics. BioRxiv. https://doi.org/10.1101/2024.04.19.590214
Garbuz DG, Zatsepina OG, Evgenev MB (2021) Beta Amyloid, tau protein, and neuroinflammation: an attempt to integrate different hypotheses of alzheimer’s disease pathogenesis. Mol Biol 55:670–682. https://doi.org/10.1134/S002689332104004X
Garnier-Crussard A, Bougacha S, Wirth M, Dautricourt S, Sherif S, Landeau B et al (2022) White matter hyperintensity topography in Alzheimer’s disease and links to cognition. Alzheimers Dement 18:422–433. https://doi.org/10.1002/alz.12410
Gate D, Saligrama N, Leventhal O, Yang AC, Unger MS, Middeldorp J et al (2020) Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577:399–404. https://doi.org/10.1038/s41586-019-1895-7
Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA et al (2013) Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an alzheimer’s mouse model. J Neurosci 33:5053–5064. https://doi.org/10.1523/JNEUROSCI.4361-12.2013
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S et al (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845. https://doi.org/10.1126/science.1194637
Giusti V, Kaur G, Giusto E, Civiero L (2024) Brain clearance of protein aggregates: a close-up on astrocytes. Mol Neurodegener 19:5. https://doi.org/10.1186/s13024-024-00703-1
Gonzales MM, Garbarino VR, Marques Zilli E, Petersen RC, Kirkland JL, Tchkonia T et al (2022) Senolytic therapy to modulate the progression of alzheimer’s disease (SToMP-AD): a pilot clinical trial. J Prev Alzheimers Dis 9:22–29. https://doi.org/10.14283/jpad.2021.62
Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C et al (2019) Cellular senescence: defining a path forward. Cell 179:813–827. https://doi.org/10.1016/j.cell.2019.10.005
Guijarro-Muñoz I, Compte M, Álvarez-Cienfuegos A, Álvarez-Vallina L, Sanz L (2014) Lipopolysaccharide activates Toll-like receptor 4 (TLR4)-mediated NF-κB Signaling pathway and proinflammatory response in human pericytes. J Biol Chem 289:2457–2468. https://doi.org/10.1074/jbc.M113.521161
Haage V, De Jager PL (2022) Neuroimmune contributions to Alzheimer’s disease: a focus on human data. Mol Psychiatry 27:3164–3181. https://doi.org/10.1038/s41380-022-01637-0
Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R et al (2020) Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci 23:701–706. https://doi.org/10.1038/s41593-020-0624-8
Haim LB, Ceyzériat K, Sauvage MAC, Aubry F, Auregan G, Guillermier M et al (2015) The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in alzheimer’s and huntington’s diseases. J Neurosci 35:2817–2829. https://doi.org/10.1523/JNEUROSCI.3516-14.2015
Han C, Yang Y, Guan Q, Zhang X, Shen H, Sheng Y et al (2020) New mechanism of nerve injury in Alzheimer’s disease: β-amyloid-induced neuronal pyroptosis. J Cell Mol Med 24:8078–8090. https://doi.org/10.1111/jcmm.15439
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–185. https://doi.org/10.1126/science.1566067
Hardy S (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356. https://doi.org/10.1126/science.1072994
Hasel P, Rose IVL, Sadick JS, Kim RD, Liddelow SA (2021) Neuroinflammatory astrocyte subtypes in the mouse brain. Nat Neurosci 24:1475–1487. https://doi.org/10.1038/s41593-021-00905-6
Hecimovic S, Wang J, Dolios G, Martinez M, Wang R, Goate AM (2004) Mutations in APP have independent effects on Aβ and CTFγ generation. Neurobiol Dis 17:205–218. https://doi.org/10.1016/j.nbd.2004.04.018
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL et al (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14:388–405. https://doi.org/10.1016/S1474-4422(15)70016-5
Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14:463–477. https://doi.org/10.1038/nri3705
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A et al (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493:674–678. https://doi.org/10.1038/nature11729
Heneka MT, McManus RM, Latz E (2018) Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci 19:610–621. https://doi.org/10.1038/s41583-018-0055-7
Heneka MT, Sastre M, Dumitrescu-Ozimek L, Dewachter I, Walter J, Klockgether T et al (2005) Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J Neuroinflammation 2:22. https://doi.org/10.1186/1742-2094-2-22
Heslegrave A, Heywood W, Paterson R, Magdalinou N, Svensson J, Johansson P et al (2016) Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol Neurodegener 11:3. https://doi.org/10.1186/s13024-016-0071-x
Hickman SE, Allison EK, El Khoury J (2008) Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci Off J Soc Neurosci 28:8354–8360. https://doi.org/10.1523/JNEUROSCI.0616-08.2008
Hollville E, Romero SE, Deshmukh M (2019) Apoptotic cell death regulation in neurons. FEBS J 286:3276–3298. https://doi.org/10.1111/febs.14970
Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL et al (2019) Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol 15:565–581. https://doi.org/10.1038/s41582-019-0244-7
Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS et al (1986) Defective brain microtubule assembly in Alzheimer’s disease. Lancet Lond Engl 2:421–426. https://doi.org/10.1016/s0140-6736(86)92134-3
Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A et al (2019) NLRP3 inflammasome activation drives tau pathology. Nature 575:669–673. https://doi.org/10.1038/s41586-019-1769-z
Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D (1989) Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 24:173–182. https://doi.org/10.1016/0165-5728(89)90115-X
Jack CR, Petersen RC, Xu YC, O’Brien PC, Smith GE, Ivnik RJ et al (1999) Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology 52:1397–1403. https://doi.org/10.1212/wnl.52.7.1397
Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FRJ et al (2015) Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA 313:1924–1938. https://doi.org/10.1001/jama.2015.4668
Jellinger KA (2010) Basic mechanisms of neurodegeneration: a critical update. J Cell Mol Med 14:457–487. https://doi.org/10.1111/j.1582-4934.2010.01010.x
Jiang S, Maphis NM, Binder J, Chisholm D, Weston L, Duran W et al (2021) Proteopathic tau primes and activates interleukin-1β via myeloid-cell-specific MyD88- and NLRP3-ASC-inflammasome pathway. Cell Rep 36:109720. https://doi.org/10.1016/j.celrep.2021.109720
Jones VC, Atkinson-Dell R, Verkhratsky A, Mohamet L (2017) Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis 8:e2696. https://doi.org/10.1038/cddis.2017.89
Jorfi M, Park J, Hall CK, Lin C-CJ, Chen M, von Maydell D et al (2023) Infiltrating CD8+ T cells exacerbate Alzheimer’s disease pathology in a 3D human neuroimmune axis model. Nat Neurosci 26:1489–1504. https://doi.org/10.1038/s41593-023-01415-3
Karran E, De Strooper B (2022) The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat Rev Drug Discov 21:306–318. https://doi.org/10.1038/s41573-022-00391-w
Katsouri L, Birch AM, Renziehausen AWJ, Zach C, Aman Y, Steeds H et al (2020) Ablation of reactive astrocytes exacerbates disease pathology in a model of Alzheimer’s disease. Glia 68:1017–1030. https://doi.org/10.1002/glia.23759
Kim H, Leng K, Park J, Sorets AG, Kim S, Shostak A et al (2022) Reactive astrocytes transduce inflammation in a blood-brain barrier model through a TNF-STAT3 signaling axis and secretion of alpha 1-antichymotrypsin. Nat Commun 13:6581. https://doi.org/10.1038/s41467-022-34412-4
Kim J, Yoo ID, Lim J, Moon J-S (2024) Pathological phenotypes of astrocytes in Alzheimer’s disease. Exp Mol Med 56:95–99. https://doi.org/10.1038/s12276-023-01148-0
Kim J-H, Afridi R, Han J, Jung H-G, Kim S-C, Hwang EM et al (2021) Gamma subunit of complement component 8 is a neuroinflammation inhibitor. Brain J Neurol 144:528–552. https://doi.org/10.1093/brain/awaa425
Kim WX, Ragonnaud E, Bodogai M, Illouz T, DeLuca M, McDevitt RA et al (2021) Therapeutic B-cell depletion reverses progression of Alzheimer’s disease. Nat Commun 12:2185. https://doi.org/10.1038/s41467-021-22479-4
Kirby L, Jin J, Cardona JG, Smith MD, Martin KA, Wang J et al (2019) Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat Commun 10:3887. https://doi.org/10.1038/s41467-019-11638-3
Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V et al (1950) LaFerla FM (2011) Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J Immunol Baltim Md 187:6539–6549. https://doi.org/10.4049/jimmunol.1100620
Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT et al (2021) Alzheimer disease Nat Rev Dis Primer 7:1–21. https://doi.org/10.1038/s41572-021-00269-y
Koper MJ, Van Schoor E, Ospitalieri S, Vandenberghe R, Vandenbulcke M, Arnim CAF et al (2020) Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer’s disease. Acta Neuropathol (Berl) 139:463–484. https://doi.org/10.1007/s00401-019-02103-y
Kosoy R, Fullard JF, Zeng B, Bendl J, Dong P, Rahman S et al (2022) Genetics of the human microglia regulome refines Alzheimer’s disease risk loci. Nat Genet 54:1145–1154. https://doi.org/10.1038/s41588-022-01149-1
Kovac A, Erickson MA, Banks WA (2011) Brain microvascular pericytes are immunoactive in culture: cytokine, chemokine, nitric oxide, and LRP-1 expression in response to lipopolysaccharide. J Neuroinflammation 8:139. https://doi.org/10.1186/1742-2094-8-139
Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U et al (2013) Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 8:e60921. https://doi.org/10.1371/journal.pone.0060921
Kurz C, Walker L, Rauchmann B-S, Perneczky R (2022) Dysfunction of the blood–brain barrier in Alzheimer’s disease: Evidence from human studies. Neuropathol Appl Neurobiol 48:e12782. https://doi.org/10.1111/nan.12782
Kwon HS, Koh S-H (2020) Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener 9:42. https://doi.org/10.1186/s40035-020-00221-2
Lau S-F, Fu AKY, Ip NY (2023) Receptor–ligand interaction controls microglial chemotaxis and amelioration of Alzheimer’s disease pathology. J Neurochem 166:891–903. https://doi.org/10.1111/jnc.15933
Lau V, Ramer L, Tremblay M-È (2023) An aging, pathology burden, and glial senescence build-up hypothesis for late onset Alzheimer’s disease. Nat Commun 14:1670. https://doi.org/10.1038/s41467-023-37304-3
LeBlanc AC, Xue R, Gambetti P (1996) Amyloid precursor protein metabolism in primary cell cultures of neurons, astrocytes, and microglia. J Neurochem 66:2300–2310. https://doi.org/10.1046/j.1471-4159.1996.66062300.x
Lee J-H, Kim J, Noh S, Lee H, Lee SY, Mun JY et al (2021) Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 590:612–617. https://doi.org/10.1038/s41586-020-03060-3
Leng F, Hinz R, Gentleman S, Hampshire A, Dani M, Brooks DJ et al (2023) Neuroinflammation is independently associated with brain network dysfunction in Alzheimer’s disease. Mol Psychiatry 28:1303–1311. https://doi.org/10.1038/s41380-022-01878-z
Li Y, Xu P, Shan J, Sun W, Ji X, Chi T et al (2020) Interaction between hyperphosphorylated tau and pyroptosis in forskolin and streptozotocin induced AD models. Biomed Pharmacother 121:109618. https://doi.org/10.1016/j.biopha.2019.109618
Lian H, Litvinchuk A, Chiang AC-A, Aithmitti N, Jankowsky JL, Zheng H (2016) Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of alzheimer’s disease. J Neurosci 36:577–589. https://doi.org/10.1523/JNEUROSCI.2117-15.2016
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. https://doi.org/10.1038/nature21029
Lim D, Iyer A, Ronco V, Grolla AA, Canonico PL, Aronica E et al (2013) Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia 61:1134–1145. https://doi.org/10.1002/glia.22502
Lindberg C, Selenica M-LB, Westlind-Danielsson A, Schultzberg M (2005) Beta-amyloid protein structure determines the nature of cytokine release from rat microglia. J Mol Neurosci MN 27:1–12. https://doi.org/10.1385/JMN:27:1:001
Liu C, Cui G, Zhu M, Kang X, Guo H (2014) Neuroinflammation in Alzheimer’s disease: chemokines produced by astrocytes and chemokine receptors. Int J Clin Exp Pathol 7:8342–8355
Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B et al (1950) Fassbender K (2012) TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J Immunol Baltim Md 188:1098–1107. https://doi.org/10.4049/jimmunol.1101121
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H et al (2016) Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535:153–158. https://doi.org/10.1038/nature18629
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD et al (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523:337–341. https://doi.org/10.1038/nature14432
Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR et al (2014) Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156:1193–1206. https://doi.org/10.1016/j.cell.2014.02.008
Lue L-F, Rydel R, Brigham EF, Yang L-B, Hampel H, Murphy GM Jr et al (2001) Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 35:72–79. https://doi.org/10.1002/glia.1072
Mallach A, Zielonka M, van Lieshout V, An Y, Khoo JH, Vanheusden M et al (2024) Microglia-astrocyte crosstalk in the amyloid plaque niche of an Alzheimer’s disease mouse model, as revealed by spatial transcriptomics. Cell Rep. https://doi.org/10.1016/j.celrep.2024.114216
Mancuso R, Fattorelli N, Martinez-Muriana A, Davis E, Wolfs L, Van Den Daele J et al (2024) Xenografted human microglia display diverse transcriptomic states in response to Alzheimer’s disease-related amyloid-β pathology. Nat Neurosci 27:886–900. https://doi.org/10.1038/s41593-024-01600-y
Mancuso R, Fryatt G, Cleal M, Obst J, Pipi E, Monzón-Sandoval J et al (2019) CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain J Neurol 142:3243–3264. https://doi.org/10.1093/brain/awz241
Mangalmurti A, Lukens JR (2022) How neurons die in Alzheimer’s disease: Implications for neuroinflammation. Curr Opin Neurobiol 75:102575. https://doi.org/10.1016/j.conb.2022.102575
Mangiafico SP, Tuo Q-Z, Li X-L, Liu Y, Haralambous C, Ding X-L et al (2023) Tau suppresses microtubule-regulated pancreatic insulin secretion. Mol Psychiatry 28:3982–3993. https://doi.org/10.1038/s41380-023-02267-w
Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M (2010) Alzheimer’s disease: clinical trials and drug development. Lancet Neurol 9:702–716. https://doi.org/10.1016/S1474-4422(10)70119-8
Maté de Gérando A, d’Orange M, Augustin E, Joséphine C, Aurégan G, Gaudin-Guérif M et al (2021) Neuronal tau species transfer to astrocytes and induce their loss according to tau aggregation state. Brain 144:1167–1182. https://doi.org/10.1093/brain/awab011
Matsumoto J, Takata F, Machida T, Takahashi H, Soejima Y, Funakoshi M et al (2014) Tumor necrosis factor-α-stimulated brain pericytes possess a unique cytokine and chemokine release profile and enhance microglial activation. Neurosci Lett 578:133–138. https://doi.org/10.1016/j.neulet.2014.06.052
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC et al (2010) Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330:1774. https://doi.org/10.1126/science.1197623
McAleese KE, Miah M, Graham S, Hadfield GM, Walker L, Johnson M et al (2021) Frontal white matter lesions in Alzheimer’s disease are associated with both small vessel disease and AD-associated cortical pathology. Acta Neuropathol (Berl) 142:937–950. https://doi.org/10.1007/s00401-021-02376-2
McAlpine CS, Park J, Griciuc A, Kim E, Choi SH, Iwamoto Y et al (2021) Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature 595:701–706. https://doi.org/10.1038/s41586-021-03734-6
McCarthy JV, Twomey C, Wujek P (2009) Presenilin-dependent regulated intramembrane proteolysis and γ-secretase activity. Cell Mol Life Sci 66:1534–1555. https://doi.org/10.1007/s00018-009-8435-9
Medzhitov R (2010) Inflammation 2010: new adventures of an old flame. Cell 140:771–776. https://doi.org/10.1016/j.cell.2010.03.006
Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P (2009) Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol 210:3–12. https://doi.org/10.1016/j.jneuroim.2009.02.003
Miners JS, Kehoe PG, Love S, Zetterberg H, Blennow K (2019) CSF evidence of pericyte damage in Alzheimer’s disease is associated with markers of blood-brain barrier dysfunction and disease pathology. Alzheimers Res Ther 11:1–6. https://doi.org/10.1186/s13195-019-0534-8
Mittelbronn M, Dietz K, Schluesener HJ, Meyermann R (2001) Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol (Berl) 101:249–255. https://doi.org/10.1007/s004010000284
Moonen S, Koper MJ, Van Schoor E, Schaeverbeke JM, Vandenberghe R, von Arnim CAF et al (2023) Pyroptosis in Alzheimer’s disease: cell type-specific activation in microglia, astrocytes and neurons. Acta Neuropathol (Berl) 145:175–195. https://doi.org/10.1007/s00401-022-02528-y
Mothes T, Portal B, Konstantinidis E, Eltom K, Libard S, Streubel-Gallasch L et al (2023) Astrocytic uptake of neuronal corpses promotes cell-to-cell spreading of tau pathology. Acta Neuropathol Commun 11:97. https://doi.org/10.1186/s40478-023-01589-8
Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP et al (2018) High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity 48:380-395.e6. https://doi.org/10.1016/j.immuni.2018.01.011
Mudher A, Colin M, Dujardin S, Medina M, Dewachter I, Alavi Naini SM et al (2017) What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun 5:99. https://doi.org/10.1186/s40478-017-0488-7
Murray CA, Lynch MA (1998) Evidence that increased hippocampal expression of the cytokine interleukin-1 beta is a common trigger for age- and stress-induced impairments in long-term potentiation. J Neurosci Off J Soc Neurosci 18:2974–2981. https://doi.org/10.1523/JNEUROSCI.18-08-02974.1998
Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang K-C, Wegiel J (2004) Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol Aging 25:663–674. https://doi.org/10.1016/j.neurobiolaging.2004.01.007
Nichols E, Steinmetz JD, Vollset SE, Fukutaki K, Chalek J, Abd-Allah F et al (2022) Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the global burden of disease study 2019. Lancet Public Health 7:e105–e125. https://doi.org/10.1016/S2468-2667(21)00249-8
Nicoll JAR, Weller RO (2003) A new role for astrocytes: beta-amyloid homeostasis and degradation. Trends Mol Med 9:281–282. https://doi.org/10.1016/s1471-4914(03)00109-6
Niewiadomska G, Niewiadomski W, Steczkowska M, Gasiorowska A (2021) Tau oligomers neurotoxicity. Life Basel Switz 11:28. https://doi.org/10.3390/life11010028
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318. https://doi.org/10.1126/science.1110647
Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z et al (2019) Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 365:eaav9518. https://doi.org/10.1126/science.aav9518
Novikova G, Kapoor M, Tcw J, Abud EM, Efthymiou AG, Chen SX (2021) Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat Commun 12:1610. https://doi.org/10.1038/s41467-021-21823-y
Ogrodnik M, Evans SA, Fielder E, Victorelli S, Kruger P, Salmonowicz H et al (2021) Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 20:e13296. https://doi.org/10.1111/acel.13296
Opland CK, Bryan MR, Harris B, McGillion-Moore J, Tian X, Chen Y et al (2023) Activity-dependent tau cleavage by caspase-3 promotes neuronal dysfunction and synaptotoxicity. iScience 26:106905. https://doi.org/10.1016/j.isci.2023.106905
Orre M, Kamphuis W, Osborn LM, Melief J, Kooijman L, Huitinga I et al (2014) Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol Aging 35:1–14. https://doi.org/10.1016/j.neurobiolaging.2013.07.008
Palacino JJ, Berechid BE, Alexander P, Eckman C, Younkin S, Nye JS et al (2000) Regulation of amyloid precursor protein processing by presenilin 1 (PS1) and PS2 in PS1 knockout cells. J Biol Chem 275:215–222. https://doi.org/10.1074/jbc.275.1.215
Pampuscenko K, Morkuniene R, Krasauskas L, Smirnovas V, Brown GC, Borutaite V (2023) Extracellular tau stimulates phagocytosis of living neurons by activated microglia via Toll-like 4 receptor–NLRP3 inflammasome–caspase-1 signalling axis. Sci Rep 13:10813. https://doi.org/10.1038/s41598-023-37887-3
Park G, Nhan HS, Tyan S-H, Kawakatsu Y, Zhang C, Navarro M et al (2020) Caspase activation and caspase-mediated cleavage of APP is associated with amyloid β-protein-induced synapse loss in alzheimer’s disease. Cell Rep 31:107839. https://doi.org/10.1016/j.celrep.2020.107839
Perea JR, López E, Díez-Ballesteros JC, Ávila J, Hernández F, Bolós M (2019) Extracellular monomeric tau is internalized by astrocytes. Front Neurosci 13:442. https://doi.org/10.3389/fnins.2019.00442
Perez-Nievas BG, Serrano-Pozo A (2018) Deciphering the astrocyte reaction in alzheimer’s disease. Front Aging Neurosci 10:114. https://doi.org/10.3389/fnagi.2018.00114
Pham C, Hérault K, Oheim M, Maldera S, Vialou V, Cauli B et al (2021) Astrocytes respond to a neurotoxic Aβ fragment with state-dependent Ca2+ alteration and multiphasic transmitter release. Acta Neuropathol Commun 9:44. https://doi.org/10.1186/s40478-021-01146-1
Potter R, Patterson BW, Elbert DL, Ovod V, Kasten T, Sigurdson W et al (2013) Increased in vivo amyloid-β42 production, exchange, and loss in presenilin mutation carriers. Sci Transl Med 5:77. https://doi.org/10.1126/scitranslmed.3005615
Prater KE, Green KJ, Mamde S, Sun W, Cochoit A, Smith CL et al (2023) Human microglia show unique transcriptional changes in Alzheimer’s disease. Nat Aging 3:894–907. https://doi.org/10.1038/s43587-023-00424-y
Prinz M, Masuda T, Wheeler MA, Quintana FJ (2021) Microglia and central nervous system-associated macrophages-from origin to disease modulation. Annu Rev Immunol 39:251–277. https://doi.org/10.1146/annurev-immunol-093019-110159
Qian Z, Qin J, Lai Y, Zhang C, Zhang X (2023) Large-scale integration of single-cell RNA-Seq data reveals astrocyte diversity and transcriptomic modules across six central nervous system disorders. Biomolecules 13:692. https://doi.org/10.3390/biom13040692
Rajani RM, Ellingford R, Hellmuth M, Harris SS, Taso OS, Graykowski D et al (2024) Selective suppression of oligodendrocyte-derived amyloid beta rescues neuronal dysfunction in Alzheimer’s Disease. PLoS Biol. https://doi.org/10.1101/2024.06.21.600003
Ravichandran KA, Heneka MT (2024) Inflammasomes in neurological disorders—mechanisms and therapeutic potential. Nat Rev Neurol 20:67–83. https://doi.org/10.1038/s41582-023-00915-x
Réu P, Khosravi A, Bernard S, Mold JE, Salehpour M, Alkass K et al (2017) The lifespan and turnover of microglia in the human brain. Cell Rep 20:779–784. https://doi.org/10.1016/j.celrep.2017.07.004
Richetin K, Steullet P, Pachoud M, Perbet R, Parietti E, Maheswaran M et al (2020) Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat Neurosci 23:1567–1579. https://doi.org/10.1038/s41593-020-00728-x
Ringman JM, O’Neill J, Geschwind D, Medina L, Apostolova LG, Rodriguez Y et al (2007) Diffusion tensor imaging in preclinical and presymptomatic carriers of familial Alzheimer’s disease mutations. Brain 130:1767–1776. https://doi.org/10.1093/brain/awm102
Rodríguez JJ, Olabarria M, Chvatal A, Verkhratsky A (2009) Astroglia in dementia and Alzheimer’s disease. Cell Death Differ 16:378–385. https://doi.org/10.1038/cdd.2008.172
Rodríguez JJ, Zallo F, Gardenal E, Cabot J, Busquets X (2023) Prominent and conspicuous astrocyte atrophy in human sporadic and familial Alzheimer’s disease. Brain Struct Funct 228:2103–2113. https://doi.org/10.1007/s00429-023-02707-x
Rodríguez-Arellano JJ, Parpura V, Zorec R, Verkhratsky A (2016) Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 323:170–182. https://doi.org/10.1016/j.neuroscience.2015.01.007
Rong Y, Ji C, Wang Z, Ge X, Wang J, Ye W et al (2021) Small extracellular vesicles encapsulating CCL2 from activated astrocytes induce microglial activation and neuronal apoptosis after traumatic spinal cord injury. J Neuroinflammation 18:196. https://doi.org/10.1186/s12974-021-02268-y
Rostami J, Holmqvist S, Lindström V, Sigvardson J, Westermark GT, Ingelsson M et al (2017) Human Astrocytes Transfer Aggregated Alpha-Synuclein via Tunneling Nanotubes. J Neurosci Off J Soc Neurosci 37:11835–11853. https://doi.org/10.1523/JNEUROSCI.0983-17.2017
Rostami J, Mothes T, Kolahdouzan M, Eriksson O, Moslem M, Bergström J et al (2021) Crosstalk between astrocytes and microglia results in increased degradation of α-synuclein and amyloid-β aggregates. J Neuroinflammation 18:124. https://doi.org/10.1186/s12974-021-02158-3
Rothhammer V, Borucki DM, Tjon EC, Takenaka MC, Chao C-C, Ardura-Fabregat A et al (2018) Microglial control of astrocytes in response to microbial metabolites. Nature 557:724–728. https://doi.org/10.1038/s41586-018-0119-x
Rustenhoven J, Jansson D, Smyth LC, Dragunow M (2017) Brain Pericytes As Mediators of Neuroinflammation. Trends Pharmacol Sci 38:291–304. https://doi.org/10.1016/j.tips.2016.12.001
Sadick JS, O’Dea MR, Hasel P, Dykstra T, Faustin A, Liddelow SA (2022) Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer’s disease. Neuron 110:1788-1805.e10. https://doi.org/10.1016/j.neuron.2022.03.008
Saha P, Guha S, Biswas SC (2020) P38K and JNK pathways are induced by amyloid-β in astrocyte: implication of MAPK pathways in astrogliosis in Alzheimer’s disease. Mol Cell Neurosci 108:103551. https://doi.org/10.1016/j.mcn.2020.103551
Salvadores N, Moreno-Gonzalez I, Gamez N, Quiroz G, Vegas-Gomez L, Escandón M et al (2022) Aβ oligomers trigger necroptosis-mediated neurodegeneration via microglia activation in Alzheimer’s disease. Acta Neuropathol Commun 10:31. https://doi.org/10.1186/s40478-022-01332-9
Sasmita AO, Depp C, Nazarenko T, Sun T, Siems SB, Yu X et al (2023) Oligodendrocytes and neurons contribute to amyloid-β deposition in Alzheimer’s disease. bioRxiv. https://doi.org/10.1101/2023.12.11.570514
Schmidt R, Schmidt H, Curb JD, Masaki K, White LR, Launer LJ (2002) Early inflammation and dementia: a 25-year follow-up of the Honolulu-Asia aging study. Ann Neurol 52:168–174. https://doi.org/10.1002/ana.10265
Schultz N, Nielsen HM, Minthon L, Wennström M (2014) Involvement of matrix metalloproteinase-9 in amyloid-β 1–42–induced shedding of the pericyte proteoglycan NG2. J Neuropathol Exp Neurol 73:684–692. https://doi.org/10.1097/NEN.0000000000000084
Sebastian Monasor L, Müller SA, Colombo AV, Tanrioever G, König J, Roth S et al (2020) Fibrillar Aβ triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. Elife 9:54083. https://doi.org/10.7554/eLife.54083
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8:595–608. https://doi.org/10.15252/emmm.201606210
Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, Zlokovic BV (2013) Deficiency in mural vascular cells coincides with blood-brain barrier disruption in alzheimer’s disease. Brain Pathol 23:303–310. https://doi.org/10.1111/bpa.12004
Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB et al (2013) CD36 coordinates NLRP3 inflammasome activation by facilitating the intracellular nucleation from soluble to particulate ligands in sterile inflammation. Nat Immunol 14:812–820. https://doi.org/10.1038/ni.2639
Shen H, Han C, Yang Y, Guo L, Sheng Y, Wang J et al (2021) Pyroptosis executive protein GSDMD as a biomarker for diagnosis and identification of Alzheimer’s disease. Brain Behav 11:e02063. https://doi.org/10.1002/brb3.2063
Shi H, Koronyo Y, Rentsendorj A, Regis GC, Sheyn J, Fuchs D-T et al (2020) Identification of early pericyte loss and vascular amyloidosis in Alzheimer’s disease retina. Acta Neuropathol (Berl) 139:813–836. https://doi.org/10.1007/s00401-020-02134-w
Shirai Y (1921) On the transplantation of the rat sarcoma in adult heterogenous animals. Jap Med World 1:14–15
Sidoryk-Wegrzynowicz M, Gerber YN, Ries M, Sastre M, Tolkovsky AM, Spillantini MG (2017) Astrocytes in mouse models of tauopathies acquire early deficits and lose neurosupportive functions. Acta Neuropathol Commun 5:89. https://doi.org/10.1186/s40478-017-0478-9
Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K (2007) Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55:412–424. https://doi.org/10.1002/glia.20468
Simard AR, Soulet D, Gowing G, Julien J-P, Rivest S (2006) Bone marrow-derived microglia play a critical role in restricting senile plaque formation in alzheimer’s disease. Neuron 49:489–502. https://doi.org/10.1016/j.neuron.2006.01.022
Skaper SD, Evans NA, Soden PE, Rosin C, Facci L, Richardson JC (2009) Oligodendrocytes are a novel source of amyloid peptide generation. Neurochem Res 34:2243–2250. https://doi.org/10.1007/s11064-009-0022-9
Smith AM, Davey K, Tsartsalis S, Khozoie C, Fancy N, Tang SS et al (2022) Diverse human astrocyte and microglial transcriptional responses to Alzheimer’s pathology. Acta Neuropathol (Berl) 143:75–91. https://doi.org/10.1007/s00401-021-02372-6
Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32:638–647. https://doi.org/10.1016/j.tins.2009.08.002
Sofroniew MV (2014) Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist 20:160–172. https://doi.org/10.1177/1073858413504466
Sosna J, Philipp S, Albay R, Reyes-Ruiz JM, Baglietto-Vargas D, LaFerla FM et al (2018) Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol Neurodegener 13:11. https://doi.org/10.1186/s13024-018-0244-x
Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA et al (2019) Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun 10:3758. https://doi.org/10.1038/s41467-019-11674-z
Spanos F, Liddelow SA (2020) An overview of astrocyte responses in genetically induced alzheimer’s disease mouse models. Cells 9:2415. https://doi.org/10.3390/cells9112415
Stadelmann C, Bruck W, Bancher C, Jellinger K, Lassmann H (1998) Alzheimer disease: DNA fragmentation indicates increased neuronal vulnerability, but not apoptosis. J Neuropathol Exp Neurol 57:456–464. https://doi.org/10.1097/00005072-199805000-00009
Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Brück W, Jellinger K et al (1999) Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in alzheimer’s disease. Am J Pathol 155:1459–1466
Stancu IC, Lodder C, Botella Lucena P, Vanherle S, Gutiérrez de Ravé M, Terwel D et al (2022) The NLRP3 inflammasome modulates tau pathology and neurodegeneration in a tauopathy model. Glia 70:1117–1132. https://doi.org/10.1002/glia.24160
Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A et al (2010) CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 11:155–161. https://doi.org/10.1038/ni.1836
Sudwarts A, Thinakaran G (2023) Alzheimer’s genes in microglia: a risk worth investigating. Mol Neurodegener 18:90. https://doi.org/10.1186/s13024-023-00679-4
Sun N, Victor MB, Park YP, Xiong X, Scannail AN, Leary N et al (2023) Human microglial state dynamics in Alzheimer’s disease progression. Cell 186:4386-4403.e29. https://doi.org/10.1016/j.cell.2023.08.037
Taipa R, Ferreira V, Brochado P, Robinson A, Reis I, Marques F et al (2018) Inflammatory pathology markers (activated microglia and reactive astrocytes) in early and late onset Alzheimer disease: a post mortem study. Neuropathol Appl Neurobiol 44:298–313. https://doi.org/10.1111/nan.12445
Takahashi H, Klein ZA, Bhagat SM, Kaufman AC, Kostylev MA, Ikezu T et al (2017) Opposing effects of progranulin deficiency on amyloid and tau pathologies via microglial TYROBP network. Acta Neuropathol (Berl) 133:785–807. https://doi.org/10.1007/s00401-017-1668-z
Tan M-S, Tan L, Jiang T, Zhu X-C, Wang H-F, Jia C-D et al (2014) Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis 5:e1382. https://doi.org/10.1038/cddis.2014.348
Tan ZS, Beiser AS, Vasan RS, Roubenoff R, Dinarello CA, Harris TB et al (2007) Inflammatory markers and the risk of Alzheimer disease. Neurology 68:1902–1908. https://doi.org/10.1212/01.wnl.0000263217.36439.da
Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E et al (2015) Clearance systems in the brain—implications for Alzheimer disease. Nat Rev Neurol 11:457–470. https://doi.org/10.1038/nrneurol.2015.119
Taylor X, Cisternas P, You Y, You Y, Xiang S, Marambio Y et al (2020) A1 reactive astrocytes and a loss of TREM2 are associated with an early stage of pathology in a mouse model of cerebral amyloid angiopathy. J Neuroinflammation 17:223. https://doi.org/10.1186/s12974-020-01900-7
Taylor X, Clark IM, Fitzgerald GJ, Oluoch H, Hole JT, DeMattos RB et al (2023) Amyloid-β (Aβ) immunotherapy induced microhemorrhages are associated with activated perivascular macrophages and peripheral monocyte recruitment in Alzheimer’s disease mice. Mol Neurodegener 18:1–16. https://doi.org/10.1186/s13024-023-00649-w
Thal DR, Gawor K, Moonen S (2024) Regulated cell death and its role in Alzheimer’s disease and amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 147:69. https://doi.org/10.1007/s00401-024-02722-0
Tzioras M, Daniels MJD, Davies C, Baxter P, King D, McKay S et al (2023) Human astrocytes and microglia show augmented ingestion of synapses in Alzheimer’s disease via MFG-E8. Cell Rep Med 4:101175. https://doi.org/10.1016/j.xcrm.2023.101175
Uekawa K, Hattori Y, Ahn SJ, Seo J, Casey N, Anfray A et al (2023) Border-associated macrophages promote cerebral amyloid angiopathy and cognitive impairment through vascular oxidative stress. Mol Neurodegener 18:1–20. https://doi.org/10.1186/s13024-023-00660-1
Ungerleider K, Beck J, Lissa D, Turnquist C, Horikawa I, Harris BT et al (2021) Astrocyte senescence and SASP in neurodegeneration: tau joins the loop. Cell Cycle 20:752–764. https://doi.org/10.1080/15384101.2021.1909260
United Nations (2022) World population prospects Highlights, 2019 revision Highlights, 2019 revision
Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S et al (2019) A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 22:1021–1035. https://doi.org/10.1038/s41593-019-0393-4
Vaquer-Alicea J, Diamond MI (2019) Propagation of protein aggregation in neurodegenerative diseases. Annu Rev Biochem 88:785–810. https://doi.org/10.1146/annurev-biochem-061516-045049
Vasile F, Dossi E, Rouach N (2017) Human astrocytes: structure and functions in the healthy brain. Brain Struct Funct 222:2017–2029. https://doi.org/10.1007/s00429-017-1383-5
Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D et al (2017) Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552:355–361. https://doi.org/10.1038/nature25158
Verdile G, Gandy SE, Martins RN (2007) The Role of Presenilin and its Interacting Proteins in the Biogenesis of Alzheimer’s Beta Amyloid. Neurochem Res 32:609–623. https://doi.org/10.1007/s11064-006-9131-x
Verkhratsky A, Butt A, Li B, Illes P, Zorec R, Semyanov A et al (2023) Astrocytes in human central nervous system diseases: a frontier for new therapies. Signal Transduct Target Ther 8:1–37. https://doi.org/10.1038/s41392-023-01628-9
Verkhratsky A, Zorec R, Parpura V (2017) Stratification of astrocytes in healthy and diseased brain. Brain Pathol Zurich Switz 27:629–644. https://doi.org/10.1111/bpa.12537
Viejo L, Noori A, Merrill E, Das S, Hyman BT, Serrano-Pozo A (2022) Systematic review of human post-mortem immunohistochemical studies and bioinformatics analyses unveil the complexity of astrocyte reaction in Alzheimer’s disease. Neuropathol Appl Neurobiol 48:e12753. https://doi.org/10.1111/nan.12753
Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LBJ, Tiwari-Woodruff S et al (2009) Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci 29:11511–11522. https://doi.org/10.1523/JNEUROSCI.1514-09.2009
Wachter A, Woodbury ME, Lombardo S, Abdourahman A, Wuest C, McGlame E et al (2024) Landscape of brain myeloid cell transcriptome along the spatiotemporal progression of Alzheimer’s disease reveals distinct sequential responses to Aβ and tau. Acta Neuropathol (Berl) 147:1–20. https://doi.org/10.1007/s00401-024-02704-2
Walker DG, Link J, Lue L-F, Dalsing-Hernandez JE, Boyes BE (2006) Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J Leukoc Biol 79:596–610. https://doi.org/10.1189/jlb.0705377
Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W et al (2007) Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol 20:947–956. https://doi.org/10.1159/000110455
Wang C, Fan L, Khawaja RR, Liu B, Zhan L, Kodama L et al (2022) Microglial NF-κB drives tau spreading and toxicity in a mouse model of tauopathy. Nat Commun 13:1969. https://doi.org/10.1038/s41467-022-29552-6
Wang H, Luo W, Zhang Y, Li Y-M, Thinakaran G, Greengard P et al (2004) Presenilins and γ-secretase inhibitors affect intracellular trafficking and cell surface localization of the γ-secretase complex components*. J Biol Chem 279:40560–40566. https://doi.org/10.1074/jbc.M404345200
Wang L, Nykänen N-P, Western D, Gorijala P, Timsina J, Li F et al (2024) Proteo-genomics of soluble TREM2 in cerebrospinal fluid provides novel insights and identifies novel modulators for Alzheimer’s disease. Mol Neurodegener 19:1. https://doi.org/10.1186/s13024-023-00687-4
Wang P, Ye Y (2021) Filamentous recombinant human Tau activates primary astrocytes via an integrin receptor complex. Nat Commun 12:95. https://doi.org/10.1038/s41467-020-20322-w
Wang T, Perera ND, Chiam MDF, Cuic B, Wanniarachchillage N, Tomas D et al (2020) Necroptosis is dispensable for motor neuron degeneration in a mouse model of ALS. Cell Death Differ 27:1728–1739. https://doi.org/10.1038/s41418-019-0457-8
Wang X, Deckert M, Xuan NT, Nishanth G, Just S, Waisman A et al (2013) Astrocytic A20 ameliorates experimental autoimmune encephalomyelitis by inhibiting NF-κB- and STAT1-dependent chemokine production in astrocytes. Acta Neuropathol (Berl) 126:711–724. https://doi.org/10.1007/s00401-013-1183-9
Weiskopf K, Schnorr PJ, Pang WW, Chao MP, Chhabra A, Seita J et al (2017) Myeloid cell origins, differentiation, and clinical implications. Myeloid cells in health and disease. Wiley, pp 857–875
Wendt S, Maricos M, Vana N, Meyer N, Guneykaya D, Semtner M et al (2017) Changes in phagocytosis and potassium channel activity in microglia of 5xFAD mice indicate alterations in purinergic signaling in a mouse model of Alzheimer’s disease. Neurobiol Aging 58:41–53. https://doi.org/10.1016/j.neurobiolaging.2017.05.027
Wheeler MA, Clark IC, Lee H-G, Li Z, Linnerbauer M, Rone JM et al (2023) Droplet-based forward genetic screening of astrocyte–microglia cross-talk. Science 379:1023–1030. https://doi.org/10.1126/science.abq4822
Wilson DM, Cookson MR, Van Den Bosch L, Zetterberg H, Holtzman DM, Dewachter I (2023) Hallmarks of neurodegenerative diseases. Cell 186:693–714. https://doi.org/10.1016/j.cell.2022.12.032
Wilson EN, Wang C, Swarovski MS, Zera KA, Ennerfelt HE, Wang Q et al (2024) TREM1 disrupts myeloid bioenergetics and cognitive function in aging and Alzheimer disease mouse models. Nat Neurosci. https://doi.org/10.1038/s41593-024-01610-w
Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S et al (2019) Complement C3 is activated in human ad brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep 28:2111-2123.e6. https://doi.org/10.1016/j.celrep.2019.07.060
Xu J, Chen S, Ahmed SH, Chen H, Ku G, Goldberg MP et al (2001) Amyloid-β peptides are cytotoxic to oligodendrocytes. J Neurosci 21:RC118
Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH et al (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11:251–253. https://doi.org/10.1038/nn2047
Yang F, Zhao K, Zhang X, Zhang J, Xu B (2016) ATP induces disruption of tight junction proteins via IL-1 beta-dependent MMP-9 activation of human blood-brain barrier in vitro. Neural Plast 2016:8928530. https://doi.org/10.1155/2016/8928530
Yang Y, Andersson P, Hosaka K, Zhang Y, Cao R, Iwamoto H et al (2016) The PDGF-BB-SOX7 axis-modulated IL-33 in pericytes and stromal cells promotes metastasis through tumour-associated macrophages. Nat Commun 7:11385. https://doi.org/10.1038/ncomms11385
Yap JKY, Pickard BS, Chan EWL, Gan SY (2019) The role of neuronal NLRP1 inflammasome in alzheimer’s disease: bringing neurons into the neuroinflammation game. Mol Neurobiol 56:7741–7753. https://doi.org/10.1007/s12035-019-1638-7
Ye H, Han Y, Li P, Su Z, Huang Y (2022) The role of post-translational modifications on the structure and function of tau protein. J Mol Neurosci 72:1557–1571. https://doi.org/10.1007/s12031-022-02002-0
Yoshiyama Y, Higuchi M, Zhang B, Huang S-M, Iwata N, Saido TC et al (2007) Synapse loss and microglial activation precede tangles in a P301s tauopathy mouse model. Neuron 53:337–351. https://doi.org/10.1016/j.neuron.2007.01.010
Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X (2021) Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther 6:1–21. https://doi.org/10.1038/s41392-021-00507-5
Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA et al (2013) Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153:707–720. https://doi.org/10.1016/j.cell.2013.03.030
Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S et al (2019) Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci 22:719–728. https://doi.org/10.1038/s41593-019-0372-9
Zhang X, Wang R, Hu D, Sun X, Fujioka H, Chan E et al (2020) Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer’s disease. Sci Adv. https://doi.org/10.1126/sciadv.abb8680
Zhao J, O’Connor T, Vassar R (2011) The contribution of activated astrocytes to Aβ production: implications for Alzheimer’s disease pathogenesis. J Neuroinflammation 8:150. https://doi.org/10.1186/1742-2094-8-150
Zhao J, Paganini L, Mucke L, Gordon M, Refolo L, Carman M et al (1996) β-secretase processing of the β-amyloid precursor protein in transgenic mice is efficient in neurons but inefficient in astrocytes *. J Biol Chem 271:31407–31411. https://doi.org/10.1074/jbc.271.49.31407
Zhao J, Wu H, Tang X (2021) Tau internalization: a complex step in tau propagation. Ageing Res Rev 67:101272. https://doi.org/10.1016/j.arr.2021.101272
Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR et al (2020) Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26:131–142. https://doi.org/10.1038/s41591-019-0695-9
Zhu K, Liang W, Ma Z, Xu D, Cao S, Lu X et al (2018) Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis 9:1–16. https://doi.org/10.1038/s41419-018-0524-y
Zindel J, Kubes P (2020) DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol 15:493–518. https://doi.org/10.1146/annurev-pathmechdis-012419-032847
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Botella Lucena, P., Heneka, M.T. Inflammatory aspects of Alzheimer’s disease. Acta Neuropathol 148, 31 (2024). https://doi.org/10.1007/s00401-024-02790-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00401-024-02790-2