
Abstract
The current range of the red ruffed lemur (Varecia rubra) population is primarily restricted to forests of the Masoala Peninsula on the northeastern coast of Madagascar. Whereas much of the peninsula is protected as Masoala National Park, parts of the forest are at risk from anthropogenic pressures and habitat fragmentation. We sampled 32 individual red ruffed lemur from two sites: Ambatoledama (DAMA), a narrow forest corridor across an area of degraded habitat connecting larger blocks of forest in the northwestern reaches of the park, and Masiaposa (MAS) forest, a largely pristine forest on the lower western side of the peninsula. Population genetic parameters were estimated for these two populations employing 15 microsatellite loci derived from the V. variegata genome. We found that by exceeding the expected heterozygosity at mutation-drift equilibrium, the DAMA population has undergone a recent population bottleneck. Population structure analysis detected individuals harboring genotypic admixture of the DAMA genetic cluster in the MAS population, suggesting a possibility of unilateral gene flow or movement between these populations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The island of Madagascar has been identified as one of the world’s top biodiversity hotspots (Myers 1988; Myers et al. 2000; Mittermeier et al. 2004) with global conservation priority (Hannah et al. 1998; Myers 1998). As its natural resources are rich in endemism (Jenkins 1990; Nicoll and Lagrand 1993), there is an increased awareness and concern over the alarming rate of deforestation (Green and Sussman 1990; Nelson and Horning 1993). With 40% of the Malagasy forest lost since 1950, all species are under increasing pressure from continued forest degradation and habitat fragmentation (Allnutt et al. 2004). This can be attributed to a two-fold attack on the forest habitats: a loss in forest coverage and an increase in forest fragmentation (Harper et al. 2007).
Masoala National Park (MasNP), 2,300 km2, established in 1997, and Makira Natural Park (MakNP), established in 2004, are the only protected areas harboring populations of the red ruffed lemur, Varecia rubra (Geoffroy-Saint Hilare 1812; Patel and Andrianandrasana 2008). The majority of red ruffed lemur populations is found on the Masoala Peninsula [Conservation Breeding Specialist Group (CBSG) 2002; Vasey and Tattersall 2002]. Sparse remnants occur in the region north of Antongil Bay where MasNP and MakNP are loosely connected by forest fragments (Hekkala et al. 2007). These northern populations have been drastically reduced within a geographic range that stretches into forests south of Andapa (Tattersall 1977; Petter et al. 1977; Hekkala et al. 2007). The northern distribution limit of the species is considered to be the Lokoho River (Goodman and Ganzhorn 2004), and the western limit is the Antainambalana River (Hekkala et al. 2007). The historic range of V. rubra at one time was in contact with V. variegata north of Antongil Bay (Vasey and Tattersall 2002), possibly resulting in a hybrid zone, but this cannot be confirmed, as the zone is now heavily degraded and primarily contains remnant, isolated ruffed lemur populations (Simons and Lindsay 1987; Hekkala et al. 2007).
Ruffed lemurs are considered among the most arboreal and frugivorous of all lemurs (Vasey 1997a, 2002) and are thus sensitive to forest health (Balko and Underwood 2005), requiring mature forests for their survival (Morland 1991; White 1991; Rigamonti 1993). Human–animal conflict arises as about half of the tree species preferred by red ruffed lemurs as dietary sources are also harvested preferentially by local villagers on the Masoala Peninsula (Merenlender et al. 1998). A little more than a decade ago, forest restoration was begun in the northwestern reaches of the MasNP to establish forest corridors between blocks of forest (Holloway 2000).
Vasey (2003) compiled many studies of the two Varecia species, noting that populations at low elevation have higher densities than do populations at high elevation, putting aside populations living in anthropogenically isolated forest fragments. Varecia tend to range in groups of two to five individuals within fission−fusion societies of up to 31 individuals and spend most of their day feeding, traveling, and resting high up in the forest canopy (Vasey 2003). Their territoriality varies from location to location, and overall, the two species appear to practice rather flexible social behavior. Both sexes are known to disperse, although few instances of dispersal have been observed (Ratsimbazafy 2002). Their social organization seems to respond to reproductive cycles, season, and food availability (Balko 1998; Vasey 2003, 2006).
We investigated the population structure of red ruffed lemurs (V. rubra) sampled from two forests differing in degrees of anthropogenic disturbance. Fifteen informative microsatellite markers generated from the V. variegata genome (Louis et al. 2005) were amplified in 32 individual lemurs from the Ambatoledama (DAMA) and Masiaposa (MAS) forests of MasNP. The MAS forest is considered to be largely pristine and not severely affected by anthropogenic pressures. On the other hand, as with many forests located north of Antongil Bay, the DAMA forest has undergone significant fragmentation. However, this forest was one of three localities in the northwestern part of the park where a reforestation project began more than a decade ago (Holloway 2000). This effort augmented the DAMA forest to form a narrow corridor approximately 1-km wide, designed to connect the MakNP to the northwestern corner of the MasNP (Hekkala et al. 2007).
Limited studies on the genera Varecia (Louis et al. 2005), Propithecus (Lawler 2008; Quéméré et al. 2009), Microcebus (Olivieri et al. 2008; Radespiel et al. 2008; Gligor et al. 2009), and Mirza (Markolf et al. 2008) have been published in which baseline population genetic estimates have been established for lemur species. Our genetic analysis provides the first information on the genetic architecture of red ruffed lemur populations.
Methods
Sample collection
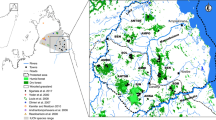
We collected samples from two forests in the MasNP: MAS (S15°40′ E049°58′), located on the western side of the Masoala Peninsula, and DAMA (S15°17′ E050°01′) in the northern part of the park (Fig. 1). We selected lemur individuals by proximity to the immobilization team while considering the safety of the lemur with respect to position and dart placement. We sampled n = 10 individuals from the five resident social groups (Total census: n = 25 individuals) observed in the DAMA forest. The groups comprised adult pairs with two or three nursing juveniles each, or 40% of the red ruffed lemur population known to inhabit the 2-km2 DAMA forest. Vasey (1997b) estimated the red ruffed lemur density for the Masoala Peninsula to be between 31.2 and 53.73 ind/km2. Using these estimates, we would expect 125–215 individuals to inhabit the 4-km2 sampling area of the MAS forest. The 22 individuals we sampled would constitute 10.2–17.6% of the red ruffed lemurs expected to live in the sampling area. We sampled the forests on two separate expeditions (MAS 25 October to 11 November 2002 and DAMA 20–25 November 2006). All lemurs were immobilized, handled, and released as described in Louis et al. (2005) for DAMA, and Dutton et al. (2008) for biomedical evaluation in MAS. We exported all samples with appropriate permits issued by the Ministère des Eaux et Forêts of Madagascar and imported with the appropriate US Fish and Wildlife Service permit to Henry Doorly Zoo (HDZ), Omaha, NE, USA.

The boundaries of Masoala National Park and Makira Natural Park outlined in black and the two study areas, Masiaposa Forest and Ambatoledama Forest, marked by asterisks
DNA extraction and microsatellite genotyping
We extracted whole genomic DNA from each individual from a quartered 2-mm biopsy ear punch. DNA extraction and polymerase chain reaction (PCR) protocols were followed in accordance with Louis et al. (2005) to generate genotype data using 24 microsatellite loci developed from the V. variegata genome. PCR products were electrophoresed on an ABI 3100 automated DNA analyzer (Applied Biosystems, Foster City, CA, USA). Fragment lengths were assigned with Genescan-500 (ROX), and allele scoring was conducted with Genescan software (Applied Biosystems).
Data analysis
We checked the genotype file with both Micro-Checker (van Oosterhout et al. 2004) and Microsatellite Analyzer (MSA; Dieringer and Schlötterer 2002), which detect typographical errors, significant null allele problems, and scoring errors due to allelic stuttering and large allele dropout that can significantly affect Hardy–Weinberg equilibrium (HWE) estimates (Morin et al. 2009). Next, CERVUS 2.0 (Slate et al. 2000) was used to estimate null allele frequencies for each locus and the utility of the markers as polymorphic information content (PIC). We opted to delete the few loci with moderate null allele frequencies (nf > 0.1) to reduce the bias from misclassifications of null heterozygotes as homozygotes (Callen et al. 1993; Hoffman and Amos 2005) and thus control the variance of parameter estimates that would be caused by excessive null allele frequencies in the analysis according to the result of Chapuis and Estoup (2007) who demonstrated the effects of moderate (0.05 < nf < 0.20) and high (0.20 < nf) null frequencies on population-genetic parameter estimates.
We verified the accepted loci for independence of linkage disequilibrium (LD), with Bonferroni-adjusted P values, in FSTAT (Goudet 1995, 2001). We tested the set of microsatellite loci globally and by population for deviation from HWE via Fisher’s exact probability test in the web-based Genepop3.4 (Raymond and Rousset 1995). Initially, we used the default settings for the Markov chain Monte Carlo (MCMC) estimation of HWE and then increased the batch size from 100 to 250 to reduce the standard error (SE) of the P value to ≤0.01. We tested genic differentiation to determine whether or not allelic distributions were the same in both populations, then we examined genotypic differentiation to determine whether or not genotype distributions were the same. Genic differentiation was tested with Fisher’s exact test. We used an unbiased estimate of the P value as G (log likelihood of an exact test) to test genotypic differentiation. Both were performed by locus and pair-wise differences between populations in GenePop 3.4. We estimated H S (gene diversity) and AR (rarefaction allelic richness) by locus and population, f as Wright’s F IS (within population f statistic) and θ as Wright’s F ST (among population f statistic) using Weir and Cockerham (1984) and G ST (analogous g statistic) with FSTAT. We estimated gene flow between populations as N M (migrants per generation) using the private alleles method in Genepop 3.4. We tested for sex-biased dispersal of individuals from social groups using the methodology of Goudet et al. (2002) as implemented in FSTAT. The test assumes even sex dispersal so that if the dispersal is biased, the dispersing sex or group is expected to have a positive and higher F IS, a lower F ST, and lower H S than the philopatric sex.
We estimated the number of effective breeders (N eb) using NeEstimator (Peel et al. 2004) to establish a range of N eb (Waples 1991). The two most commonly used estimators, LD (Hill 1981) and heterozygote excess (HEx) (Pudovkin et al. 1996), are applicable to single-sampled populations as opposed to temporally sampled populations. In using the latter method, Pudovkin et al. (1996) and Luikart and Cornuet (1999) found that it may overestimate N eb, whereas Waples (1991) found several flaws in the basic assumptions of the model. However, Balloux (2004) found that despite the limitations, HEx is advantageous for populations sampled once only rather than multiple times on a temporal scale. Therefore, as suggested by Waples (1991), we report both estimators in this study and compare them with the population census estimates based on density (Vasey 1997b) and our observations (MAS and DAMA, respectively).
Having met the minimum statistical threshold required by Bottleneck 2.0 (Cornuet and Luikart 1996; Cornuet et al. 1999; Piry et al. 1999), 20 alleles and 10 individuals per population, we examined both populations for evidence of a recent genetic bottleneck (Nei et al. 1975). We used the three models available: the infinite alleles model (IAM), the stepwise mutation model (SMM), and the two-phase model (TPM)—the latter permitting stepwise as well as multi-step mutation events (Di Rienzo et al. 1994). Additionally, under neutrality, alleles at low frequency (<0.01) are expected to outnumber those with intermediate frequencies, regardless of the underlying mutation model (Chakraborty et al. 1980; Luikart et al. 1998). Mutation-drift equilibrium is characterized by an L-shaped distribution, whereas a recent genetic bottleneck can be inferred by a shift in allele counts from low to intermediate frequency classes (Luikart et al. 1998) disrupting the expected L-shaped distribution.
As an exploratory measure to test for cryptic substructure in the populations, we used the model-based clustering program STRUCTURE 2.0 (Pritchard et al. 2000). The procedure uses a Bayesian clustering method to determine whether the two populations should be further subdivided into genetically distinct groups to reflect the population substructure. STRUCTURE endeavors to identify population subsets that are at HWE and minimize LD via multilocus genotypes (Pritchard et al. 2000). We used the ancestry model, correlated allele frequencies, different F ST values assumed for each subpopulation, a uniform prior for alpha [maximum 10, standard deviation (SD) for updating 0.025], constant lambda value of 1, prior F ST mean (0.01), and SD (0.05). We analyzed data for K = 1–6 genetic clusters, as Evanno et al. (2005) suggests, estimating a range of at least three clusters more than the number of sampling locations. We set the initial burn-in period at 105 iterations to reduce noise before launching 106 MCMC repetitions to obtain genetic cluster proportion estimates (q) for each individual. Occasionally, STRUCTURE overestimates the number of genetic clusters in a data set by generating similar likelihood values for adjacent values. We used the ad hoc test statistic ΔK (Evanno et al. 2005), developed to overcome that limitation, to aid in elucidating the most likely number of genetic clusters. We then compared the model with the highest posterior probability and the model with the maximum ΔK to the sampled demographic populations. Whereas Cornuet and Luikart (1996) and Paetkau et al. (2004) warn of small sample size bias in their simulations, Pritchard et al. (2000) demonstrated the utility of the program with small data sets (n = 4 individuals typed with 7 loci).
We used GeneClass 2.0 (Piry et al. 2004) to co-analyze the substructure for migrant probabilities. This nonequilibrium approach uses Bayesian methodology to identify migrants (Rannala and Mountain 1997) and assign individuals of unknown population affinity to potential source populations using multilocus genotypes (Paetkau et al. 1995; Rannala and Mountain 1997; Cornuet et al. 1999). To identify first-generation migrants, we computed the L value or likelihood ratio from the population from which the individual was sampled (L-home) over the highest likelihood value among all populations sampled (L-max; Paetkau et al. 2004). We calculated the logarithm of the likelihood odds ratio (LOD) for each individual’s assignment to the most likely population of origin by jackknifing the data set in the WHICHRUN population assignment program (Banks and Eichert 2000).
We acknowledge that implicit theoretical assumptions built into analysis programs may be somewhat violated. We tested for HWE at the locus and population levels and for independence between the marker loci assuming equal sex ratios, random mating, and discrete generations, though in most in situ studies, these assumptions are often overlooked in the absence of pedigree information. Finally, the minimum sample size (n = 10) and locus number (n = 10) were met for the most stringent program, Bottleneck 2.0 with n = 10 individuals (DAMA) and n = 15 loci.
Results
Of the 24 loci amplified, we found that two markers that were polymorphic in V. variegata were monomorphic among the V. rubra samples and an additional seven loci had moderately high or high null allele frequencies (nf > 0.10, data not shown). The seven loci were dropped from the genotyping suite, resulting in 15 informative microsatellites (Table 1). None of the locus pairs tested significant for LD, thus, marker independence was assumed. We report the relative quality of the loci as PIC values in Table 2 for the use of researchers who might evaluate the application of these markers for future studies. The mean number of alleles was 6.2, ranging in count from 3 (51HDZ339) to 10 (51HDZ20) alleles per locus. None of the loci and neither of the two sample sites deviated by exact tests from HWE, thus meeting the equilibrium assumptions set in the analysis programs. Several loci varied considerably in AR between the two populations, though the difference in the population averages was not significant (Table 2).
We present population genetic parameter estimates by locus and population in Table 2 to establish baseline data for V. rubra. We estimated the genetic differentiation between the MAS and DAMA populations by f and g statistics: θ as 0.077 and g as 0.039. We found both genic and genotypic differentiations to be highly significant (P < 0.00001) between the two sampling locations. In both populations, we found that the observed heterozygosity was slightly but not significantly higher (0.651 and 0.681) than the expected heterozygosity (0.629 and 0.656, P = 0.09 and P = 0.24, MAS and DAMA, respectively). We also found that the F IS estimate was negligible and nonsignificant in both populations (−0.034 and −0.035, MAS and DAMA, respectively).
We estimated the N eb for the MAS (31.7, 151.4; LD and HEx models, respectively) and DAMA (10.2, 22.4; LD and HEx models, respectively) populations. We found no evidence in the MAS population of a genetic bottleneck under any model. However, we found in the DAMA population a significant heterozygosity excess departing from mutation-drift equilibrium under the IAM (P < 0.001) with continuing significance using the TPM up to a 68% SMM contribution (P < 0.05) and a shift in the modal allele distribution from low to intermediate frequency classes disrupting the expected L-shaped allele frequency distribution.
From the test for sex-biased dispersal, estimates for F IS, F ST, and H S were −0.022, 0.063, and 0.626 for males and −0.040, 0.087, and 0.649 for females, respectively. Neither sex had a significantly higher (or positive) estimator for F IS (P = 0.40), lower estimator for F ST (P = 0.23), or lower H S (P = 0.80). Thus, dispersal was not favored in either sex. We estimated an effective gene flow of 1.09 migrant exchanges per generation following a correction for sample size, indicating movement or gene flow between the populations.
We found no evidence of substructure or significant admixture of ancestral genetic architectures in the DAMA population. The posterior likelihood of the data maximized at K = 2 (log likelihood = −1,349.9) in the samples as supported by the ad hoc test statistic, Evanno et al.’s (2005) ΔK (data not shown). In the MAS population, 67.6% of the genetic pool was attributed to cluster 1 and 32.4% to cluster 2. In the DAMA population, only 5.9% of the genetic pool was attributed to cluster 1 and 94.1% to cluster 2. Two types of individuals with marked differences in ancestral genetic architecture were detected in MAS: one representative of DAMA (white) and the second clustering (grey) only in MAS (Fig. 2).

Genotypic proportion of the Masiaposa (MAS) and Ambatoledama (DAMA) Varecia rubra individuals with individual identification on the x-axis and the proportion of admixture (q) depicted on the y-axis
Two individuals (MAS8 and MAS17) in the MAS sample set were identified as first-generation migrants from the DAMA population with GENECLASS2 (P < 0.05). WHICHRUN marginally assigned MAS8 (f, LOD 2), MAS15 (m, LOD 2), and MAS17 (m, LOD 1) to DAMA and MAS1 (f, LOD 2), MAS14 (f, LOD 2), MAS24 (f, LOD 2) and MAS32 (m, LOD 2) to MAS. No DAMA-sampled individuals were assigned to the MAS population (LOD ≥ 3). Correct assignment in this data set was 84%.
Discussion
Red ruffed lemur populations have been drastically reduced (Tattersall 1977; Petter et al. 1977; Hekkala et al. 2007; Goodman and Ganzhorn 2004). Substantial populations are found only on the Masoala Peninsula (CBSG 2002; Vasey 2003). Whereas most of the peninsula is protected as MasNP, anthropogenic pressures such as fuel-wood harvesting, tavy (slash-and-burn clearing), and local subsistence hunting affect red ruffed lemur populations, with hunting among the top threats (Vasey 1997b; Hekkala et al. 2007).
From this study, we established baseline population genetic parameters for red ruffed lemurs from DAMA and MAS forests. Although the sampling sites are approximately 40 km apart, a significant habitat corridor connects them. The two genetic clusters represented by the MAS and DAMA populations are not clearly defined. The genetic imprint represented by the DAMA genetic cluster in the MAS population may be an indication of the utilization of the reforested DAMA corridor. Based on the size of the MAS sampling area, Vasey’s (1997b) red ruffed lemur (N eb) density estimates were used to calculate an expected population size of 124.8–214.8 individuals. In this study, the number of N eb of the MAS forest was estimated to be 31.7 by the LD method and 151.4 by the HEx method. The DAMA population was the focus of a long-term study in which population demographics were well known to the research team. A total of five social groups were observed, each composed of an adult pair with two to three nursing juveniles for a total population census at the time of our sample collection of 25 individuals (B. Martinez, personal communication). We estimate the N eb of the DAMA population to be 10.2 by the LD method and 22.4 by the HEx method. The introduction of sampling bias due to small sample size is often considered problematic. In this case, the sample size consisted of all of the adults, representing 40% of the total population, in the DAMA forest corridor. Moreover, for the estimation of N eb, precision of estimation is more affected by the number of loci used. The potential for bias in the estimate and the noise or sampling variance decreases by increasing the number of loci from 8 or 10 to 15 (Waples 1991). In this case, we feel confident that the sample size is not adversely affecting the parameter estimates, as the LD method accurately estimated N eb, which was the same as the census number of adults in the population. The HEx method, on the other hand, overestimated N eb, which is often the case.
Data indicates that DAMA suffers from a demographic bottleneck or reduction of the effective population size, implying potential genetic consequences for future generations (Garza and Williamson 2001), whereas MAS does not. The negative F IS suggests that despite the reduction in effective population size, the population is likely not undergoing detectable inbreeding (Pollack 1987; Frankham et al. 2004).
The sister species, V. variegata, disperses before reproduction and is not sex-biased according to the few records available (Ratsimbazafy 2002). In the MAS and DAMA populations, no significant differences in estimators for F IS, F ST, or H S between the sexes were identified. Thus, there was no evidence to reject the null hypothesis of equal sex dispersal. However, we did detect a genetic signal of gene flow between the DAMA and MAS populations at an effective migrant (N M) estimate of one individual per generation. From the STRUCTURE analysis, we found two genetic clusters, supported by both the asymptote of the likelihood curve and ΔK. We found individuals of both sexes in MAS populations, with admixture representative of the genetic cluster found in the DAMA population. This is supported by the GENECLASS2 analysis, which identifies two individuals as DAMA migrants (MAS8 and MAS17). The WHICHRUN population assignment misassigned the same two individuals and MAS15 to DAMA (Fig. 2) and four more MAS-sampled individuals to the MAS population but without significant support (MAS1, MAS14, MAS24, and MAS32; Fig. 2). From the samples we collected, the movement appears to be unilateral, as we found no evidence of individuals harboring admixture representative of the MAS population in the DAMA sample set.
At least two hypotheses could be entertained that might generated these results. One thought could be that a founding group of individuals with genetic architecture more similar to the minority of individuals in the MAS forest population might have left the pristine forest and migrated into the disturbed DAMA forest region to inhabit the newly planted forest corridor. The second might be that individuals in the DAMA forest region could be moving out of this highly disturbed region and beginning to encroach upon the pristine MAS forest habitat. Either could result in a recent population bottleneck in the DAMA forest region and a higher proportion of one genetic cluster in the DAMA compared with the MAS forest.
As red ruffed lemurs are now confined to such a restricted region of the island, populations are especially imperiled by current anthropocentric habitat fragmentation and from stochastic events such as the major cyclones that devastated Manombo Special Reserve on Madagascar’s southeastern coast in 1997 (Louis et al. 2005). The results from this study provide a sound baseline for future population studies of this species on the Masoala Peninsula. We suggest that the detection of unilateral movement from DAMA into MAS, coupled with evidence of a population bottleneck in DAMA, suggests our hypothesis of movement by DAMA individuals into the far less human populated MAS forest. Whereas evidence supports that anthropogenic pressure may be more adversely affecting the populations of the northern part of the park, additional populations between the DAMA and MAS forests should be sampled and studied to confirm, reject, or expand on this hypothesis.
References
Allnutt TF, Powell GVN, Ferrier S, Ricketts TH, Steininger MK, Manion G (2004) Quantifying biodiversity loss in Madagascar using biological inventories, environmental data and a 50-year record of deforestation. Society for Conservation Biology Meetings, New York, 30 July–22 August 2004
Balko EA (1998) A behaviorally plastic response to forest composition and logging disturbance by Varecia variegata in Ranomafana national park, Madagascar. PhD dissertation, Syracuse University
Balko EA, Underwood HB (2005) Effects of forest structure and composition on food availability for Varecia variegata at Ranomafana National Park, Madagascar. Am J Primatol 66(1):45–70
Balloux F (2004) Heterozygote excess in small populations and the heterozygote-excess effective population size. Evolution 58(9):1891–1900
Banks MA, Eichert W (2000) WHICHRUN (version 3.2) a computer program for population assignment of individuals based on multilocus genotype data. J Hered 91:87–89
Callen DF, Thompson AD, Shen Y, Phillips HA, Richards RI, Mulley JC, Sutherland GR (1993) Incidence and origin of “null” alleles in the (AC)n microsatellite markers. Am J Hum Genet 52:922–927
Chakraborty R, Fuerst PA, Nei M (1980) Statistical studies on protein polymorphism in natural populations. III. Distribution of allele frequencies and the number of alleles per locus. Genetics 94:1039–1063
Chapuis M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24(3):621–631
Conservation Breeding Specialist Group (SSC/IUCN) (2002) Evaluation et Plans de Gestion pour la Conservation (CAMP) de la Fauna de Madagascar: Lémuriens, Autre Mammifères, Reptiles et Amphibiens, Poissons d’eau douce et Evaluation de la Viabilitiédes Populations et des Habitats de Hypogeomys antimena (Vositse). CBSG, Apple Valley
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Cornuet JM, Piry S, Luikart G, Estoup A, Solignac M (1999) New methods employing multi-locus genotypes to select and exclude populations as origins of individuals. Genetics 153:1989–2000
Di Rienzo A, Peterson AC, Garza JC, Valdez AM, Slatkin M, Freimer NB (1994) Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci USA 91:3166–3170
Dieringer D, Schlötterer C (2002) Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes 1(3):167–169
Dutton CJ, Junge RE, Louis EE (2008) Biomedical evaluation of free-ranging red ruffed lemurs (Varecia rubra) within Masoala National Park, Madagascar. J Zool Wildl Med 39(1):76–85
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Frankham R, Ballou JA, Briscoe DA (2004) Introduction to conservation genetics. Cambridge University Press, Cambridge, p 536
Garza JC, Williamson E (2001) Detection and reduction population size using data from microsatellite DNA. Mol Ecol 10:305–318
Geoffroy-Saint Hilare E (1812) Notes sur trios dessins de Commerçon représentant des Quadrumanes d’un genre inconnu. Ann Mus Hist Nat Paris 19:171–175
Gligor M, Ganzhorn JU, Radotondravony D, Ramilijaona OR, Razafimhatratra E, Zischler H, Hapke A (2009) Hybridization between mouse lemur species in an ecological transition zone in southern Madagascar. Mol Ecol 18:520–533
Goodman SM, Ganzhorn JU (2004) Biogeography of lemurs in the humid forests of Madagascar: the role of elevational distribution and rivers. J Biogeogr 31(1):47–55
Goudet J (1995) FSTAT, a computer program to test F-statistics. J Hered 86:485–486
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Updated from Goudet 1995
Goudet J, Perrin N, Wasser P (2002) Tests for sex-biased dispersal using bi-parentally inherited genetic markers. Mol Ecol 11:1103–1114
Green C, Sussman RW (1990) Deforestation history of the eastern rain forests of Madagascar from satellites images. Science 248:212–215
Hannah L, Rakotosamimanana B, Ganzhorn J, Mittermeier RA, Olivieri S, Iyer L, Rajaobelina S, Hough J, Andramialisoa F, Bowles I, Tilkin G (1998) Participatory planning, scientific priorities and landscape conservation in Madagascar. Environ Conserv 25:30–36
Harper GJ, Steininger MK, Tucker CJ, Juhn D, Hawkins F (2007) Fifty years of deforestation and forest fragmentation in Madagascar. Environ Conserv 34(4):1–9
Hekkala ER, Rakotondratsima M, Vasey N (2007) Habitat and distribution of the ruffed lemur, Varecia, north of the Bay of Antongil in northwestern Madagascar. Primate Conserv 22:89–95
Hill WG (1981) Estimation of effective population size from data on linkage disequilibrium. Genet Res 38:209–216
Hoffman JI, Amos W (2005) Microsatellite genotyping errors: detection approaches, common sources and consequences for parental exclusion. Mol Ecol 14:599–612
Holloway L (2000) Catalysing rainforest restoration in Madagascar. In: Lorenço WR, Goodman SM (eds) Diversité et Endémisme à Madagascar. Mémoires, Société de Biogéographie, Paris, pp 115–124
Jenkins MD (ed) (1990) Madagascar: an environmental profile. IUCN Conservation Monitoring Centre, Gland
Lawler R (2008) Testing for a historical bottleneck in wild Verreaux’s sifaka (Propithecus verreauxi verreauxi) using microsatellite data. Am J Primatol 70:990–994
Louis EE Jr, Ratsimbazafy JH, Razakamaharavo VR, Pierson DJ, Barber RC, Brenneman RA (2005) Conservation genetics of black and white ruffed lemurs (Varecia variegata variegata) from Southeastern Madagascar. Anim Conserv 8:105–111
Luikart G, Cornuet J-M (1999) Estimating the effective number of breeders from heterozygote excess progeny. Genetics 151:1211–1216
Luikart G, Allendorf FW, Cornuet J-M, Sherwin WB (1998) Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89:238–247
Markolf M, Roos C, Kappeler P (2008) Genetic and demographic consequences of a rapid reduction in population size in a solitary lemur (Mirza coquereli). Open Conserv Biol J 2:21–29
Merenlender A, Kremen C, Rakotondratsima M, Weiss A (1998) Monitoring impacts of natural resource extraction on lemurs of the Masoala peninsula, Madagascar. Conserv Ecol 2(2):5
Mittermeier RA, Langrand O, Lowry PP II, Schatz G, Gerlasch J, Goodman SM, Steininger M, Hawkins F, Raminosoa N, Ramilijaona OAL, Andriamaro O, Randrianasolo H, Rabarison H, Rakotobe ZL (2004) Hotspots revisited: earth’s biologically richest and most endangered terrestrial ecosystems. CEMEX, USA
Morin PA, Leduc RG, Archer FI, Martien KK, Huebinger R, Bickham JW, Taylor BL (2009) Significant deviations from Hardy–Weinberg equilibrium caused by low levels of microsatellite genotyping errors. Mol Ecol Res 9:498–504
Morland HS (1991) Social organization and ecology of black and white ruffed lemurs (Varecia variegata variegata) in lowland rain forest, Nosy Mangabe, Eastern Madagascar. Folia Primatol 56:253–265
Myers N (1988) Threatened biotas: “hotspots” in tropical forests. Environmentalist 8:1–12
Myers N (1998) Lifting the veil on perverse subsidies. Nature 392:327–328
Myers N, Mittermeier RA, Mittermeier CG, da Fonseca GAB, Kent J (2000) Biodiversity hotspots for conservation priorities. Conserv Biol 12:516–520
Nei M, Maryuama T, Chakraborty R (1975) The bottleneck effect and genetic variability in population size using data from microsatellite DNA. Mol Ecol 10:305–318
Nelson R, Horning N (1993) Cover forest/non-forest classification of Madagascar from AVHRR data. Int J Remote Sens 14(8):1445–1446
Nicoll ME, Lagrand O (1993) AVHRR-LAC estimates of forested area in Madagascar, 1990. Int J Remote Sens 14(8):1463–1475
Olivieri GI, Sousa V, Chikhi L, Radespiel U (2008) From genetic diversity and structure to conservation: genetic signature of recent population declines in three mouse lemur species (Microcebus spp.). Biol Conserv 141:1257–1271
Paetkau DW, Calvert W, Stirling I, Strombeck C (1995) Microsatellite analysis of population structure in Canadian polar bears. Mol Ecol 4:347–354
Paetkau D, Slade R, Burden M, Estoup A (2004) Direct real-time estimation of migration rate using assignment methods: a simulation-based exploration of accuracy and power. Mol Ecol 13:55–65
Patel E, Andrianandrasana LH (2008) Low elevation silky sifakas (Propithecus candidus) in the Makira Conservation Site at Andaparaty-Rabeson: ranging, demography, and possible sympatry with red ruffed lemurs (Varecia rubra). Lemur News 13:18–22
Peel D, Ovenden JR, Peel SL (2004) NeEstimator: software for estimating effective population size, version 1.3. Queensland Government, Department of Primary Industries and Fisheries
Petter JJ, Albignac R, Rumpler Y (1977) Faunne de Madagascar 44: Mammifères lémuriens (Primates prosimiens). ORSTOM/CNRS, Paris
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503
Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L, Estoup A (2004) GeneClass2: a software for genetic assignment and first generation migrants detection. J Hered 95:536–539
Pollack E (1987) On the theory of partially inbreeding finite populations. I. Partial selfing. Genetics 117:353–360
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pudovkin AI, Zaykin DV, Hedgecock D (1996) On the potential for estimating the effective number of breeders from heterozygote-excess progeny. Genetics 144:383–387
Quéméré E, Louis EE Jr, Ribéron L, Chikhi L, Crouau-Roy B (2009) Non-invasive conservation genetics of the critically endangered golden-crowned sifaka (Propithecus tattersalli): high diversity and significant genetic differentiation over a small range. Conserv Genet. doi:10.1007/s10592-009-9837-9
Radespiel U, Rakotondravony R, Chikhi L (2008) Natural and anthropogenic determinants of genetic structure in the largest remaining population of the endangered golden-brown mouse lemur, Microcebus ravelobensis. Am J Primatol 70:860–870
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA 94:9197–9201
Ratsimbazafy JH (2002) On the brink of extinction and the process of recovery: responses of black and white ruffed lemurs (Varecia variegata variegata) to disturbances in Manombo. PhD dissertation, State University of New York, Stony Brook
Raymond M, Rousset F (1995) GENEPOP (Version3.1): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rigamonti MM (1993) Home range and diet in red ruffed lemurs (Varecia variegata rubra) on the Masoala peninsula, Madagascar. In: Kappeler PM, Ganzhorn JU (eds) Lemur social systems and their ecological basis. Plenum Press, New York, pp 25–39
Simons HJ, Lindsay NB (1987) Survey work on ruffed lemurs (Varecia variegata) and other primates in the northeastern rainforests of Madagascar. Primate Conserv 8:88–91
Slate J, Marshall TC, Pemberton JM (2000) A retrospective assessment of the accuracy of the paternity inference program CERVUS. Mol Ecol 9(6):801–808
Tattersall I (1977) Distribution of Malagasy lemurs. Part 1: the lemurs of northern Madagascar. Annal N Y Acad Sci 293:160–169
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Vasey N (1997a) Community ecology and behavior of Varecia variegata rubra and Lemur fulvus albifrons on the Masoala Peninsula, Madagascar. PhD dissertation, Washington University, St. Louis
Vasey N (1997b) How many red ruffed lemurs are left? Int J Primatol 18(2):207–216
Vasey N (2002) Niche separation in Varecia variegata rubra and Eulemur fulvus albifrons: II. Intraspecific patterns. Am J Phys Anthropol 118(2):169–183
Vasey N (2003) Varecia, ruffed lemurs. In: Goodman SM, Benstead JP (eds) The natural history of Madagascar. University of Chicago Press, Chicago, pp 1332–1336
Vasey N (2006) Impact of seasonality and reproduction on social structure, ranging patterns, and fission–fusion social organization in red ruffed lemurs. In: Gould L, Sauther ML (eds) Lemurs: ecology and adaptation. Springer, New York, pp 275–304
Vasey N, Tattersall I (2002) Do ruffed lemurs form a hybrid zone?: distribution and discovery of Varecia with systematic and conservation implications. Am Mus Novit 3376:1–26
Waples RS (1991) Genetic methods for estimating the effective size of Cetacean populations. In: Hoezel AR (ed) Genetic ecology of whales and dolphins, special issue 13. International Whaling Commission, London, pp 279–300
Weir B, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38(6):1358–1370
White FG (1991) Social organization, feeding ecology and reproductive strategy of ruffed lemurs (Varecia variegata). In: Ehara A, Takenaka O (eds) Primatology, proceedings of the XIII congress of the IPS, Iwamoto, Nagoya and Kyoto. Elsevier Science, Amsterdam
Acknowledgments
This project would not have been possible without the support of the staff of the Institute for Conservation of Tropical Environments, Madagascar (ICTE-MICET), as well as, the National Parks Madagascar (NPM), US Fish and Wildlife Service, and the Ministère des Eaux et Forêts of Madagascar. We thank Bill and Berniece Grewcock for their support of student interns, the Ahmanson Foundation for their generous support of DNA sequencers and other laboratory components, and the Theodore F. and Claire M. Hubbard Family Foundation for supporting conservation projects undertaken by the HDZ Madagascar Biodiversity and Biogeography Project. We thank the late Prof. Randria Ravololonarivo Gisele for her support of graduate students and the Department of Anthropology and Paleontology at the University of Antananarivo. Finally, we thank the three reviewers whose helpful comments greatly improved the manuscript. No animals were injured in this study, and the research protocols were performed within the guidelines of the Primate Society of Japan, the HDZ-IACUC #97-001, and in compliance with the care and expectations of local, national, and international laws and standards.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Razakamaharavo, V.R., McGuire, S.M., Vasey, N. et al. Genetic architecture of two red ruffed lemur (Varecia rubra) populations of Masoala National Park. Primates 51, 53–61 (2010). https://doi.org/10.1007/s10329-009-0171-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10329-009-0171-0