
Abstract
Population decline and fragmentation often lead to reduced genetic diversity and population differentiation. Habitat destruction throughout Madagascar has caused population decline and extinction of many endemic species. Lemur populations, including those of the largest extant lemur, Indri indri, have been fragmented into remaining forest patches. We assessed the level of genetic diversity in indri populations in three protected reserves by genotyping a total of 43 individuals at 17 microsatellite loci. Genetic diversity in terms of heterozygosity was high in all three reserves, with no differences between reserves. Population structure and F ST analyses revealed Analamazaotra Forest Station and the Torotorofotsy Conservation Area, which are separated by ca. 18 km to be genetically differentiated from each other with some admixture. Betampona Strict Nature Reserve, which is separated from the other reserves by ca. 130 km, exhibited clear population genetic differentiation, with no signs of admixture with the other reserves. Our genetic diversity estimates are similar to those for other Indridae in similar habitats and may reflect past rather than current population processes, given that populations have declined recently. Our results suggest that Betampona may be genetically isolated and that it is important to maintain gene flow between remaining populations to prevent loss of genetic diversity for the future conservation of Indri indri.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Habitat fragmentation and population declines are critical threats to biodiversity and have been correlated with genetic diversity loss in numerous species (Bellemain et al. 2007; Curtis and Taylor 2004; Goossens et al. 2006). Reduction of genetic diversity results in decreased effective population size and a general increase in homozygosity, often leading to decreased fitness (Templeton et al. 1990). Population genetic studies are useful in evaluating current levels of diversity, assessing existing habitat connectivity, and identifying putative corridors that should be maintained or established for population conservation (Cushman et al. 2009). They also can provide insight into population processes, including identification and timing of population size changes (Cornuet and Luikart 1996; Storz and Beaumont 2002). For example, isolation through reduced dispersal of offspring associated with habitat fragmentation can cause an accumulation of relatives in habitat patches, putting populations at risk for inbreeding (Frankham et al. 2002). Local extinctions are also more common in small fragmented populations, making migration and recolonization of habitat patches critical for long-term persistence (Kindlmann and Burel 2008).
Madagascar is valued for its high level of biodiversity, but has faced massive habitat destruction, with ca. 40% of its forest cover lost from 1950 to 2000 (Harper et al. 2007; Mittermeier et al. 2010). More than 100 species of lemurs are endemic to Madagascar, with most facing extinction threat due to this habitat loss (Mittermeier et al. 2010). The genetic consequences of this fragmentation have been studied for several lemur species (Craul et al. 2009; Olivieri et al. 2008; Quéméré et al. 2010). These studies showed that lemurs have variable responses to habitat loss, with some species displaying high genetic diversity despite habitat fragmentation (Quéméré et al. 2010), while others show signatures of genetic diversity loss in fragmented habitats (Craul et al. 2009; Olivieri et al. 2008). All of these studies found strong genetic differentiation between sampling sites.
The indri, Indri indri, is the largest of all extant Malagasy primates and the sole member of its genus, and is currently listed as Endangered on the IUCN Red List (Andriaholinirina et al. 2014). Indris live in territorial groups consisting of an adult pair and their offspring, with both males and females dispersing when mature (Pollock 1979; Powzyk 1997). Indris are distributed throughout the northeastern rainforests of Madagascar, north from Anjanaharibe-Sud and Antohaka Lava, and south to Anosibe An-ala Classified Forest (Mittermeier et al. 2010). They live in isolated patches of remaining forest at low population densities (5.2–22.9 individuals km2; Glessner and Britt 2005; Powzyk and Thalmann 2003). Subfossil indris have been found in the far northern and central highlands of Madagascar, but indris have been extirpated from much of their historical range, likely due to long-term climate change and/or human disturbance (Jungers et al. 1995). Indris are typically found at elevations from near sea level to 900 m, but also inhabit forests up to 1800 m (Goodman and Ganzhorn 2004). Although two subspecies of indri have been proposed based on pelage color, genetic research has indicated this is clinal variation in color form and not indicative of distinct lineages (Andriaholinirina et al. 2014; Groves 2001). Indri populations have declined by >50% over the past 50 year, with the greatest current threat to remaining populations being habitat destruction through human deforestation (Andriaholinirina et al. 2014; Mittermeier et al. 2010). Habitat destruction and degradation have reduced indri population sizes, decreased their home range sizes and increased population densities in disturbed forests (Glessner and Britt 2005; Pollock 1979; Powzyk 1997). Hunting is also becoming a significant threat to populations as taboos against hunting the indri have diminished (Britt et al. 2003).
We investigated the genetic diversity and differentiation of indri populations in three reserves in Madagascar that have all experienced habitat fragmentation and loss due to development of surrounding forests. We aimed to examine 1) genetic variation within populations and 2) genetic differentiation between populations. Because of the observed population declines and habitat fragmentation of the indri range, we hypothesized that genetic diversity would be low and genetic differentiation would be high between reserves.
Materials and Methods
Study Populations and Sampling
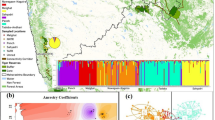
We sampled indris from the Betampona Strict Nature Reserve (Betampona), the Torotorofotsy Conservation Area (Torotorofotsy), and the Analamazaotra Forest Station (Analamazaotra), a secondary-growth forest managed by the Mitsinjo Association (Table I). Betampona (2228 ha) is an isolated, relatively pristine, low-altitude forest habitat that has received the highest level of protection in Madagascar, including restricted human access. It has an estimated population of 77–147 indris (Glessner and Britt 2005). Torotorofotsy (9900 ha) and Analamazaotra (700 ha) are located in the Analamazaotra Forest Complex, a fragmented mid-altitude forest habitat, with an estimated population of 21–32 indris (Junge et al. 2011). Betampona is isolated from other indri populations, while Torotorofotsy and Analamazaotra are surrounded by fragmented forest that may allow for migration (Fig. 1).

Locations of the three study sites where we sampled indris between August 2007 and October 2009. Reserve sizes are in parentheses. Forested areas are shaded. Map was created in Arcmap 10.2, with forest cover estimates taken from Harper et al. (2007). The forested areas are not intended to represent the boundaries of conservation areas or reserves, the boundaries that were not available as data layers.
We darted and anesthetized 43 indris between August 2007 and October 2009 using Type “C” Disposable Darts (Pneu-Dart, Williamsport, PA; Junge et al. 2011) and tiletamine and zolazepam (Fort Dodge Animal Health, Overland Park, KS; 15 mg/kg, i.m.) (Table I). After darting, we processed all individuals on a blanket on the ground at the darting site. Recovery times varied by body size, but would take anywhere from 1 to 4 h. We kept indri warm with a sheet and project staff held them until fully recovered, at which point we released them at the darting site. If captured at twilight, we held indri overnight for recovery and released at the darting site in the morning. No indri was darted unsuccessfully, and there were no visible injuries on the individuals. We collected eight samples from Betampona as blood samples preserved on Whatman FTA® cards (Table I). We collected all other samples as 1 × 1 mm ear snips preserved in 95% ethanol.
Microsatellite Genotyping
DNA isolation techniques varied with sample preservation. For blood samples, we took a 2 × 2 mm punch from the FTA card, washed it three times with 200 μl of FTA purification reagent, and then washed it twice with 200 μl of TE buffer (10 mM Tris–HCl, 0.1 mM EDTA, pH 8.0). We used the FTA card punch as the DNA template in all polymerase chain reactions (PCRs). We extracted DNA from tissue samples using a Qiagen DNeasy Blood and Tissue Kit.
We genotyped individuals at 19 indri-specific microsatellite loci using primers identified by Zaonarivelo et al. (2007; Table II). We optimized PCR annealing temperatures for different primer pairs with thermal gradient PCR and carried out PCR reactions in 20-μl reactions (10× buffer, 10 mM dNTPs, 10 μM of each primer, 5 units of Taq polymerase, and 100 ng of template DNA) using the protocol in Schuelke (2000). Thermocycling consisted of an initial denaturation of 5 min at 95 °C followed by 35 cycles of 95 °C for 30 s, 50°–65° for 45 s, and 72 °C for 45 s with a final elongation at 72 °C for 10 min. We used negative PCR controls to rule out contamination. We ran PCR products on a capillary sequencer (Applied Biosystems 3730) and visualized and manually genotyped them using Peak Scanner (Applied Biosystems). To assess the repeatability of genotypes from individuals, we randomly selected two microsatellite loci from each individual and repeated the PCR and genotyping process. All loci were included in at least one genotyping repetition. Repeated genotypes were consistent in all but two loci (67HDZ27 and 67HDZ28), and we removed these loci from all analyses.
Genetic Analyses
We screened for typographical and genotyping errors using Micro-Checker (Van Oosterhout et al. 2004). We assessed the effect of null alleles on results using FreeNA (Chapuis and Estoup 2007). To determine if the full data set produced results that were similar to the data set that excluded null alleles, we calculated F ST measures of population differentiation from the full data set and a data set that excluded potential null alleles with 95% confidence intervals (CI) using 1000 bootstrapped replicates. We focused our assessment of genetic diversity on allelic richness and H E, which are expected to be less biased than H O in response to differing sample sizes and null alleles (Chapuis et al. 2008).
We tested for deviations from linkage disequilibrium between all pairs of loci and for deviations from Hardy–Weinberg equilibrium (HWE) with Bonferroni corrections using GenePop v 4.0.1 (Raymond and Rousset 1995). We estimated observed (H O) and expected (H E) heterozygosity, and standardized allelic richness (calculated using rarefaction based on the minimum number of samples per population) in FSTAT v 2.9.3 (Goudet 1995). We calculated F IS and estimated 95% confidence intervals by bootstrapping loci 10,000 times using GENETIX v 4.05 (Belkhir et al. 1996). We evaluated differences in genetic variation among sampling sites using independent-samples Kruskal–Wallis tests in Systat v12.02 (©SYSTAT Software Inc. 2007).
To assess population genetic structure across sampling sites, we used a Bayesian assignment analysis implemented in Structure v 2.3.2 (Pritchard et al. 2000) to estimate the number of genetically distinct clusters (K). We used a model that allowed for admixture and correlation of allele frequencies among clusters for all analyses. We used K ranging from 1 to 10, with 20 replicate runs, a burn-in value of 100,000 generations, and 1,000,000 generations used to estimate the posterior distribution. To estimate the optimum value of K, we calculated the statistic ∆K using the estimated natural log probability of the data across replicates for each K (Evanno et al. 2005). To account for possible biases due to unequal sample sizes, we repeated the above Structure procedure using a subsample of six individuals from each population.
To test for genetic distance between sampling sites, we calculated pairwise F ST (Weir and Cockerham 1984) between all three sampling sites, and with Analamazaotra and Torotorofotsy combined, in Arlequin v 3.5.1.2 using permutation tests for significance (1000 permutations; Excoffier and Lischer 2010). We examined hierarchical partitioning of genetic variance using an analysis of molecular variance (AMOVA) to examine the distribution of genetic variation in GenAlEx (Peakall and Smouse 2006).
Population Size Changes
We tested for signatures of population bottlenecks with BOTTLENECK v 1.2.02 (Cornuet and Luikart 1996) and Wilcoxon’s test (Piry et al. 1999) under three different mutational models: infinite allele mutation model (IAM), the stepwise mutation model (SMM), and the two-phase model (TPM) with 70% SMM and 30% multistep mutation events (Di Rienzo et al. 1994). This test identifies heterozygosity excess under an assumption of mutation-drift equilibrium, a characteristic of populations that have recently experienced a decline in effective population size (N e, Cornuet and Luikart 1996). We also tested for mode-shift in allele frequencies as populations that have experienced a bottleneck will lose rare alleles and have increased frequency of common alleles (Cornuet and Luikart 1996). We repeated these analyses using various sampling strategies: 1) all samples pooled, 2) the geographically close Analamazaotra and Torotorofotsy combined, and 3) all sampling sites assessed separately. Gene flow and/or population structure can generate false signals of bottlenecks (Chikhi et al. 2010; Städler et al. 2009). By pooling samples from multiple sites we aimed to assess whether the metapopulation as a whole has experienced a change in N e, possibly through overall habitat fragmentation.
To further test for population size changes, we used a full likelihood-based Bayesian approach implemented in MSVAR v 1.3 to quantify and date changes in N e. For MSVAR analysis, we analyzed Betampona separately and pooled Analamazoatra and Torotorofotsy (which are geographically close to one another) to increase sample size. This method assumes a stable ancestral population size (N 1) that experienced an exponential population size change T years ago relative to the current population size (N 0) (Storz and Beaumont 2002). Prior distributions for N 0, N 1, T, and μ are assumed to lognormal. We used different prior distributions for each run to test their influence on posteriors (Electronic Supplementary Material [ESM] Table SI). We performed four independent Markov chain Monte Carlo (MCMC) runs for each population with 4 × 1010 iterations and a thinning value of 5 × 104, with the first 10% of each run discarded as burn-in. We checked convergence of MCMC chains visually and tested this with the Gelman and Rubin convergence statistic in the coda package in R v 3.20 (Gelman and Rubin 1992; R Core Team 2015). We defined convergence as values <1.1 (Gelman and Hill 2006). We estimated of the mode and 90% highest posterior density (HPD) intervals for each parameter using the boa package in R.
Ethical Note
All research adhered to the American Society of Primatologists’ Principles for the Ethical Treatment of Non-Human Primates, and complied with protocols approved by the St. Louis Zoo and Duke University’s Institutional Animal Care and Use Committee (Permit # A282-07-10-(1)), as well as all research requirements in Madagascar and Convention on International Trade in Endangered Species of Wild Flora and Fauna (CITES) regulations (Permit ID #US182626/9).
Results
Marker Performance and Genetic Variation
Micro-Checker detected no evidence of large allele dropout or stuttering but detected null alleles in each sampling locality: Betampona (67HDZ39, 67HDZ3, 67HDZ16, 67HDZ177, 67HDZ72, and 67HDZ41), Analamazaotra (67HDZ76, 67HDZ16, 67HDZ41, and 67HDZ161), and Torotorofotsy (67HDZ76, 67HDZ18, 67HDZ180, 67HDZ72, and 67HDZ114). These null alleles were not consistent across localities. Pairwise F ST estimates between the three sampling localities produced slightly lower F ST estimates based on a data set that excluded null alleles, relative to F ST estimates based on the full data set (ESM Table SII). However, the 95% bootstrapped CIs for both the uncorrected and corrected F ST estimates largely overlapped, indicating that null alleles have minimal impact on results. Therefore, we used the full data set with all loci in all subsequent analyses.
There was evidence of linkage disequilibrium in one locus pair in Betampona (67HDZ39 and 67HDZ18; P < 0.0001). We observed deviations from HWE at six loci in Betampona and one locus in Torotorofotsy (Table II). There were no significant differences between sample sites in H E, or allelic richness (Table II). At Betampona, F IS was significantly greater than zero, while at the other sites CIs overlapped zero (Table II). Results for Betampona were similar with null alleles removed (F IS = 0.14156; 95% CI: 0.07033–0.18517).
Population Structure
Structure analysis revealed two distinct population genetic groups using the statistic ∆K. Mean log probability (lnPr(K)) values plateaued at K = 5 but ∆K calculations favored K = 2. At the K = 2 level all individuals from Betampona were placed in one cluster, with all individuals from Analamazaotra and Torotorofotsy placed in a second cluster (Fig. 2). The division between Analamazaotra and Torotorofotsy was less clear, with analysis of K = 3 suggesting admixture between the two populations (Fig. 2). ∆K calculations from Structure analyses that included only Analamazaotra and Torotorofotsy similarly identified them as separate genetic clusters. The subsampled data set identified the same genetic clusters, with ∆K calculations favoring K = 2 and the analysis of K = 3 suggesting admixture between Analamazaotra and Torotorofotsy (ESM Fig. S1).

Results of Structure analyses for indri at Betampona Strict Nature Reserve, Analamazaotra Forest Station, and Torotorofotsy Conservation Area sampled between August 2007 and October 2009. The upper plot presents individual assignment results of analyses assuming a model of two genetic clusters (K = 2). The lower plot presents individual assignment results of analyses assuming a model of three genetic clusters (K = 3). Vertical bars indicate the posterior probability of each sampled individual’s assignment to a distinct genetic cluster.
AMOVA analyses indicated that the majority of genetic variation is accounted for at the intraindividual level (Table III); however, significant F ST values indicated genetic structure among populations (Table III). This result was similar when we treated all three populations as separate groups or when we treated Analamazaotra and Torotorofotsy populations as a single group. Pairwise F ST estimates involving Betampona (vs. Analamazaotra, F ST = 0.113; vs. Torotorofotsy, F ST = 0.129) were larger than that between Analamazaotra and Torotorofotsy (F ST = 0.0746). All values of F ST were significantly different from zero (P < 0.05).
Population Size Changes
Using BOTTLENECK each sampling strategy and population showed a bottleneck signature under the IAM model (Table IV). The bottleneck signature was less clear using TPM and SMM models. Only Analamazaotra showed a mode shift in allele frequencies with a disproportionate loss of rare alleles.
Using MSVAR, the Gelman–Rubin diagnostic indicated poor convergence across the independent MCMC runs. The Gelman–Rubin convergence statistics were M N0 = 1.10 (1.20) and M N1 = 1.06 (1.13) for Betampona and M N0 = 1.15 (upper 97.5% quantile = 1.33) and M N1 = 1.05 (1.13) for Analamazaotra and Torotorofotsy combined. The marginal posterior distributions for N 1 (ancestral population) overlapped or were nearly identical to those for N 0 (Fig. 3). However, the marginal posterior distributions for N 1 were broad and largely tracked those of the prior distributions (Fig. 3), suggesting a lack of information for ancestral population size. Analyses of the combined Analamazaotra and Torotorofotsy populations resulted in modes of log10 N 0 ranging 3.51–5.27, log10 N 1 ranging −0.87 to 4.67, and log10 T ranging 2.86–8.49. Analyses of the Betampona population resulted in modes of log10 N 0 ranging 2.67–6.22, log10 N 1 ranging −0.68 to 5.85, and log10 T ranging −0.22 to 10.64.

Results of MSVAR analyses for indri at Betampona Strict Nature Reserve, Analamazaotra Forest Station, and Torotorofotsy Conservation Area sampled between August 2007 and October 2009. The upper plot presents results from Betampona and the lower plot presents results for pooled samples from Analamazaotra and Torotorofotsy. The solid black lines represent posterior distribution of the present (log N 0); dashed black lines represent past (log N 1) population size. Red lines represent the prior distributions.
Discussion
Genetic Variation Within Populations
The three indri populations we surveyed had measures of heterozygosity (global H E = 0.85; population range H E = 0.75–0.79) similar to those for other Indriidae species in similarly disturbed habitats (Propithecus coquereli HE = 0.77 in Rakotoarisoa et al. 2006; P. verreauxi HE = 0.71 in Lawler et al. 2001; P. tattersalli H E = 0.72 in Quéméré et al. 2010). These measures are high, and may relate to the slow decline in heterozygosity in previously large populations after a bottleneck (Cornuet and Luikart 1996). Because allele number is reduced faster than heterozygosity after a bottleneck, heterozygosity may remain elevated until a new mutation-drift equilibrium is reached (Nei et al. 1975). The three indri populations also showed similar levels of genetic diversity, although they differ in density and anthropogenic impact levels (Junge et al. 2011). These genetic diversity estimates may be more reflective of past, rather than current, landscape patterns as populations may not have reached equilibrium (Keyghobadi et al. 2005).
Bottleneck analysis were sensitive to different mutation models, with all sites and pooling strategies showing a bottleneck under the IAM, and more varied signatures under the SMM and TPM. These results indicate a lack of power to detect robust signatures of population decline. MSVAR analysis also yielded no meaningful results and we were unable to draw conclusions about the magnitude and timing of population size change. These results may be due to the low sample size. Future studies of indris may benefit from next generation sequencing (NGS) methods, which can generate more comprehensive surveys of genome-wide variation and provide the level of genetic information needed to validate potential bottlenecks (Allendorf et al. 2010). Other lemur species show bottleneck signatures in response to human deforestation (Craul et al. 2009; Holmes et al. 2013; Lawler 2011; Olivieri et al. 2008; Quéméré et al. 2012; Razakamaharavo et al. 2010) and such signatures may become more apparent in indris with additional data, as habitat fragmentation and human disturbance have resulted in contraction of indri populations in the past 50 year (Andriaholinirina et al. 2014; Mittermeier et al. 2010).
The deforestation surrounding our sampling sites has occurred relatively recently, within the past century (Harper et al. 2007). Given the long life span and generation time (ca. 12 years) of indris, only a limited number of generations have occurred during this time period (Andriaholinirina et al. 2014; Mittermeier et al. 2010; Pollock 1977). Long-lived individuals in our samples may buffer the loss of genetic diversity (Lippe et al. 2006). Betampona is the oldest reserve of the three (established in 1927), but the surrounding land is largely agricultural, likely isolating this indri population (R. Junge pers. obs.), which may explain the elevated F IS in comparison to the other sites. The more recently established Torotorofotsy conservation area (established 2005) and Analamazaotra forest station (established 1970) may be connected by the Analamazoatra Special Reserve and Mantadia National Park, and the Ankeniheny–Zahamena Corridor (425,000 ha), a stretch of somewhat contiguous forest that has been protected since 2005 (Fig. 1).
Population Structure
We observed genetic differentiation among the three sampling sites. The geographically close Analamazaotra and Torotorofotsy populations (separated by ca. 18 km) were moderately genetically differentiated from each other, and both showed higher levels of genetic differentiation from the Betampona population (ca. 130 km away). Given that populations are likely not in drift-migration equilibrium, some of the detected genetic differentiation may represent historical patterns prior to forest fragmentation (Whitlock and McCauley 1999). However, the extent of genetic differentiation is not surprising given the current patchy distribution of indris, and the greater geographic isolation of Betampona from the other forest reserves. Although Analamazaotra and Torotorofotsy are geographically close, contiguous forest is fragmented and the land separating these sites is affected by human land use, which may further explain their population differentiation.
The results of our study indicate isolation of indri populations separated by nonforest on a relatively small spatial scale compared to their overall range. However, given the wide variation in density of indri populations (Pollock 1975), improved sampling is needed to understand fully the population structure across the entire indri range. The structure we detected may also reflect past, not current, population structure due to the lack of drift-migration equilibrium, furthering the need for more intense sampling.
Conservation Implications
Our results suggest that indri populations in Madagascar possess moderate to high genetic diversity in terms of heterozygosity, which is encouraging for the conservation of the species, but may reflect past, rather than current, population processes. Indri display plasticity in their ability to live in a range of forest sizes, with varying degrees of fragmentation (Glessner and Britt 2005). However, we observed population differentiation between sites and Betampona may be genetically isolated. Further studies are needed to elucidate the demographic history of populations and to estimate gene flow and migration rates, which influence the chances of future inbreeding. Conservation efforts should aim to protect large continuous blocks of forest with migration corridors between them in order to maintain genetic diversity and avoid further genetic isolation of populations.
References
Allendorf, F. W., Hohenlohe, P. A., & Luikart, G. (2010). Genomics and the future of conservation genetics. Nature Reviews Genetics, 11(10), 697–709.
Andriaholinirina, N., Baden, A., Blanco, M., Chikhi, L., Cooke, A., et al. (2014). Indri indri. The IUCN Red List of Threatened Species. Version 2014.3. www.iucnredlist.org. Accessed 24 Nov 2014.
Belkhir, K., Borsa, P., Chikhi, L., Raufaste, N., & Bonhomme, F. (1996). GENETIX 4.05, logiciel sous Windows® pour la génétique des populations. Laboratoire génome, populations, interactions, CNRS UMR, 5000, 1996–2004.
Bellemain, E., Nawaz, M. A., Valentini, A., Swenson, J. E., & Taberlet, P. (2007). Genetic tracking of the brown bear in northern Pakistan and implications for conservation. Biological Conservation, 134(4), 537–547.
Britt, A., Randriamandratonirina, N. J., Glasscock, K. D., & Iambana, B. R. (2003). Diet and feeding behaviour of Indri indri in a low-altitude rain forest. Folia Primatologica, 73(5), 225–239.
Chapuis, M. P., & Estoup, A. (2007). Microsatellite null alleles and estimation of population differentiation. Molecular Biology and Evolution, 24(3), 621–631.
Chapuis, M. P., Lecoq, M., Michalakis, Y., Loiseau, A., Sword, G. A., et al. (2008). Do outbreaks affect genetic population structure? A worldwide survey in Locusta migratoria, a pest plagued by microsatellite null alleles. Molecular Ecology, 17(16), 3640–3653.
Chikhi, L., Sousa, V. C., Luisi, P., Goossens, B., & Beaumont, M. A. (2010). The confounding effects of population structure, genetic diversity and the sampling scheme on the detection and quantification of population size changes. Genetics, 186(3), 983–995.
Cornuet, J. M., & Luikart, G. (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics, 144(4), 2001–2014.
Craul, M., Chikhi, L., Sousa, V., Olivieri, G. L., Rabesandratana, A., et al. (2009). Influence of forest fragmentation on an endangered large-bodied lemur in northwestern Madagascar. Biological Conservation, 142(12), 2862–2871.
Curtis, J. M., & Taylor, E. B. (2004). The genetic structure of coastal giant salamanders (Dicamptodon tenebrosus) in a managed forest. Biological Conservation, 115(1), 45–54.
Cushman, S. A., McKelvey, K. S., & Schwartz, M. K. (2009). Use of empirically derived source-destination models to map regional conservation corridors. Conservation Biology, 23, 368–376.
Di Rienzo, A., Peterson, A. C., Garza, J. C., Valdes, A. M., Slatkin, M., & Freimer, N. B. (1994). Mutational processes of simple-sequence repeat loci in human populations. Proceedings of the National Academy of Sciences of the United States of America, 91(8), 3166–3170.
Evanno, G., Regnaut, S., & Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology, 14(8), 2611–2620.
Excoffier, L., & Lischer, H. E. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3), 564–567.
Frankham, R., Briscoe, D. A., & Ballou, J. D. (2002). Introduction to conservation genetics. Cambridge: Cambridge University Press.
Gelman, A., & Hill, J. (2006). Data analysis using regression and multilevel/hierarchical models. Cambridge: Cambridge University Press.
Gelman, A., & Rubin, D. B. (1992). Inference from iterative simulation using multiple sequences. Statistical Science 7(4), 457–472.
Glessner, K. D., & Britt, A. (2005). Population density and home range size of Indri indri in a protected low altitude rain forest. International Journal of Primatology, 26(4), 855–872.
Goodman, S. M., & Ganzhorn, J. U. (2004). Elevational ranges of lemurs in the humid forests of Madagascar. International Journal of Primatology, 25(2), 331–350.
Goossens, B., Chikhi, L., Ancrenaz, M., Lackman-Ancrenaz, I., Andau, P., & Bruford, M. W. (2006). Genetic signature of anthropogenic population collapse in orang-utans. PLoS Biology, 4(2), e25.
Goudet, J. (1995). FSTAT (version 1.2): a computer program to calculate F-statistics. Journal of Heredity, 86(6), 485–486.
Groves, C. P. (2001). Primate taxonomy. Washington, DC: Smithsonian Institution Press.
Harper, G. J., Steininger, M. K., Tucker, C. J., Juhn, D., & Hawkins, F. (2007). Fifty years of deforestation and forest fragmentation in Madagascar. Environmental Conservation, 34(4), 325–333.
Holmes, S. M., Baden, A. L., Brenneman, R. A., Engberg, S. E., Louis, E. E., Jr., & Johnson, S. E. (2013). Patch size and isolation influence genetic patterns in black-and-white ruffed lemur (Varecia variegata) populations. Conservation Genetics, 14(3), 615–624.
Junge, R. E., Barrett, M. A., & Yoder, A. D. (2011). Effects of anthropogenic disturbance on indri (Indri indri) health in Madagascar. American Journal of Primatology, 73(7), 632–642.
Jungers, W. L., Godfrey, L. R., Simons, E. L., & Chatrath, P. S. (1995). Subfossil Indri indri from the Ankarana Massif of northern Madagascar. American Journal of Physical Anthropology, 97(4), 357–366.
Keyghobadi, N., Roland, J., Matter, S. F., & Strobeck, C. (2005). Among-and within-patch components of genetic diversity respond at different rates to habitat fragmentation: an empirical demonstration. Proceedings of the Royal Society B: Biological Sciences, 272(1562), 553–560.
Kindlmann, P., & Burel, F. (2008). Connectivity measures: a review. Landscape Ecology, 23(8), 879–890.
Lawler, R. R. (2011). Historical demography of a wild lemur population (Propithecus verreauxi) in southwest Madagascar. Population Ecology, 53(1), 229–240.
Lawler, R. R., Richard, A. F., & Riley, M. A. (2001). Characterization and screening of microsatellite loci in a wild lemur population (Propithecus verreauxi verreauxi). American Journal of Primatology, 55(4), 253–259.
Lippe, C., Dumont, P., & Bernatchez, L. (2006). High genetic diversity and no inbreeding in the endangered copper redhorse, Moxostoma hubbsi (Catostomidae, Pisces): the positive sides of a long generation time. Molecular Ecology, 15(7), 1769–1780.
Mittermeier, R. A., Louis, E., Hawkins, F., Langrand, O., Ganzhorn, J., et al. (2010). Lemurs of Madagascar (3rd ed.). Arlington, VA: Conservation International.
Nei, M., Maruyama, T., & Chakraborty, R. (1975). The bottleneck effect and genetic variability in populations. Evolution, 29, 1–10.
Olivieri, G. L., Sousa, V., Chikhi, L., & Radespiel, U. (2008). From genetic diversity and structure to conservation: Genetic signature of recent population declines in three mouse lemur species (Microcebus spp.). Biological Conservation, 141, 1257–1271.
Peakall, R. O. D., & Smouse, P. E. (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes, 6(1), 288–295.
Piry, S., Luikart, G., & Cornuet, J. M. (1999). BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. The Journal of Heredity, 90(4), 502–503.
Pollock, J. (1977). The ecology and sociology of feeding in Indri indri. In T. Clutton-Brock (Ed.), Primate ecology: Studies of feeding and ranging behaviour in lemurs, monkeys and apes (pp. 37–69). London: Academic.
Pollock, J.I. (1975). Field observations on Indri indri: A preliminary report. In Lemur biology (pp. 287–311). New York: Springer-Verlag.
Pollock, J. I. (1979). Spatial distribution and ranging behavior in lemurs. In G. A. Doyle (Ed.), The study of prosimian behavior (pp. 359–409). New York: Academic.
Powzyk, J. A. (1997). The socio-ecology of two sympatric indrids, Propithecus diadema diadema and Indri indri: A comparison of feeding strategies and their possible repercussions on species-specific behaviors. Ph. D. dissertation, Duke University.
Powzyk, J., & Thalmann, U. (2003). Indri indri, indri. In S. M. Goodman & J. P. Benstead (Eds.), The natural history of Madagascar (pp. 1342–1345). Chicago: University of Chicago Press.
Pritchard, J. K., Stephens, M., & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945–959.
Quéméré, E., Amelot, X., Pierson, J., Crouau-Roy, B., & Chikhi, L. (2012). Genetic data suggest a natural prehuman origin of open habitats in northern Madagascar and question the deforestation narrative in this region. Proceedings of the National Academy of Sciences of the United States of America, 109(32), 13028–13033.
Quéméré, E., Louis, E. E., Jr., Ribéron, A., Chikhi, L., & Crouau-Roy, B. (2010). Non-invasive conservation genetics of the critically endangered golden-crowned sifaka (Propithecus tattersalli): high diversity and significant genetic differentiation over a small range. Conservation Genetics, 11(3), 675–687.
R Core Team (2015). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/.
Rakotoarisoa, G., Shore, G., Mcguire, S., Engberg, S., Louis, E., & Brenneman, R. (2006). Characterization of 20 microsatellite marker loci in Coquerel’s sifaka (Propithecus coquereli). Molecular Ecology Notes, 6(4), 1119–1121.
Raymond, M., & Rousset, F. (1995). GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. Journal of Heredity, 86(3), 248–249.
Razakamaharavo, V. R., McGuire, S. M., Vasey, N., Louis, E. E., Jr., & Brenneman, R. A. (2010). Genetic architecture of two red ruffed lemur (Varecia rubra) populations of Masoala National Park. Primates, 51(1), 53–61.
Schuelke, M. (2000). An economic method for the fluorescent labeling of PCR fragments. Nature Biotechnology, 18(2), 233–234.
Städler, T., Haubold, B., Merino, C., Stephan, W., & Pfaffelhuber, P. (2009). The impact of sampling schemes on the site frequency spectrum in nonequilibrium subdivided populations. Genetics, 182(1), 205–216.
Storz, J. F., & Beaumont, M. A. (2002). Testing for genetic evidence of population expansion and contraction: an empirical analysis of microsatellite DNA variation using a hierarchical Bayesian model. Evolution, 56(1), 154–166.
Templeton, A. R., Shaw, K., Routman, E., & Davis, S. K. (1990). The genetic consequences of habitat fragmentation. Annals of the Missouri Botanical Garden, 77(1), 13–27.
Van Oosterhout, C., Hutchinson, W. F., Wills, D. P., & Shipley, P. (2004). MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Molecular Ecology Notes, 4(3), 535–538.
Weir, B.S., & Cockerham, C.C. (1984). Estimating F-statistics for the analysis of population structure. Evolution, 38(6), 1358–1370.
Whitlock, M. C., & McCauley, D. E. (1999). Indirect measures of gene flow and migration: FST ≠ 1/(4Nm + 1). Heredity, 82, 117–125.
Zaonarivelo, J. R., Sommer, J. A., Shore, G. E., McGuire, S. M., Engberg, S. E., et al. (2007). Isolation and characterization of 20 microsatellite marker loci from the Indri (Indri indri) genome. Molecular Ecology Notes, 7(1), 25–28.
Acknowledgments
We thank two anonymous reviewers for their constructive suggestions. We thank Madagascar National Parks (MNP) for permission to conduct this research, the Madagascar Institute for the Conservation of Tropical Ecosystems (MICET) and Madagascar Fauna Group (MFG) for logistical assistance in Madagascar, the Madagascar Biodiversity Project Field Team for field support, and the Duke Lemur Center for planning assistance. We thank Dr. Charles Faulkner (College of Veterinary and Comparative Medicine, Lincoln Memorial University) who has completed much of the parasitological identification over several years. We thank the team at Betampona Strict Nature Reserve, R. Dolch and the Mitsinjo Association, C. Williams, C. Welch, A. Katz, S. Zehr, F. Rasambainarivo, K. Freeman, G. Kett, B. Iambana, A. Junge, A. Greven, T. Rakotonanahary, H. Rafalinirina, and B. Allen for assistance in the field and in preparation. We also thank G. Stillings for help with ArcGIS. This work was supported by the St. Louis Zoo Field Research for Conservation Fund, the Duke University Center for International Studies, the Duke University Graduate School, the Nicholas School of the Environment, the Commonwealth of Kentucky, and the National Science Foundation (DEB-0949532). M. A. Barrett was a Robert Wood Johnson Foundation Health & Society Scholar at the University of California (UC) San Francisco and UC Berkeley; she thanks the program for its financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: Joanna M. Setchell
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
Supporting information with priors used for MSVAR analysis (Table SI), and pairwise F ST 95% confidence interval values calculated from the full data set and a data set that excluded potential null alleles (Table SII) are available online. (DOCX 89 kb)
Rights and permissions
About this article

Cite this article
Nunziata, S.O., Wallenhorst, P., Barrett, M.A. et al. Population and Conservation Genetics in an Endangered Lemur, Indri indri, Across Three Forest Reserves in Madagascar. Int J Primatol 37, 688–702 (2016). https://doi.org/10.1007/s10764-016-9932-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10764-016-9932-y