
Abstract
Cyanidin-3-O-glucoside (C3G), an anthocyanin belonging to the flavonoid family and commonly present in food and vegetables in human diet, has exhibited anti-inflammatory and anti-oxidant effects. This study aimed to investigate the protective ability of C3G against inflammatory and oxidative injuries, as well as to clarify the possible mechanism in lipopolysaccharide (LPS)-stimulated human umbilical vein endothelial cells (HUVECs) in vitro and acute respiratory distress syndrome mouse model in vivo. HUVECs or male Kunming mice were pretreated with C3G 1 h before LPS stimulation. C3G significantly inhibited the production of pro-inflammatory cytokines (tumor necrosis factor-α, interleukin (IL) -6, and IL-1β) in cell supernatants and bronchoalveolar lavage fluid (BALF) as determined by enzyme-linked immunosorbent assay. Histopathologic examination with hematoxylin and eosinstaining showed that C3G pretreatment substantially suppressed inflammatory cell infiltration, alveolar wall thickening, and interstitial edemain lung tissues. C3G markedly prevented LPS-induced elevation of malondialdehyde and myeloperoxidase levels in lung tissue homogenates, wet to dry ratio of lung tissues, total cells, and inflammatory cells (neutrophils and macrophages) in BALF. Moreover, C3G reduced superoxide dismutase activity in the lung tissue homogenates. Western blot assay also showed that C3G pretreatment significantly suppressed LPS-induced activation of nuclear factor-kappaB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways by blocking the phosphorylation of inhibitor κB-α, NF-κB/P65, extracellular signal-regulated kinase, p38, and c-Jun NH2-terminal kinase in the lung tissues. In summary, C3G may ameliorate LPS-induced injury, which results from inflammation and oxidation, by inhibiting NF-κB and MAPK pathways and playing important anti-inflammatory and anti-oxidative roles.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Acute respiratory distress syndrome (ARDS) is the more severe form of acute lung injury (ALI). ARDS is a complex devastating clinical syndrome characterized by increased production of pro-inflammatory cytokines, neutrophil accumulation, disruption of pulmonary endothelial and epithelial cell capillary barriers, and leakage of proteins into alveolar space [1, 2]. Diverse predisposing factors participate in ARDS development. Such factors include sepsis, shock, and pneumonia. Sepsis and sepsis-related ARDS significantly contribute to worldwide morbidity and mortality [3, 4]. As a cell wall component of gram-negative bacteria, lipopolysaccharide (LPS) is an important pathogen leading to ARDS [5]. Experimental administration of LPS has also been used to induce ARDS in animal models. Activation of novel therapeutic targets, namely, nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinase (MAPK), is an important step in ARDS development [6]. Upon exposure to LPS, NF-κB will be activated and will release numerous cytokines, including tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6, as well as reactive oxygen species, which play a critical role in ARDS [7, 8]; thus, MAPK pathways, such as p38 MAPK, c-Jun NH2-terminal kinase (JNK), and extracellular signal-regulated kinase (ERK), also participate in ARDS development.
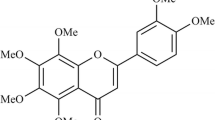
Anthocyanins, which belong to the family of flavonoids in nature, exhibit various health benefits, primarily anti-oxidative and anti-inflammatory properties. Cyanidin-3-O-glucoside (C3G; Fig. 1), which is one of the most abundant anthocyanins [9] commonly present in food and vegetables in human diet, exhibits anti-inflammatory and anti-oxidative effects [10, 11]. Based on recent studies, we hypothesized that C3G might exert preventive and therapeutic effects on ARDS. Thus, our main design assessed the protection afforded by C3G in an ARDS model in vivo, as well as in human umbilical vein endothelial cells (HUVECs) in vitro, to further investigate the possible protective mechanisms related to the suppression of NF-κB and MAPK pathways.

Chemical structures of cyanidin-3-O-glucoside.
MATERIALS AND METHODS
Animals
Adult male Kunming mice (age 8 to 10 weeks, weighing 18 to 20 g) were purchased from Shandong Lvye Pharmaceutical Company (Yantai, PR China). All animals were housed individually at 22 ± 2 °C and 50 ± 10 % relative humidity with a 12 h light/dark cycle. They were allowed with free access to food and water. The animals were used in complete compliance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) revised 1996. The mice were allowed to adapt to the environment for 2 days to 3 days before experimentation.
Chemicals and Reagents
C3G was purchased from Dalian Meilun Biology Technology Co., Ltd. (Liaoning, China) with purity >98 %. LPS (Escherichia coli LPS, 055:B5), dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-y1)-2,5-diphenyltetrazolium bromide (MTT), sodium dodecyl sulfate (SDS), phenylmethylsulfonyl fluoride, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and streptomycin/penicillin were purchased from Sigma Aldrich Co. Mouse TNF-α, IL-6, and IL-1β enzyme-linked immune sorbent assay (ELISA) kits were purchased from BioLegend (San Diego, CA, USA). Rabbit mAb, NF-κB/P65, inhibitor κB (IκB)-α, p44/42 Erk, phospho-p44/42 Erk, p38, phospho-p38, SAPK/JNK, and phospho-SAPK/JNK were purchased from Cell Signaling Technology, Inc. (MA, USA). Horseradish peroxidase -conjugated goat anti-rabbit antibodies were provided by GE Healthcare (Buckinghamshire, UK).
In Vitro Study
Cell Culture and Treatment
HUVECs were purchased from the China Cell Line Bank (Beijing, China). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % heat-inactivated fetal bovine serum (Invitrogen/Gibco Life Technologies, Carlsbad, CA), penicillin (100 U/mL), and streptomycin (100 g/mL) in a humidified atmosphere of 5 % CO2 and 95 % air at 37 °C. Cells were sub-cultured at confluence and used between the third and eighth passages for all experiments. When the cells were grown to confluence, the culture medium was replaced with the serum-free medium for an additional 24 h incubation to render the cells quiescent before adding stimulus. For all experiments, C3G was consistently dissolved in DMSO and used immediately. The final concentration of DMSO in the culture medium upon different treatments was <0.1 %. All the endothelial cells were incubated with or without various concentrations of C3G, which were always added 1 h prior to LPS (1 μg/mL) stimulation. Control cells were not exposed to LPS.
MTT Assay for Cell Viability
Cell viability was assessed by MTT formazan, which is directly proportional to the number of living cells. HUVECs were plated at a density of 4 × 105 cells/mL onto 96-well plates in a 37 °C, 5 % CO2 incubator for 1 h. The cells were then treated with different concentrations of C3G (0 to 200 μg/mL and 50 μL/well) for 1 h, followed by stimulation with LPS (1 μg/mL and 50 μL/well). After 18 h, 20 μL of MTT (5 mg/mL) was added to each well. The cells were further incubated for 4 h at 5 % CO2 and 95 % air at 37 °C. The supernatant was then removed, and formazan formation was resolved with 150 μL/well of DMSO. The optical density was measured at 570 nm on a microplate reader. Concentrations were determined for three wells of each sample, and each experiment was performed in triplicate.
Cytokine Assays In Vitro
Cells were plated onto 24-well plates (105 cells per well) and added with diverse concentrations of C3G (0, 12.5, 25, and 50 μg/mL). After 1 h, cells were incubated with or without LPS (1 μg/mL) in a 37 °C, 5 % CO2 incubator for 6 h. The control group was added with equal volume of DMEM. The collected cell supernatants were assayed with cytokines by ELISA for TNF-α, IL-6, and IL-1β in accordance with the manufacturer’s instructions.
In Vivo Study
Experimental Design
A pilot study was conducted with C3G at a single dose of 12.5,25,50, and 100 mg/kg to determine the dose-dependent effect on LPS-induced ARDS mice. At 6 h after, the mice were induced with LPS (12.5 to 50 mg/kg); C3G significantly reduced the elevated concentrations of TNF-α, IL-6, and IL-1β levels in bronchoalveolar lavage fluid (BALF), as well as the superoxide dismutase (SOD), and malondialdehyde (MDA) levels in lung tissue homogenates. By contrast, C3G improved the lung tissue homogenate level of myeloperoxidase (MPO). Hence, 50 mg/kg of C3G was the selected dose for a follow-up study.
Experimental animals were randomly allocated into three groups, namely, the control group, LPS group, and C3G + LPS group. C3G (50 mg/kg) was intraperitoneally injected to animals. After 1 h, the LPS and C3G + LPS groups were intraperitoneally injected with LPS (20 mg/kg) to induce lung injury. The control group received equal volume of normal saline instead of LPS in the same manner. All the mice were alive after 6 h LPS stimulation.
BALF Collection
At 6 h after LPS stimulation, BALF collection was performed thrice through a tracheal cannula with autoclaved PBS and instilled up to a total volume of 1.5 mL. The selected doses of these drugs were based on our previous studies and preliminary experiments. The recovery rate of BALF was >90 %. The BALF recovered from each sample was immediately centrifuged (4 °C, 3,000 rpm, 10 min).
Inflammatory Cell Counts and Measurement of Cytokines in BALF
The sediment cells were resuspended in 50 μL of PBS, and cell differentiation was determined using a hemocytometer. Differences in cell numbers were examined by counting on a smear prepared by Wright-Giemsa staining. TNF-α, IL-1β, and IL-6 in the BALF were evaluated with the corresponding ELISA kitsin accordance with the manufacturer’s instructions.
Measurement of Lung Wet/Dry Weight Ratio
After the mice were euthanized and the lungs were excised by blunt dissection, the lungs were blotted dry, weighed to obtain the “wet” weight, and then placed in an oven at 80 °C for 48 h to acquire the “dry” weight. The lung wet/dry (W/D) weight ratio was calculated to assess tissue edema.
Histological Assessment
Histopathologic examination was performed on mice that were not subjected to BALF collection. After excision blunt dissection, the pulmonary tissue samples were fixed in normal 10 % neutral buffered formalin for 48 h, followed by dehydration in ascending series of alcohol and embedding in paraffin wax, then sliced. Sections (5 μm thick) were stained with hematoxylin and eosin. Pathological changes of lung tissues were observed under a light microscope. The rest of pulmonary tissues were flash frozen in liquid nitrogen and stored at −80 °C for analyses of MPO, MDA, and SOD.
Measurement of MPO, MDA, and SOD
Mice were sacrificed 6 h after LPS challenge under diethyl ether anesthesia. Lung tissues were frozen in liquid nitrogen and homogenized in PBS. The MPO activity in 10 % of lung tissue homogenates was determined using test kits purchased from Nanjing Jiancheng Bioengineering Institute (Jiangsu, China) in accordance with the manufacturer’s instructions. The rest of the homogenate dilution was centrifuged at 4,000×g/10 min at 4 °C. MDA and SOD levels were detected using the supernatants in accordance with the manufacturer’s instructions (Nanjing Jiancheng Bioengineering Institute, China).
Western Blot Analysis
At 6 h after LPS induction, the lung tissues were harvested and frozen in liquid nitrogen immediately until homogenization and stored at −80 °C until use. The extraction of nuclear proteins from lung tissues was performed using a nuclear and cytoplasmic protein extraction kit (Beyotime Institute of Biotechnology, China) following the manufacturer’s protocol. To extract the total protein from the lung tissues, protein concentrations were determined by BCA protein assay kit, and equal amounts of protein were loaded per well on a 10 % SDS polyacrylamide gel. Theproteins were then transferred onto polyvinylidene difluoride membranes. These membranes were washed in Tris-buffered saline with Tween 20 (TBST) and incubated in 5 % skim milk for 2 h at room temperature on a rotary shaker to reduce non-specific bindingand then washed again in TBST. Subsequently, the samples were probed overnight at 4 °C on the shaker with primary antibodies, including nuclear and cytoplasmic NF-κB/P65 and phosphorylated and non-phosphorylated forms of IκB-α, ERK, JNK, or p38 dilution in 1:1,000 primary antibody diluent. GAPDH Western blot was performed as an internal control of protein loading. The membrane was then washed with TBST followed by incubation with peroxidase-conjugated secondary antibody at room temperature for 1 h. Protein bands were detected by an ECL detection kit (Thermo Fisher Scientific, Inc.) in accordance with the manufacturer’s instructions.
Statistical Analysis
All values were expressed as means ± SEM. Data were analyzed using one-way ANOVA (Dunnett’s t test) and two-tailed Student’s t test. Two-tailed P < 0.05 or P < 0.01 was considered statistically significant. Statistical analyses were conducted with SPSS 13.0.
RESULTS
C3G at Some Concentrations Showed Nontoxic effect on HUVECs
After 18 h of cell incubation, the cell viability was evaluated by MTT assay to determine whether C3G exhibits potential cytotoxicity on HUVECs. Cells were incubated at concentrations ranging from 0 to 200 μg/mL with C3G. The results showed that C3G did not display any cellular toxicity against HUVECs at concentrations of 0, 12.5, 25, and 50 μg/mL. However, at 100 μg/mL, C3G exhibited cellular toxicity. The results showed that C3G at concentrations from 12.5 to 50 mg/L induced no cytotoxic effect on HUVECs (Fig. 2).

Effect of C3G on the viability of HUVECs. Cells were cultured with C3G (0 to 200 μg/mL). Cell viability was evaluated by MTT reduction assays. Data were presented as mean ± SEM of three independent experiments.
C3G Could Inhibit the Induction of TNF-α, IL-1β, and IL-6 After LPS Stimulation
TNF-α, IL-1β, and IL-6 concentrations in the culture supernatant of HUVECs were measured using the ELISA kits (Fig. 3a–c). Endothelial cells treated with LPS alone significantly increased TNF-α, IL-1β, and IL-6 compared with the control group. However, C3G pretreatment considerably inhibited the induction of TNF-α, IL-1β, and IL-6 upon LPS stimulation.

Effects of C3G on expression of pro-inflammatory cytokines in LPS-stimulated HUVECs. TNF-α (a), IL-1β (b), and IL-6 (c) concentrations in culture supernatants of HUVECs were measured using enzyme-linked immunosorbent assay (ELISA) kits. Values represented as mean ± SEM of three independent experiments, and the differences between mean values were assessed by Student’s t test. **P < 0.01 versus LPS group; *P < 0.05 versus LPS group; ##P < 0.01 versus control group.
C3G Could Decrease the Number of Cells in BALF of LPS-Induced mice
In the current study, we analyzed the total cells, neutrophils, and macrophages in BALF 6 h after LPS administration. Compared with the control group, the number of total cells, neutrophils, and macrophages were markedly increased after treatment with LPS alone (Fig. 4a–c). By contrast, C3G pretreatment significantly decreased the number of total cells, neutrophils, and macrophages in a dose-dependent manner.

Effects of C3G on inflammatory cell count in BALF of LPS-induced mice. Mice were intraperitoneally injected with C3G (12.5, 25, or 50 mg/kg) 1 h prior to LPS induction. BALF was collected at 6 h following LPS challenge to measure the numbers of total cells (a), neutrophils (b), and macrophages (c) in BALF 6 h after LPS administration. Values presented were mean ± SEM (n = 6 in each group). **P < 0.01 versus LPS group; ##P < 0.01 versus control group.
C3G Down-Regulated the Production of Cytokines in BALF in Different Concentrations
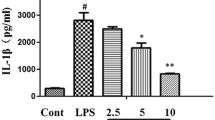
The levels of the cytokines TNF-α, IL-6, and IL-1β in BALF were analyzed 6 h after intraperitoneal injection of LPS to ensure the anti-inflammatory effects of C3G on pulmonary inflammation initiated by endotoxin. The levels of TNF-α and IL-6 were markedly elevated after LPS stimulation (Fig. 5a-c). However, after acute LPS stimulation, C3G downregulated the IL-6, IL-1β, and TNF-α in a dose-dependent manner at 12.5, 25, and 50 mg/kg, respectively, compared with the group treated with LPS alone.

Effects of C3G on concentrations of cytokine production in BALF. Levels of cytokines TNF-α (a), IL-1β (b), and IL-6 (c) in BALF were analyzed 6 h after intraperitoneal injection of LPS with corresponding ELISA kits. Values presented were mean ± SEM (n = 6 in each group). **P < 0.01 versus LPS group; ##P < 0.01 versus control group.
C3G Reduced the Severity of Histopathologic Changes in Lung Tissues
At 6 h after intraperitoneal injection of LPS, the sections of lung tissues were subjected to hematoxylin and eosin staining to evaluate the C3G effects on histopathologic changes in LPS-induced mice. In contrast with the lung specimens of the control group (Fig. 6a), those of the LPS group (Fig. 6b) were significantly damaged with inflammatory cell infiltration, alveolar wall thickening, and interstitial edema. For the C3G-treated groups (Fig. 6c), the severity of histopathologic changes in lung tissues were significantly reduced at the degree of inflammatory cell infiltration.

C3G Decreased the W/D Weight Ratio in LPS-Stimulated Mice Model
Lung tissue edema is one of the typical features of ARDS. In this study, we used lung W/D weight ratios to independently evaluate the LPS-induced changes in pulmonary vascular permeability to water (Fig. 7). LPS instilled for 6 h significantly increased the lung wet to dry weight ratio compared with the control group. By contrast, pretreatment with C3G could efficiently decrease the W/D weight ratio (Fig. 7).

Effects of C3G on lung W/D ratio in LPS-stimulated mice model. Mice were intraperitoneally injected with C3G (12.5, 25, or 50 mg/kg) 1 h prior to LPS induction. Lung W/D ratio was determined at 6 h after LPS challenge. Values presented were mean ± SEM (n = 6 in each group). **P < 0.01 versus LPS group; ##P < 0.01 versus control group.
C3G Increased the Level of SOD and Decreased the Content of MPO and MDA in Lung Tissues
Compared with the lung tissues of the control group, those of the LPS group exhibited significant increases in MPO (Fig. 8a) and MDA (Fig. 8b) levels. However, the MPO and MDA activities were reduced in the C3G-treatment group compared with those in the LPS group. After LPS administration, the SOD activity (Fig. 8c) in lung tissue was markedly decreased compared with that in the control group. However, the level of SOD increased in C3G-treated mice compared with that in the LPS group.

Effects of C3G on MPO, MDA, and SOD activities in LPS-induced mice. At 6 h after LPS instillation, lung homogenates were prepared for determination of MPO (a), MDA (b), and SOD (c) activities. Data were presented as mean ± SEM (n = 6 in each group). **P < 0.01 versus LPS group; ##P < 0.01 versus control group.
C3G Inhibited the Activation of NF-κB and MAPK Signal Pathway in LPS-Induced Mice
NF-κB (Fig. 9a) and MAPK (Fig. 9b) signaling pathways play important roles in the regulation of inflammatory process. Thus, we performed Western blot assay to investigate the effects of C3G on the activation of NF-κB-p65 subunits in the nucleus, as well as phosphor-IκB-α, phosphor-ERK, phosphor-JNK, phosphor-p38, and IκB-α in the lung tissues 6 h after LPS treatment. NF-κB-p65 and phosphor-IκB-α were markedly increased (Fig. 9) compared with the control group. The results also showed that LPS stimulation increased the phosphorylation of p38, JNK, and ERK after LPS administration. Pretreatment with C3G (50 mg/kg) inhibited the phosphorylation of IκB-α, p38, JNK, and ERK. Simultaneously, the C3G group markedly inhibited NF-κB-p65 translocation.


Effects of C3G on NF-κB activation in LPS-induced mice. Mice were intraperitoneally injected with C3G (12.5, 25, or 50 mg/kg) 1 h prior to LPS induction. Nuclear proteins from lung tissues were extracted using a nuclear and cytoplasmic protein extraction kit (Beyotime Institute of Biotechnology, China) following the manufacturer’s protocol. Similar results were obtained in three independent experiments, and one of the three representative experiments was shown. **P < 0.01 versus LPS group; ##P < 0.01 versus control group. a IκBα, phosphor-IκBα, and cellular nucleus NF-κB-p65 after pretreatment with C3G and LPS challenge. b Phosphorylation of ERK, JNK, and p38 after pretreatment with C3G and LPS challenge. Data represented three independent experiments and were expressed as mean ± SEM. **P <0.01 versus LPS group; ##P < 0.01 versus control group.
DISCUSSION
LPS is a glycolipid that constitutes the major portion of the outer membrane of gram-negative bacteria. LPS can bind to the cell membrane receptor of the monocytes/macrophages, which plays an important role in the development of most ARDS cases [12, 13], increases endothelial permeability and tissue damage, enters the bloodstream, and elicits inflammatory response with excessive production of inflammatory cytokines and edema in the lungs [14, 15]. Endothelial injury or dysfunction is an acknowledged finding among patients and experimental animals with ARDS. As a remarkable pro-inflammatory factor, LPS is relevant to the pathogenesis of endothelial cell injury or dysfunction by eliciting a wide array of endothelial responses [16]. Neutrophils excrete MPO, which reflects the infiltration of neutrophils and correlates with the number of neutrophils [17] into the extracellular medium under LPS stimulation, thereby generating MPO-derived oxidants and leading to tissue damage. In the development and manifestations of ARDS, oxidative stress causes serious damage to cellular structure and function [18, 19]. Both MDA [20] and SOD [21] are endogenous agents that can eliminate ROS. Moreover, alveolar macrophages phagocytize and kill invading pathogens actively, thereby releasing cellular mediators to induce inflammatory and immune responses.
Our study showed that exposure to LPS could induce MPO production and increase the total cells, neutrophils, macrophages in BALF, and MDA activity in lung tissues. Our study also demonstrated that LPS markedly decreased the SOD activity in lung tissues, which is consistent with the decreased SOD activity in ARDS patients [22]. As expected, intraperitoneal injection of C3G to mice significantly reduced the LPS-induced increment of MPO activity which is consistent with research of Fu [23] and decreased the total cells, neutrophils, macrophages in BALF, and MDA activity. By contrast, SOD level was increased. Effects of C3G on MDA activity and SOD level are consistent with research in streptozotocin-diabetic rats by Nasri S [24]. These findings suggest that C3G may decrease the LPS-induced pulmonary inflammation and injury by reducing inflammatory cell accumulation and MPO activity and elevating antioxidant levels.
Pro-inflammatory cytokines of TNF-α, IL-1β, and IL-6 play critical roles in amplifying the inflammatory responses and injuries in the development of LPS-induced ARDS. Other pro-inflammatory compounds initiate, amplify, and perpetuate the inflammatory response [25, 26]. As the earliest and primary pro-inflammatory factor, TNF-α can elicit the inflammatory cascade, cause damage to the vascular endothelial cells, and induce alveolar epithelial cells to produce other cellular factors, such as IL-6 [7]. TNF-a and IL-6 amplify the inflammatory cascade, cause inflammatory injury, and recruit neutrophils into the lung and induce the production of IL-1β [26]. Therefore, inhibiting the overproduction and expression of pro-inflammatory cytokines is very indispensable in reducing LPS-induced pulmonary injuries. As a typical symptom of the inflammatory process, edema also plays a key role in ARDS development [27, 28].
In our present research, the expression of TNF-α, IL-1β, and IL-6 was markedly overproduced after LPS administration in both the culture supernatant of HUVEC and the BALF of mice. However, these cytokines were significantly inhibited by C3G treatment 1 h before LPS administration, which is consistent with the research entitled “Cyanidin-3-O-glucoside ameliorates lipopolysaccharide-induced acute lung injury by reducing TLR4 recruitment into lipid rafts.” The results indicated that the effects of C3G might be attributed to the inhibition of pro-inflammatory cytokine release on LPS-induced ALI. The W/D weight ratio of the lung tissues was determined to quantify the magnitude of pulmonary edema, which was significantly improved by C3G in LPS-challenged mice. The histopathological changes in the lung tissues of the model group, such as pro-inflammatory cell infiltration, hyaline membrane formation, and alveolar edema, occurred relatively rare in the C3G groups; This result confirmed that C3G could alleviate the severity of histopathologic changes in LPS-challenged mice. In summary, our results showed that C3G could attenuate lung injury severity in LPS-stimulated mice.
To further investigate the inhibitory effects of C3G on LPS-induced lung tissues, we examined the effects of C3G on the activation of MAPK and NF-κB signaling pathways. NF-κB, which consists of p50, p65, and IκB-α [29], is sequestered in the cytoplasm by binding to the members of the inhibitor κB protein. After stimulation with LPS or ROS, the degradation and phosphorylation of IκB-α will increase. NF-κB P65 is then released from the NF-κB/IκB complex and is translocated from the cytoplasm into the nucleus, leading to the transcription of specific target genes, such as TNF-α, IL-1β, IL-6, and ROS [30, 31]. MAPKs also play an important role in inducing cytokine production. Inhibition of MAPK family pathways, such as ERK, p38, and JNK, can alleviate the production of pro-inflammatory cytokines [32]. Research has shown that C3G can inhibit LPS-induced inflammatory response in mouse mastitis model [33]. C3G also inhibits TNF-α-induced oxidative damage in HUVECs by nuclear translocation of NF-κB [11]. Thus, in our study, the effects of C3G on the activation of NF-κB-p65 subunits in the nucleus, phosphor-IκB-α, phosphor-ERK, phosphor-JNK, and phosphor-p38 were activated in the lung tissues by Western blot assay. C3G significantly inhibited the activities of these kinases. C3G treatment could also decrease LPS-induced cytokine production, which might be associated with the effects on the NF-κB and MAPK signaling pathways.
In summary, we demonstrate the preventive and therapeutic effects of C3G in LPS-induced HUVEC and mouse models; These effects are indicated by decreased pro-inflammatory cytokines in cell supernatants and BALF, attenuation of lung edema, and improvement of the pathologic changes and oxidative stress injury in the lung tissues. C3G can also inhibit the activation of phosphor-IκB-α, phosphor-ERK, phosphor-JNK, and phosphor-p38. These findings suggest that C3G exhibits a protective effect on LPS-induced injury in vivo and in vitro, and the most possible mechanism may be related to the anti-inflammatory and anti-oxidative effects by suppression of the NF-κB and MAPK signaling pathways.
References
Ware, L.B., and M.A. Matthay. 2000. The acute respiratory distress syndrome. The New England Journal of Medicine 342: 1334–1349.
Schuepbach, R.A., C. Feistritzer, J.A. Fernandez, J.H. Griffin, and M. Riewald. 2009. Protection of vascular barrier integrity by activated protein C in murine models depends on protease-activated receptor-1. Thrombosis and Haemostasis 101: 724–733.
Herridge, M.S., C.M. Tansey, A. Matté, G. Tomlinson, N. Diaz-Granados, A. Cooper, et al. 2011. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 364: 1293–1304.
Matthay, M.A., L.B. Ware, and G.A. Zimmerman. 2012. The acute respiratory distress syndrome. Journal of Clinical Investigation 122(8): 2731–2740.
Conti, G., S. Tambalo, G. Villetti, et al. 2010. Evaluation of lung inflammation induced by intratracheal administration of LPS in mice: comparison between MRI and histology. Magma 23(2): 93–101.
Liu, Y., H. Wu, Y.C. Nie, et al. 2011. Naringin attenuates acute lung injury in LPS-treated mice by inhibiting NF-κB pathway. International Immunopharmacology 11(10): 1606–1612.
Bhatia, M., and S. Moochhala. 2004. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. Journal of Pathology 202: 145.
Zhou, E., Y. Li, Z. Wei, Y. Fu, et al. 2014. Schisantherin A protects lipopolysaccharide-induced acute respiratory distress syndrome in mice through inhibiting NF-κB and MAPKs signaling pathways. International Immunopharmacology 22(1): 133–140.
Speciale, A., S. Anwar, R. Canali, et al. 2013. Cyanidin-3-O-glucoside counters the response to TNF-alpha of endothelial cells by activating Nrf2 pathway. Molecular Nutrition & Food Research 57(11): 1979–1987.
Wang, Q., M. Xia, C. Liu, H. Guo, Q. Ye, Y. Hu, Y. Zhang, M. Hou, H. Zhu, J. Ma, W. Ling. 2008. Cyanidin-3-O-beta-glucoside inhibits iNOS and COX-2 expression by inducing liver X receptor alpha activation in THP-1 macrophages. Life Sciences 176–184.
Speciale, A., R. Canali, J. Chirafisi, A. Saija, et al. 2010. Cyanidin-3-O-glucoside protection against TNF-α-induced endothelial dysfunction: involvement of nuclear factor-κB signaling. Journal of Agricultural and Food Chemistry 58: 12048–12054.
Faffe, D.S., V.R. Seidl, P.S. Chagas, et al. 2000. Respiratory effects of lipopolysaccharide-induced inflammatory lung injury in mice. European Respiratory Journal 15: 85.
Zarbock, A., K. Singbartl, and K. Ley. 2006. Complete reversal of acidinduced acute lung injury by blocking of platelet-neutrophil aggregation. Journal of Clinical Investigation 116: 3211.
Fenton, M.J., and D.T. Golenbock. 1998. LPS-binding proteins and receptors. Journal of Leukocyte Biology 64(1): 25–32.
Parker, J.C., and M.I. Townsley. 2004. Evaluation of lung injury in rats andmice. American Journal of Physiology - Lung Cellular and Molecular Physiology 286: L231–L246.
Bannerman, D.D., and S.E. Goldblum. 2003. American Journal of Physiology - Lung Cellular and Molecular Physiology 284(6): L899–L914.
Zhang, X., H. Huang, T. Yang, et al. 2010. Chlorogenic acid protects mice against lipopolysaccharide-induced acute lung injury. Injury 41: 746.
Mishra, V. 2007. Oxidative stress and role of antioxidant supplementation in critical illness. Clinical Laboratory 53: 199–209.
Sakaguchi, S., and S. Furusawa. 2006. Oxidative stress and septic shock: metabolic aspects of oxygen-derived free radicals generated in the liver during endotoxemia. FEMS Immunology and Medical Microbiology 47: 167–177.
Gawel, S., M. Wardas, E. Niedworok, and P. Wardas. 2004. Malondialdehyde(MDA) as a lipid peroxidation marker. Wiadomości Lekarskie 57: 453–455.
Macarthur, H., T.C. Westfall, D.P. Riley, T.P. Misko, and D. Salvemini. 2000. Inactivation of catecholamines by superoxide gives new insights on the pathogenesis of septic shock. Proceedings of the National Academy of Sciences of the United States of America 97: 9753–9758.
Ueda, J., M.E. Starr, H. Takahashi, J. Du, L.Y. Chang, J.D. Crapo, B.M. Evers, and H. Saito. 2008. Decreased pulmonary extracellular superoxide dismutase during systemic inflammation. Free Radical Biology and Medicine 45: 897–904.
Fu, Y., E. Zhou, Z. Wei, W. Wang, T. Wang, Z. Yang, and N. Zhang. 2014. Cyanidin-3-O-β-glucoside ameliorates lipopolysaccharide-induced acute lung injury by reducing TLR4 recruitment into lipid rafts. Biochemical Pharmacology 90(2): 126–134.
Nasri, S., M. Roghani, T. Baluchnejadmojarad, T. Rabani, and M. Balvardi. 2011. Vascular mechanisms of cyanidin-3-glucoside response in streptozotocin-diabetic rats. Pathophysiology 18(4): 273–278.
Cribbs, S.K., M.A. Matthay, and G.S. Martin. 2010. Stem cells in sepsis and acute lung injury. Critical Care Medicine 38: 2379–2385.
Goodman, R.B., J. Pugin, J.S. Lee, and M.A. Matthay. 2003. Cytokine-mediated inflammation in acute lung injury. Cytokine & Growth Factor Reviews 14: 523–535.
Manicone, A.M. 2009. Role of the pulmonary epithelium and inflammatory signals in acute lung injury. Expert Review of Clinical Immunology 5: 63–75.
Lucas, R., A.D. Verin, S.M. Black, and J.D. Catravas. 2009. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochemical Pharmacology 77(12): 1763–1772.
Tak, P. 2001. Firestein G.NF-kappaB: a key role in inflammatory diseases. Journal of Clinical Investigation 107: 7.
Bouwmeester, T., A. Bauch, H. Ruffner, P.O. Angrand, G. Bergamini, K. Croughton, et al. 2004. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nature Cell Biology 6: 97–105.
Urso, M.L., and P.M. Clarkson. 2003. Oxidative stress, exercise, and antioxidant supplementation. Toxicology 189: 41–54.
Chon, H., B. Choi, G. Jeong, E. Lee, and S. Lee. 2010. Suppression of proinflammatory cytokine production by specificmetabolites of Lactobacillus plantarum10hk2 via inhibiting NF-κB and p38 MAPK expressions. Comparative Immunology, Microbiology and Infectious Diseases 33(6): e41–e49.
Fu, Y., Z. Wei, E. Zhou, et al. 2014. Cyanidin-3-O-β-glucoside inhibits lipopolysaccharide-induced inflammatory response in mouse mastitismodel. Journal of Lipid Research 55(6): 1111–1119.
Acknowledgments
This work has received funding from the Natural Science Foundation of Shandong Province, (ZR2014HM112 and ZR2014HL004), the Science and Technology Development Plan of Shandong Province (2011GSF11830), and Taishan Scholar project of Shandong Province.
Conflict of Interest
None of the authors of this study has any financial or commercial conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Ming-Ming Ma and Yan Li contributed equally to this work.
Rights and permissions
About this article

Cite this article
Ma, MM., Li, Y., Liu, XY. et al. Cyanidin-3-O-Glucoside Ameliorates Lipopolysaccharide-Induced Injury Both In Vivo and In Vitro Suppression of NF-κB and MAPK Pathways. Inflammation 38, 1669–1682 (2015). https://doi.org/10.1007/s10753-015-0144-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-015-0144-y