
Abstract
Objective
This pilot study aimed to determine the efficacy of acamprosate (N-acetyl homotaurine) in reducing the pathological features of experimental autoimmune encephalomyelitis (EAE) which is an animal model for multiple sclerosis (MS).
Background
The amino acid taurine has multiple biological activities including immunomodulation and neuromodulation. The synthetic acetylated taurine derivative, acamprosate, which crosses the blood–brain barrier more readily compared to taurine, is currently being used for the prevention of alcohol withdrawal symptoms associated with enhanced glutamatergic receptor function and GABA receptor hypofunction.
Methods
EAE was induced in C57BL/6 female mice with myelin oligodendrocyte glyocoprotein, amino acid 35–55. Mice were treated with 20, 100 and 500 mg/kg acamprosate for 21 days.
Results
Neurological scores at disease peak were reduced by 21, 64 and 9% in the 20, 100 and 500 mg/kg groups, respectively. Neurological improvement in the 100 mg/kg group correlated with a reduction in numbers of inflammatory lesions and the extent of CNS demyelination. Blood TNF-α levels were significantly reduced in the 500 mg/kg group.
Discussion
Acamprosate and other taurine analogs have a potential for future MS therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is a demyelinating and neurodegenerative disorder mediated in part by the autoreactive T lymphocytes proinflammatory responses to myelin antigens in the central nervous system (CNS) (Weiner 2009). Furthermore, studies show that the N-methyl d-aspartate (NMDA) and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor (Newcombe et al. 2008) and their ligand glutamate (Srinivasan et al. 2005) are upregulated in the MS brain and plays a role in neurodegenerative damage in the course of MS (Centonze et al. 2010). Glutamate receptor antagonists have been shown to exert beneficial effects in the experimental autoimmune encephalomyelitis (EAE) model of MS (Pitt et al. 2000; Smith et al. 2000; Wallstrom et al. 1996; Bolton and Paul 1997) by limiting both oligodendrocyte and neuronal damage (Centonze et al. 2009, 2010; Pitt et al. 2000).
In addition, a lower than normal level of the neurotransmitter gamma aminobutyric acid (GABA) has been shown in the cerebrospinal fluid (CSF) of MS patients (Manyam et al. 1980) while increasing GABAergic activity led to improvement of EAE-induced paralysis (Bhat et al. 2010). These studies collectively suggest an imbalance between neuroexcitatory glutamate axis and neuroinhibitory GABA axes in MS and calls for therapeutic modalities with agents that exert not only immunomodulatory effects but also improve neuroimmunomodulatory balance of the NMDA/GABA axes.
Taurine, an amino acid, is such an agent with both immunomodulatory and neuromodulatory effects. In vitro studies show that taurine binds to the hypochlorous acid forming taurine–chloramine (tau–Cl) (Schaffer et al. 2003) which has the potential to inhibit the activation of nuclear factor kappa B (NF-kB) (Kanayama et al. 2002) and the expression of proinflammatory mediators including that of tumor necrosis factor-α (TNF-α) (Barua et al. 2001). In addition, taurine has been shown to exert neuroprotective effects by both inhibiting the action of the excitatory neurotransmitter glutamate in inducing calcium influx and NMDA receptor activation (Wu et al. 2009) and by binding to and activating the receptor for the inhibitory neurotransmitter GABA (Wang et al. 2007). Despite its beneficial effects, the use of taurine as an orally administered therapeutic agent is hampered due to its unfavorable levels attained in the CNS (Gupta et al. 2005).
Therefore, this study investigated the effect of the taurine derivative, acamprosate (calcium bis acetyl homotaurine), a structural analog of GABA on the course of EAE with a relevance to MS. The acetylation and calcium salification to form acamprosate facilitates the passage of taurine across the blood–brain barrier (Nalpas et al. 1990) with a potential to exert local effects in the CNS.
Acamprosate is a FDA approved drug for treating alcohol withdrawal symptoms in individuals with alcohol dependency (Mann et al. 2004). Symptoms of alcohol withdrawal in chronic alcoholics are mainly associated with the enhanced function of glutamatergic receptors and the hypofunction of GABA receptors (De Witte 2004). In long-term alcoholics who experience repeated cycles of withdrawal and relapse, chronic glutamatergic system hyperactivation may lead to both gray and white matter damage as well as to brain atrophy (Kril and Halliday 1999).
Acamprosate shares taurine characteristics regarding the modulation of the NMDA/GABA axes. Acamprosate interacts with the NMDA receptors reducing Ca2+ flux through the voltage-gated channels (Spanagel and Zieglgansberger 1997) as well as reducing postsynaptic NMDA-induced neuronal excitation (Zeise et al. 1993) in the rat’s brain. In addition, acamprosate has been shown to enhance the GABAA-mediated inhibition of spontaneous rhythmic activity of hypoglossal nerve rootlet (XII) in rat brainstem slices (Pierrefiche et al. 2004). Therefore, acamprosate has the potential to modify the NMDA/GABA balance.
This study investigated the effect of 3 doses of acamprosate (20, 100 and 500 mg/kg) on the course of EAE. The decision to use multiple doses is based on the results of a study showing a biphasic effect of acamprosate on the NMDA receptor. Acamprosate, at low concentrations, enhances the NMDA receptor activation when receptor activity is low but at high concentrations, the drug inhibits the NMDA receptor activation when the NMDA receptor activity is high (Dahchour and De Witte 2000).
Materials and methods
Reagents
Acamprosate was prepared from tablets of Campral ground into a fine powder. The Rat MOG35–55 peptide was obtained from UCLA. Pertussis toxin was obtained from List Biological Laboratories (Campbell, CA, USA). Complete Freunds adjuvant (CFA) (H37Ra) containing 10 mg heat-inactivated Mycobacterium tuberculosis was obtained from Voigt Global Distribution Inc. (Lawrence, KS). LPS (lot#: 050M4100, Escherichia coli strain 0111:B4) was obtained from Sigma-Aldrich (St. Louis, MO). Recombinant mouse OptEIA™ Rat TNF-α ELISA kit was purchased from BD Pharmingen (Franklin Lakes, NJ). Lithium heparin microtainers were purchased from BD Biosciences (Franklin Lakes, NJ).
Mice
Studies were performed with 6- to 8-week-old C57BL/6 female mice (Jackson Laboratory, Bar Harbor, ME). Standard housing conditions were implemented. All animals were kept at an ambient temperature of 20–22°C with a 12-h light/dark cycle and food was available ad libitum. Animal use and protocol procedures were approved by the Institutional Animal Care and Use Committee of the University of Buffalo.
EAE induction and neurological evaluation
Mice were immunized by subcutaneous injection of 250 μg MOG35–55 emulsified in PBS and CFA 1:1 volume in 2 adjacent flanks on day 0 with a similar booster 7 days later. Pertussis toxin, 250 ng, was injected intraperitoneally on the initial day of immunization and again on day 2. Each mouse body weight (BW) and neurological score was recorded daily over the duration of 21 days. The scoring was done by a trained individual blinded to the treatment group. The following grading scheme was adopted to score the neurological severity of the EAE in mice: 0, no neurological signs; 0.5, hook tail; 1, flaccid/floppy tail; 2, beginning of walking deficits; 3, hind limb paresis; 4, partial hind limb paralysis; 5, complete hind limb paralysis; 6, complete hind limb and fore limb paralysis. Walking speed and righting response time were taken into consideration to increase scoring accuracy.
Oral administration of acamprosate
Acamprosate was solubilized in distilled water. Thirty-six mice were randomly assigned to four groups of nine each. Acamprosate in total volume of 200 μl was administered to the three treatment groups in doses of 10, 50 and 250 mg/kg each, by oral gavage, twice daily (morning and evening). The control group of nine mice received equal volume of the vehicle (distilled water) twice daily. Acamprosate’s administration started on the day of disease induction (day 0) until completion of the study on d-21, at which point the animals were killed.
Furthermore, in order to examine the effect of acamprosate on mice in the absence of the EAE, an additional group of 12 healthy mice was randomly assigned to two groups of 6 each, and was administered either the vehicle or 500 mg/kg acamprosate in the absence of EAE induction.
Tumor necrosis factor-α assay in whole blood
On the d-19 PI, blood was collected for the measurement of the acamprosate’s effect on TNF-α. Since TNF-α is a proinflammatory cytokine, its measurement would be an indicator for the acamprosate’s anti-inflammatory effect. TNF-α was measured in the following 6 groups, EAE mice treated with (1) the vehicle (distilled water), (2) EAE mice treated 20 mg/kg, (3) EAE mice treated with 100 mg/kg, (4) EAE mice treated with 500 mg/kg, (5) healthy control (HC) mice treated with vehicle, and (6) and HC mice treated with acamprosate 500 mg/kg.
Mice were lightly anesthetized under isofluorene gas and bled retro-orbitally. Approximately 150 μl of blood was collected from each mouse into a microtainer containing lithium heparin. Blood cells were stimulated with a 10 μg/5 μl of LPS for 4 h at 37°C in humidified air containing 5% CO2. Subsequently, plasma was collected and stored in −80°C until assayed. The level of TNF-α in the plasma was assessed using mouse recombinant ELISA kit, following manufacturer’s instructions. The absorbance was measured at 450 nm in a μQuant Microplate reader system (BioTek Instruments, Inc).
Histology
At the termination of the experiment on d-21, mice were deeply anesthetized using ketamine (100 mg/kg) and xylazine (10 mg/kg). Brains and spinal cords were removed and embedded routinely in paraffin. Sections of 5 μm each were stained with Luxol fast blue-H&E (LHE) to demonstrate inflammatory infiltrates and demyelination. Inflammatory foci (>10 perivascular cells) were counted in brain parenchyma and meninges by a neuropathologist blinded to mice’s treatments.
Statistics
Statistical software Systat 14 was used to analyze the results on BW, neurological score, TNF-α and CNS pathology. Mean values from each experiment were compared using a one-way analysis of variance (ANOVA) followed by Newman–Keuls multiple comparisons tests. Values are mean ± SEM in each mice treatment group, *p ≤ 0.05 was considered significant.
Results
Acamprosate reduces EAE neurological scores
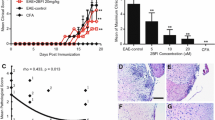
EAE mice treated with vehicle presented early neurological deficits such as hooked tail on d-13 PI which later increased in severity, peaking on d-17 PI. Acamprosate at doses 20, 100 and 500 mg/kg reduced the EAE neurological scores when compared to the treatment with the vehicle alone. When compared to the vehicle treated group, acamprosate at a dose of 20 mg/kg lowered EAE neurological scores starting on d-14 PI, reaching near statistical significance on d-21 PI (1.6 ± 0.5 vs. 2.66 ± 0.4, reduction of 38% in neurological scores, p = 0.09). Acamprosate at dose of 100 mg/kg significantly reduced neurological scores starting on d-16 until completion of the study on d-21 PI (0.089 ± 0.3 vs. 2.66 ± 0.4, reduction of 66.6% in neurological scores, ps between 0.01 and 0.001). Acamprosate at a dose of 500 mg/kg significantly reduced neurological scores from d-14 PI up to d-16 PI (ps between <0.001 and 0.05) (Fig. 1).

Dose-dependent effects of acamprosate on the daily average neurological scores. A significant reduction in EAE neurological scores was observed for 100 mg/kg acamprosate between d-16 and d-21 PI and for 500 mg/kg acamprosate between d-14 and d-16 PI, when compared to the vehicle-treated group. Each line represents mean average clinical scores ± SEM for 9 mice. Results were considered significant for *p ≤ 0.05. PI postimmunization
A comparison between acamprosate-treated and vehicle-treated mice at the peak of the disease (d-17 PI for the vehicle-treated group, d-18 PI for the 20 mg/kg group, d-19 PI for the 100 mg/kg group, d-20 PI for the 500 mg/kg group), observed a reduction of 21% (2.55 ± 0.70 vs. 3.22 ± 0.73, p = 0.5), 64% (1.16 ± −0.33 vs. 3.22 ± 0.73, p = 0.02) and 9% (2.93 ± 0.78 vs. 3.22 ± 0.73, p = 0.7) in the average mean EAE neurological scores in 20 mg/kg, 100 mg/kg and 500 mg/kg groups of mice, respectively (Table 1). When the results of mice which did not develop the disease were excluded from the analysis, the neurological scores for the 100 mg/kg dose remained statistically significant (p = 0.03).
Doses of 20, 100 and 500 mg/kg acamprosate reduced mean cumulative disease score (the area under the curve), by 34% (102.6 ± 28.9 vs. 155.3 ± 31.3, p = 0.2), 72% (43.7 ± 27.7 vs. 155.3 ± 31.3, p = 0.01) and 48% (80.4 ± 26.7 vs. 155.3 ± 31.3, p = 0.09), respectively, when compared to the vehicle-treated group (Table 1).
At the completion of the study on d-21 PI, 7/9 (77%) mice in the vehicle-treated group had neurological scores of ≥2 which indicated neurological deficits above floppy tail. This value was reduced to 4/9 (44%), 1/9 (11%) and 5/9 (50%) in group of mice treated with 20, 100 and 500 mg/kg acamprosate, respectively.
Acamprosate delays the appearance of neurological deficits and reduces the rate of EAE induction
Acamprosate delayed the appearance of neurological deficits in mice, when compared to the vehicle-treated group. The mean delay calculated from the day of the appearance of the first neurological deficits (recorded as scale of 0.5 in each mouse) was 14.7 ± 0.61 days versus 16.0 ± 1.0 days, p = 0.3, 14.7 ± 0.61 days versus 17.6 ± 0.88 days, p = 0.01 and 14.7 ± 0.61 days versus 17.1 ± 0.69 days, p = 0.02 in the 20, 100 and 500 mg/kg groups, respectively (Table 1). As a result, the mean percent of disease-free mice (from d-13 PI to d-21 PI) was higher in the group of mice treated with 20 mg/kg (35.7 ± 7.3 vs. 19.7 ± 8.0%, p = 0.1), 100 mg/kg (54.2 ± 9.7 vs. 19.7 ± 8.0%, p = 0.02) and 500 mg/kg (54.1 ± 13.1 vs. 19.7 ± 8.0%, p = 0.04) compared to the vehicle-treated group (Table 1).
In addition, at the completion of the study on d-21 PI, a reduction of 23% (2/9, p = 0.15) in MOG-induced EAE incidence was observed in groups of mice treated with 20 mg/kg and 100 mg/kg acamprosate, while the incidence of disease reached 100% (9/9) in the vehicle-treated group and the 500 mg/kg acamprosate-treated group (Table 1).
Acamprosate reduces EAE-induced body weight loss
The effect of acamprosate and EAE and on BW was determined by calculating the percent BW gain/loss relative to d-0 PI and d-13 PI, respectively. Since both acamprosate and EAE-influenced BW, for reasons of clarity, a biphasic analysis of BW was conducted measuring the percent of BW gain/loss from d-0 PI to d-12 PI (Fig. 2a) (before the appearance of neurological deficits) and from d-13 PI to d-21 PI (Fig. 2b). It is noteworthy that BW gain was calculated from d-13 PI since the average weight gain peaked on this day, despite the appearance of neurological deficit in one mouse in the vehicle-treated group.

Effects of acamprosate and EAE on body weight (BW). A biphasic analysis of BW was conducted. The percent BW gain/loss was calculated between d-1 to -12 PI (before the appearance of neurological deficits) (a) representing the effect of acamprosate, and between d-13 to -21 PI (after the appearance of neurological deficits) (b), representing EAE-induced BW loss. a Acamprosate suppresses BW gain. The effect of acamprosate on BW was determined by calculating the percent BW gain/loss relative to d-0 PI (representing 100% BW). The figure shows a significant BW gain in the group of mice treated with vehicle; this BW gain was suppressed in the acamprosate-treated mice, more markedly in the 500 mg/kg group of mice. Each line represents average percent BW gain/loss ± SEM relative to d-0 PI for 9 mice. The asterisks indicate significant BW gain in the vehicle treated group. Results were considered significant for *p ≤ 0.05. Abbreviation: as in Fig. 1. b EAE induces BW loss. The percent BW changes were calculated from d-13 PI (considered as 100% BW). The group of mice treated with the vehicle shows no significant EAE-related BW change. The group of mice treated with 20 mg/kg acamprosate shows early and significant BW changes, while the 500 mg/kg group had a relatively slower BW changes. The asterisks indicate the days of significant BW changes relative to d-13 PI. *p ≤ 0.05 was considered statistically significant. BW body weight, PI postimmunization
Between d-0 PI and d-8 PI mice of all groups experienced a degree of BW loss associated with the stress of injections required to induce EAE. The comparison of BW between d-0 PI to d-12 PI showed lack of BW gain in groups of mice treated with 20 mg/kg (1% BW loss, from 17.7 ± 0.3 g to 17.5 ± 0.3 g), 100 mg/kg (0% BW loss, from 17.4 ± 0.3 to 17.4 ± 0.5 g,) and 500 mg/kg (3.3% BW loss, from 18.1 ± 0.2 to 17.5 ± 0.4 g) acamprosate, whereas the vehicle-treated group had significant BW gain (7.1 ± 2.9% BW gain, from 16.9 ± 0.5 g to 18.1 ± 0.3 g, p = 0.04) (Fig. 2a). The 500 mg/kg acamprosate had no significant effect on BW gain in mice in the absence of EAE (results not shown).
Figure 2b shows the percent BW change relative to d-13 PI for each group. Day 13 PI was considered as 100% BW. The group of mice treated with 100 mg/kg acamprosate showed no significant EAE-related BW change; the 500 mg/kg group showed slower BW change. The delay in the EAE-induced BW change was consistent with the delay in the appearance of neurological deficits in the group treated with 500 mg/kg acamprosate. Mice in the vehicle-treated group lost an average of 8.5% (from 18.1 ± 0.3 to 16.6 ± 0.5 g, p = 0.01) of their BW (from d-13 PI to d-16 PI). This value stood at 9% (from 17.9 ± 0.3 to 16.2 ± 0.5 g, p = 0.008) (from d-13 PI to d-17 PI), 0.2% (17.7 ± 0.5–17.5 ± 0.5 g, p = 0.9) (from d-13 PI to d-14 PI), and 6% (17.9 ± 0.4–16.9 ± 0.8 g, p = 0.1) (from d-13 PI to d-19 PI), for groups of mice treated with 20, 100 and 500 mg/kg, respectively.
Acamprosate reduces LPS-induced TNF-α release in whole blood
The plasma level of LPS-induced TNF-α release in whole blood was measured on d-19 PI. Since TNF-α is a proinflammatory cytokine, its measurement would be an indicator for acamprosate’s modulation of inflammatory responses. The plasma level of LPS-induced TNF-α was significantly higher in vehicle-treated EAE mice (1,833.3 ± 117.6 pg) compared to HC mice (1,432 ± 87.2 pg, p = 0.03). TNF-α release was reduced in the group of EAE mice treated with 20 mg/kg (1,499 ± 119 pg, p = 0.07), 100 mg/kg (1,552.4 ± 177 pg, p = 0.1) and 500 mg/kg (1,318 ± 215.5 pg, p = 0.05) acamprosate compared to the vehicle-treated group. TNF-α level was similar between HC mice treated with the vehicle (1,448 ± 126 pg) and 500 mg/kg acamprosate (1,426 ± 103 pg) (Fig. 3).

Dose-dependent effects of acamprosate on LPS-stimulated TNF-α plasma levels. TNF-α plasma levels were measured on d-19 PI, in mice with EAE, treated with the vehicle, those treated with acamprosates in doses of 20, 100 or 500 mg/kg (n = 9 in each group), and HC mice w/0 500 mg/kg acamprosate (n = 6 in each group). TNF-α plasma level was significantly higher in vehicle-treated EAE mice compared to HC mice w/0 500 mg/kg acamprosate. The treatment with acamprosate normalized the TNF-α plasma level toward the level observed in HC mice, reaching statistical significance with 500 mg/kg acamprosate. Differences were considered significant at *p ≤ 0.05. HC healthy control, TNF-α tumor necrosis factor-α, w/0 with and without
Acamprosate reduces inflammatory foci and demyelination
Histopathological evaluation was performed in mice from the vehicle group and from 100 mg/kg acamprosate, the dose of the drug which showed significant efficacy in lowering neurological deficits. Mononuclear cell infiltrates were observed in the leptomeninges and parenchyma in the vehicle-treated mice which were accompanied with loss of blue staining which indicated demyelination (Fig. 4a, b). However, 100 mg/kg acamprosate reduced the total level of inflammatory foci measured in the meninges and parenchyma by 85% (30.0 ± 9.2 vs. 4.7 ± 1.8, p = 0.01) and significantly reduced the extent of demyelination (Fig. 4a, b). We did not observe inflammation or demyelination in HC mice and mice treated with 500 mg/kg acamprosate in the absence of EAE (data not shown).

Acamprosate reduces inflammatory foci spinal cord histopathology. a Inflammatory foci. Histopathological evaluations were performed on mice from the vehicle group and from the group treated with 100 mg/kg acamprosate, the dose of the drug which showed significant efficacy in lowering neurological deficits. Each bar represents mean meningeal and parenchymal inflammatory foci ± SEM for 9 mice. *p ≤ 0.05. b Spinal cord pathology. Representative sections of posterior spinal cord from vehicle-treated and 100 mg/kg acamprosate-treated mice. Mononuclear cell infiltrates are present in the leptomeninges and parenchyma in the panel on the left. Loss of blue staining indicates demyelination. Posterior columns are intact and stained blue in the treated mouse. Luxol fast blue-H&E stain. Bar 50 μm. HC mice and mice treated with 500 mg/kg acamprosate, in the absence of EAE, showed no inflammation or demyelination (data not shown)
Discussion
This is the first study examining the efficacy of three doses of acamprosate in improving the course of the EAE with a relevance to MS. The selected doses of 20, 100 and 500 mg/kg were based on earlier studies of rodent models of alcohol dependence and morphine and nicotine seeking behaviors (Czachowski and Delory 2009; Bowers et al. 2007; McGeehan and Olive 2003) and were, respectively, equivalent to 0.7 time, 3.5 times and 17.7 times the therapeutic dose of 2 g/day (equivalent to 28.5 mg/kg) acamprosate in an average 70 kg human subject. Earlier studies (Czachowski and Delory 2009; Bowers et al. 2007; McGeehan and Olive 2003) used acamprosate in wide ranges of 30–500 mg/kg. However, consistent with our results, the observed acamprosate’s efficacy in reducing alcohol craving and morphine and nicotine seeking behavior was mostly observed in doses of ≥100 mg/kg. Similarly, in animal model of stroke, intraperitoneal administration of 200 mg/kg acamprosate was required to improve neurological deficits following transient hemispheric ischemia (Engelhard et al. 2000).
Among factors for the acamprosate’s suboptimal efficacy at low doses is the poor intestinal absorption observed in both humans (11%) (Saivin et al. 1998) and rodents (20%) (Zornoza et al. 2006) as well as considerably faster elimination rate compared to its absorption rate (Zornoza et al. 2006). The poor intestinal absorption and the slow absorption rate stem in part from the passive diffusion as being the main route for the intestinal absorption of the drug (Mas-Serrano et al. 2000). Therefore, the protocol was adapted to include, twice daily (morning and evening) acamprosate administration to increase drug availability. The 10, 50 and 250 mg/kg acamprosate administered at each of the two time points was equivalent to 0.35 time, 1.75 times and 8.75 times the therapeutic dose of 2 g/day in humans, respectively.
An additional factor for the acamprosate’s suboptimal efficacy at low doses is the poor passage through the blood–brain barrier. Less than 3% of the absorbed drug ever reaches the brain despite the presence of the acetyl moiety (Courtyn et al. 2004). As a result, repeated administrations of relatively high doses of 300 mg/kg acamprosate are necessary to augment the levels of the drug in the brain relative to the baseline levels (Burattini et al. 2008).
It should be noted that the administration of as high as 4 g/day to human volunteers has not been shown to be associated with adverse events (Brasser et al. 2004) suggesting low toxicity of the drug. The 4 g/day dose (equivalent to 57 mg/kg in humans), approximates the 100 mg/kg, effective dose to reduce neurological deficits in the EAE model.
Nevertheless, one should refrain from translating drug dosage from animals to humans based on mg/kg since other factors including body surface area, blood volume, renal function and differences in bioavailability and metabolism between the two species could significantly influence the therapeutic dose in humans compared to that in other species (Reagan-Shaw et al. 2008).
Although 100 mg/kg acamprosate more effectively reduced the mean severity of the EAE neurological deficits, when compared to 20 and 500 mg/kg doses, both the 20 and 100 mg/kg doses were equally effective in reducing the incidence of the EAE induction. Furthermore, similar to the 100 mg/kg dose, the 500 mg/kg dose of acamprosate was effective in delaying disease induction and reducing mean cumulative disease scores. The relatively high dose of acamprosate necessary for reducing the neurological scores may be related to the results of an in vitro study showing the requirement for a relatively high dose (200 μM) acamprosate to modify both the NMDA and GABA axes, while the low dose of 30 μM acamprosate modified only the NMDA axis (Pierrefiche et al. 2004). These results suggest the importance of the balance between the NMDA axis and GABA axis in reducing the EAE disease severity.
The effects of acamprosate on the EAE neurological scores were also time-dependent. For the first 16 days, the 100 and 500 mg/kg acamprosate doses had similar effects on EAE severity scores, while 21 days of administration of 20 mg/kg acamprosate was required to reduce the severity of EAE scores to values resembling those of 100 mg/kg. These results suggest that a shorter duration of high doses of acamprosate or a longer duration of low doses of acamprosate is required for optimum beneficial effects, and that long-term administration of high doses of acamprosate may adversely affect EAE disease severity. This assumption is supported by a study (Bowers et al. 2007) showing that repeated administration of 500 mg/kg acamprosate, and not lower doses, had adverse effects reducing the locomotor activity in rats. Therefore, both the dose and the length of acamprosate’s administration can affect the EAE disease severity.
The observed results for the 100 mg/kg dose of acamprosate were consistent with a significant reduction in the number of inflammatory foci and the extent of CNS demyelination. The lack of correlation between the effect of 500 mg/kg dose on TNF-α plasma level and on the severity of the EAE neurological scores suggest that the two observations may not necessarily be related. The effect of acamprosate on inflammatory-related pathways, though monophasic, its effects in multiple other pathways, including NMDA/GABA axis (Pierrefiche et al. 2004) may be complex and multiphasic.
Among acamprosate’s well-characterized pharmacological effects is the modulation of NMDA/GABA axis (Spanagel and Zieglgansberger 1997; Zeise et al. 1993; Pierrefiche et al. 2004). Because of this effect, acamprosate has been suggested as a pharmacological treatment for Amyotrophic lateral sclerosis (ALS) (Kast and Altschuler 2007). In addition, glutamate/GABA imbalance has been shown to be one of the many pathological features of MS (Newcombe et al. 2008; Srinivasan et al. 2005; Centonze et al. 2010) correlating with MRI markers of inflammation and gray matter atrophy (Lenzi et al. 2007; Morgen et al. 2007). Acamprosate’s balancing of glutamate/GABA in the brain may lead to the reduction of the neurological deficits observed in the EAE model. In addition, the increase in GABA may result in augmented brain’s norepinephrine levels, with a potential to exert neuroprotective effects (Simonini et al. 2010). Nevertheless, one can not exclude the possibility that acamprosate may exert immunomodulatory effects by interacting with GABA receptors, known to be expressed on peripheral immune cells (Tian et al. 2004).
Acamprosate has been shown to increase the plasma levels of beta-endorphin in alcoholic rats (Zalewska-Kaszubska et al. 2005) and alcohol-dependent patients (Kiefer et al. 2006) following alcohol withdrawal. In addition, naltrexone, which also increases opioids and opioid receptors functional responses, delays the onset and progression of EAE (Zagon et al. 2009) suggesting that acamprosate may reduce EAE pathology via its effects on opioid system.
The chronic administration of 20–500 mg/kg acamprosate resulted in reduction in BW gain. Acamprosate-induced suppression of BW gain occurred only in the presence of EAE and was unrelated to the EAE-induced weight changes. But the administration of 500 mg/kg acamprosate in the absence of EAE had no effect on BW gain, suggesting that immune activation in the EAE mice may interact with acamprosate to suppress BW gain. Bowers et al. (2007) did not observe an effect of 100 and 300 mg/kg on BW gain in rats, but 500 mg/kg acamprosate did reduce BW gain.
Conclusion
This pilot study shows that the oral administration of acamprosate starting immediately after EAE disease induction (prevention treatment) has the potential to reduce EAE-induced neurological deficits and weight loss.
Further studies are warranted for understanding the mechanism(s) through which acamprosate exerts beneficial effects in the EAE model and whether acamprosate is still effective if administered after the appearance of the neurological deficits (suppression treatment) or during the chronic phase of the disease (delayed suppression). Additional studies will also be required to examine in detail the underlying causes for acamprosate-induced lack of weight gain in EAE.
Despite the observed encouraging results, one should note that although animal models of MS are generally suitable for a better understanding of the immune regulatory processes and histopathological features, the model is less suitable for investigation of new treatment modalities. Only a limited number of drugs have shown efficacy in both the animal model and in clinical trials in MS. Many other compounds, while promising in the EAE model, did not show efficacy in clinical trials in MS (Mix et al. 2010). Therefore, the results of this study should be taken cautiously and further studies will be needed to assess the value of acamprosate as a new treatment modality in MS clinical practice.
Abbreviations
- NMDA:
-
N-Methyl-d-aspartic acid
- GABA:
-
Gamma-aminobutyric acid
- EAE:
-
Experimental autoimmune encephalomyelitis
- MOG:
-
Myelin oligodendrocyte glycoprotein
- CFA:
-
Complete Freund’s adjuvant
- ELISA:
-
Enzyme-linked immunosorbent assay
- LPS:
-
Lipopolysaccharide
- PBS:
-
Phosphate buffer saline
- TNF-α:
-
Tumor necrosis factor-α
- PI:
-
Postimmunization
References
Barua M, Liu Y, Quinn MR (2001) Taurine chloramine inhibits inducible nitric oxide synthase and TNF-alpha gene expression in activated alveolar macrophages: decreased NF-kappaB activation and IkappaB kinase activity. J Immunol 167:2275–2281
Bhat R, Axtell R, Mitra A, Miranda M, Lock C, Tsien RW, Steinman L (2010) Inhibitory role for GABA in autoimmune inflammation. Proc Natl Acad Sci USA 107:2580–2585
Bolton C, Paul C (1997) MK-801 limits neurovascular dysfunction during experimental allergic encephalomyelitis. J Pharmacol Exp Ther 282:397–402
Bowers MS, Chen BT, Chou JK, Osborne MP, Gass JT, See RE, Bonci A, Janak PH, Olive MF (2007) Acamprosate attenuates cocaine- and cue-induced reinstatement of cocaine-seeking behavior in rats. Psychopharmacology (Berl) 195:397–406
Brasser SM, McCaul ME, Houtsmuller EJ (2004) Alcohol effects during acamprosate treatment: a dose-response study in humans. Alcohol Clin Exp Res 28:1074–1083
Burattini C, McGeehan AJ, Griffin WC 3rd, Gass JT, Kinder JR, Janak PH, Olive MF (2008) A microdialysis study of extracellular levels of acamprosate and naltrexone in the rat brain following acute and repeated administration. Addict Biol 13:70–79
Centonze D, Muzio L, Rossi S, Cavasinni F, De Chiara V, Bergami A, Musella A, D’Amelio M, Cavallucci V, Martorana A, Bergamaschi A, Cencioni MT, Diamantini A, Butti E, Comi G, Bernardi G, Cecconi F, Battistini L, Furlan R, Martino G (2009) Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J Neurosci 29:3442–3452
Centonze D, Muzio L, Rossi S, Furlan R, Bernardi G, Martino G (2010) The link between inflammation, synaptic transmission and neurodegeneration in multiple sclerosis. Cell Death Differ 17:1083–1091
Courtyn J, Cornelissen B, Oltenfreiter R, Vandecapelle M, Slegers G, Strijckmans K (2004) Synthesis and assessment of [11C]acetylhomotaurine as an imaging agent for the study of the pharmacodynamic properties of acamprosate by positron emission tomography. Nucl Med Biol 31:649–654
Czachowski CL, Delory MJ (2009) Acamprosate and naltrexone treatment effects on ethanol and sucrose seeking and intake in ethanol-dependent and nondependent rats. Psychopharmacology (Berl) 204:335–348
Dahchour A, De Witte P (2000) Ethanol and amino acids in the central nervous system: assessment of the pharmacological actions of acamprosate. Prog Neurobiol 60:343–362
De Witte P (2004) Imbalance between neuroexcitatory and neuroinhibitory amino acids causes craving for ethanol. Addict Behav 29:1325–1339
Engelhard K, Werner C, Lu H, Mollenberg O, Zieglgansberger W, Kochs E (2000) The neuroprotective effect of the glutamate antagonist acamprosate following experimental cerebral ischemia. A study with the lipid peroxidase inhibitor u-101033e. Anaesthesist 49:816–821
Gupta RC, Win T, Bittner S (2005) Taurine analogues; a new class of therapeutics: retrospect and prospects. Curr Med Chem 12:2021–2039
Kanayama A, Inoue J, Sugita-Konishi Y, Shimizu M, Miyamoto Y (2002) Oxidation of Ikappa Balpha at methionine 45 is one cause of taurine chloramine-induced inhibition of NF-kappa B activation. J Biol Chem 277:24049–24056
Kast RE, Altschuler EL (2007) Consideration of acamprosate for treatment of amyotrophic lateral sclerosis. Med Hypotheses 69:836–837
Kiefer F, Jahn H, Otte C, Nakovics H, Wiedemann K (2006) Effects of treatment with acamprosate on beta-endorphin plasma concentration in humans with high alcohol preference. Neurosci Lett 404:103–106
Kril JJ, Halliday GM (1999) Brain shrinkage in alcoholics: a decade on and what have we learned? Prog Neurobiol 58:381–387
Lenzi D, Conte A, Mainero C, Frasca V, Fubelli F, Totaro P, Caramia F, Inghilleri M, Pozzilli C, Pantano P (2007) Effect of corpus callosum damage on ipsilateral motor activation in patients with multiple sclerosis: a functional and anatomical study. Hum Brain Mapp 28:636–644
Mann K, Lehert P, Morgan MY (2004) The efficacy of acamprosate in the maintenance of abstinence in alcohol-dependent individuals: results of a meta-analysis. Alcohol Clin Exp Res 28:51–63
Manyam NV, Katz L, Hare TA, Gerber JC 3rd, Grossman MH (1980) Levels of gamma-aminobutyric acid in cerebrospinal fluid in various neurologic disorders. Arch Neurol 37:352–355
Mas-Serrano P, Granero L, Martin-Algarra RV, Guerri C, Polache A (2000) Kinetic study of acamprosate absorption in rat small intestine. Alcohol Alcohol 35:324–330
McGeehan AJ, Olive MF (2003) The anti-relapse compound acamprosate inhibits the development of a conditioned place preference to ethanol and cocaine but not morphine. Br J Pharmacol 138:9–12
Mix E, Meyer-Rienecker H, Hartung HP, Zettl UK (2010) Animal models of multiple sclerosis—potentials and limitations. Prog Neurobiol 92:386–404
Morgen K, Sammer G, Courtney SM, Wolters T, Melchior H, Blecker CR, Oschmann P, Kaps M, Vaitl D (2007) Distinct mechanisms of altered brain activation in patients with multiple sclerosis. Neuroimage 37:937–946
Nalpas B, Dabadie H, Parot P, Paccalin J (1990) Acamprosate. From pharmacology to therapeutics. Encephale 16:175–179
Newcombe J, Uddin A, Dove R, Patel B, Turski L, Nishizawa Y, Smith T (2008) Glutamate receptor expression in multiple sclerosis lesions. Brain Pathol 18:52–61
Pierrefiche O, Daoust M, Naassila M (2004) Biphasic effect of acamprosate on NMDA but not on GABAA receptors in spontaneous rhythmic activity from the isolated neonatal rat respiratory network. Neuropharmacology 47:35–45
Pitt D, Werner P, Raine CS (2000) Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 6:67–70
Reagan-Shaw S, Nihal M, Ahmad N (2008) Dose translation from animal to human studies revisited. FASEB J 22:659–661
Saivin S, Hulot T, Chabac S, Potgieter A, Durbin P, Houin G (1998) Clinical pharmacokinetics of acamprosate. Clin Pharmacokinet 35:331–345
Schaffer S, Azuma J, Takahashi K, Mozaffari M (2003) Why is taurine cytoprotective? Adv Exp Med Biol 526:307–321
Simonini MV, Polak PE, Sharp A, McGuire S, Galea E, Feinstein DL (2010) Increasing CNS noradrenaline reduces EAE severity. J Neuroimmune Pharmacol 5:252–259
Smith T, Groom A, Zhu B, Turski L (2000) Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med 6:62–66
Spanagel R, Zieglgansberger W (1997) Anti-craving compounds for ethanol: new pharmacological tools to study addictive processes. Trends Pharmacol Sci 18:54–59
Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D (2005) Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain 128:1016–1025
Tian J, Lu Y, Zhang H, Chau CH, Dang HN, Kaufman DL (2004) Gamma-aminobutyric acid inhibits T cell autoimmunity and the development of inflammatory responses in a mouse type 1 diabetes model. J Immunol 173:5298–5304
Wallstrom E, Diener P, Ljungdahl A, Khademi M, Nilsson CG, Olsson T (1996) Memantine abrogates neurological deficits, but not CNS inflammation, in Lewis rat experimental autoimmune encephalomyelitis. J Neurol Sci 137:89–96
Wang GH, Jiang ZL, Fan XJ, Zhang L, Li X, Ke KF (2007) Neuroprotective effect of taurine against focal cerebral ischemia in rats possibly mediated by activation of both GABAA and glycine receptors. Neuropharmacology 52:1199–1209
Weiner HL (2009) The challenge of multiple sclerosis: how do we cure a chronic heterogeneous disease? Ann Neurol 65:239–248
Wu JY, Wu H, Jin Y, Wei J, Sha D, Prentice H, Lee HH, Lin CH, Lee YH, Yang LL (2009) Mechanism of neuroprotective function of taurine. Adv Exp Med Biol 643:169–179
Zagon IS, Rahn KA, Turel AP, McLaughlin PJ (2009) Endogenous opioids regulate expression of experimental autoimmune encephalomyelitis: a new paradigm for the treatment of multiple sclerosis. Exp Biol Med (Maywood) 234:1383–1392
Zalewska-Kaszubska J, Cwiek W, Dyr W, Czarnecka E (2005) Changes in the beta-endorphin plasma level after repeated treatment with acamprosate in rats selectively bred for high and low alcohol preference. Neurosci Lett 388:45–48
Zeise ML, Kasparov S, Capogna M, Zieglgansberger W (1993) Acamprosate (calciumacetylhomotaurinate) decreases postsynaptic potentials in the rat neocortex: possible involvement of excitatory amino acid receptors. Eur J Pharmacol 231:47–52
Zornoza T, Cano-Cebrian MJ, Hipolito L, Granero L, Polache A (2006) Evidence of a flip-flop phenomenon in acamprosate pharmacokinetics: an in vivo study in rats. Biopharm Drug Dispos 27:305–311
Acknowledgments
The study was supported by grants from Jog For The Jake (grant # 9333-521926) and from the National Multiple Sclerosis Society (grant # RG-4278).
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s10787-012-0120-1.
Rights and permissions
About this article
Cite this article
Sternberg, Z., Cesario, A., Rittenhouse-Olson, K. et al. Acamprosate modulates experimental autoimmune encephalomyelitis. Inflammopharmacol 20, 39–48 (2012). https://doi.org/10.1007/s10787-011-0097-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-011-0097-1