
Abstract
IL-33 is a new member of the IL-1 family that plays a role in inflammation. In this study, we evaluated the potential of IL-33 inhibition as a treatment for systemic lupus erythematosus (SLE) using the lupus-prone model MRL/lpr mice and the underlying mechanisms of action. We treated mice with anti-mouse IL-33 antibody (anti-IL-33Ab) via intraperitoneal injection every other day from week 14 until week 20 for 6 weeks. A control group received the same amount of IgG control. Renal damage and mouse survival were compared. Cytokines, antibodies, immune complex, Tregs, myeloid-derived suppressor cells (MDSCs), and Th17 cells were also analyzed. Correlations between serum IL-33 and SLE disease activity index in human SLE were also investigated. MRL/lpr mice treated with anti-IL-33Ab showed reduced proteinuria and reduced serum anti-dsDNA levels. Nephritis, immune complex deposits, and the circulating antibodies and immune complex besides the mortality were significantly reduced by anti-IL-33Ab. Anti-IL-33Ab remarkably increased Tregs and MDSCs and reduced the Th17 cells and IL-1β, IL-6, and IL-17 levels in MRL/lpr mice. These results suggest that IL-33 inhibition may inhibit SLE via expansion of Tregs and MDSCs and inhibition of Th17 cells and proinflammatory responses, indicating that blockade of IL-33 has a protective effect on SLE.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
IL-33 is a member of the IL-1 family, and its molecular properties are very similar to a number of closely associated cytokines including IL-1, IL-1R, and IL-18 [1]. IL-33 and its receptor ST2 promote various activities related to the upregulation of systemic Th2 response of different cell types [1, 2]. Notably, recent data highlight the dual roles of IL-33 in protective and deleterious immune responses [3]. In the cardiovascular setting, IL-33 is protective in atherosclerosis by reducing the formation of atherosclerotic plaques and reducing foam cell formation [4, 5]. Although there is also a protective role in infections, IL-33 can be a deleterious process in other diseases inluding asthma [6]. In addition, IL-33 can drive autoantibody-induced arthritis via mast cells [7] and levels of IL-33 are increased in patients with rheumatoid arthritis [8]. However, it remains elusive whether IL-33 plays a protective or deleterious role in systemic lupus erythematosus (SLE).
Adoptive transfer of the immunosuppressive cells including myeloid-derived suppressor cells and regulatory T cells (Tregs) can inhibit the autoimmunity in autoimmune brain inflammation and SLE [9, 10]. Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of cells that have been shown to exert immunosuppressive properties during cancer, inflammation, and infections [11]. In mice, MDSCs are characterized by the coexpression of the myeloid-cell lineage differentiation Ag Gr-1 and CD11b [12]. Recently, data have demonstrated a potent role of MDSCs in suppressing experimental autoimmune encephalomyelitis in mice, an animal model for multiple sclerosis, and T cell responses in patients with multiple sclerosis in vitro [13]. Specifically, MDSCs suppress the progression of collagen induced arthritis by inhibiting the proinflammatory immune response of CD4+ T cells in autoimmune arthritis [14]. These observations suggest that MDSCs play crucial roles in the control of autoimmunity, which could be exploited in new cell-based therapies for human autoimmune diseases.
However, the relationship between IL-33 and MDSCs, and Tregs and Th17 has not been reported. SLE is characterized by abnormal T cell capacity, increased production of T cell-dependent IgG autoantibodies, and invasion of activated T cells into target tissues [15]. In the current study, we explored the IL-33 and we evaluated the potential of IL-33 inhibition as a treatment option for lupus nephritis (LN) and SLE using the lupus-prone model MRL/lpr mice and the interaction between IL-33 and MDSCs/T cells. This study reveals that IL-33 represents a potential therapeutic target in SLE disease and that anti-IL-33Ab treatment delays lupus progression.
MATERIALS AND METHODS
Animals
Female MRL/lpr mice were purchased from Shanghai SLAC Animal Company, China. The animal experimentation was conducted according to the “Principles of Laboratory Animal Care” and with the approval of our ethics committee.
SLE Patients
Blood samples were obtained from 28 Chinese patients with SLE who fulfilled the American College of Rheumatology Criteria for the disease and 20 healthy Chinese volunteers. Patients did not receive any treatment before enrollment and matched for co-morbidities, age, and sex. The patients who have other autoimmune diseases and asthma/allergies were excluded. All samples were obtained from volunteers attending the clinic of our hospital and were collected during routine clinical procedures. SLE disease activity index (SLEDAI) was routinely recorded. Informed consent was obtained from all individuals in agreement with the Helsinki declaration.
Treatment with Anti-IL-33Ab
The 14-week-old female MRL/lpr mice (n = 12/group) received anti-mouse IL-33 antibody (R&D Systems, USA) as previously described [16]. Anti-IL-33 antibody (3.6 μg/mouse) was given intraperitoneally every other day from week 14 until week 20. As the control, the same amount of rabbit control IgG (purified goat polyclonal antibody; R&D Systems) was given by intraperitoneal injection. All mice were monitored regularly for different clinical and biological parameters over 6 weeks.
Urinary Protein and Serum Creatinine
Urinary protein excretion was tested every 2 weeks on freshly obtained 24-h urine using a semi-quantitative test. Proteinuria was evaluated as 0–4 according to the manufacturer (Bayer Clinitek). At the termination of the study, serum creatinine was determined with an autoanalyzer (Beck- man Coulter Inc., USA).
Serum Anti-dsDNA Antibodies and Circulating Immune Complex
Serum anti-DNA antibody levels, circulating immune complex, and C3 were determined by ELISA as previously described [17]. Means of the triplicate OD450 values were recorded for the serum. Levels of anti-DNA and immune complex were expressed as units per milliliter, using a positive reference standard of pooled serum from 6-week-old MRL/lpr mice. For the detection of total IgG, HRP-conjugated goat anti-mouse IgG antibodies were added.
Histology
Kidneys were removed from anti-IL-33Ab-treated and control-treated mice, fixed in 10 % formaldehyde solution, and processed for paraffin embedding. Serial 5-μm tissue sections were cut and stained with hematoxylin and eosin (H&E) before examination under the light microscope. Glomerular, tubulointerstitial, and vascular damage was evaluated using a semiquantitative scoring system 0, 1, 2, and 3 as described previously [18].
Immunofluorescence Detection
For the examination of glomerular immune complex deposits, the kidneys were removed and snap frozen in liquid nitrogen. Frozen tissue was fixed in acetone, washed in phosphate-buffered saline (PBS), incubated in 3 % hydrogen peroxide for 10 min, and rinsed in PBS. Slides were incubated in fluorescein isothiocyanate–conjugated monoclonal IgG antibody to C3 or IgG (Santa Cruz) at a 1:50 dilution in PBS for 1 h in the dark at room temperature. Slides were then rinsed in PBS and distilled water and mounted with a cover slip using Vectashield hard set medium (Vector H-1400, Vector Laboratories). Slides were scored (0–3) in a masked fashion by an experienced renal pathologist for the intensity and coverage of immunofluorescence in the glomerulus (0, no fluorescence; 0.5, trace, just detectable above background; 1, fluorescence scattered and light; 2, bright but not diffuse; and 3, bright and diffuse).
Preparation of Kidney Extracts
Mice were killed by cervical dislocation at 20 weeks. Kidneys were isolated, immediately frozen in liquid N2, and stored at −80 °C. Frozen kidneys were powdered-ground in liquid nitrogen using a mortar/pestle set-up. The resulting tissue powders were homogenized with a glass–glass potter at 4 °C in the RIPA buffer. The homogenates were centrifuged at 14,000×g for 10 min at 4 °C, and the supernatants were stored as aliquots at −80 °C until use. Protein concentration was determined using a BCA method.
Measurement of Cytokines
The levels of IL-1β, IL-6, and IL-17 in the kidneys and the serum were determined by using specific ELISA kits (R&D Systems) as per the instructions.
Flow Cytometry
For intracellular cytokine staining, 5 × 105 splenocytes from individual mice were incubated in triplicates with 50 ng/ml phorbol 12-myristate 13-acetate (Sigma-Aldrich, Munich, Germany) and 1 μM ionomycin (Sigma-Aldrich) in the presence of brefeldin A (5 μg/ml, Sigma-Aldrich) in 200 μl RPMI 1640 supplemented with 10 % FCS for 4 h at 37 °C before staining. For each staining, cells of three cultures were pooled to obtain a sufficient number of cells for cytokine analysis. Staining was performed with the intracellular cytokine staining kit (eBioscience) according to the manufacturer’s instructions. For Tregs (CD4 + Foxp3+) and Th17 (CD4 + IL-17A+) cell staining freshly isolated cells from individual mice were used. The staining was performed using the Foxp3 staining set and anti-mouse IL-17A (eBioscience). For MDSCs (CD11b + Gr-1+) detection, splenocytes were stained for MDSCs markers for 20 min at 4 °C in PBS/5 % FBS. Anti-CD11b and Gr-1 antibodies (BD Pharmingen) were added thereafter for 20 min on ice.
Statistical Analysis
Statistics were performed using SPSS 15.0 software. Data are presented as means ± standard error of the mean (SEM). Data were tested for homogeneity of variances and then compared at each time interval by one-way analysis of variance or Kruskal–Wallis test. Log-rank analysis was used to compare trends in animal survival. A value of p < 0.05 was considered statistically significant.
RESULTS
Anti-IL-33Ab Reduced the Renal Damage
To determine the effect of IL-33 inhibition on renal function, we measured 24-h urinary protein excretion from the beginning of 14 weeks. Control mice developed increasing proteinuria (Fig. 1a). In contrast, mice treated with anti-IL-33Ab developed significantly less severe proteinuria than controls. And at the end of the study, serum creatinine was significantly reduced by anti-IL-33Ab treatment (Fig. 1b).

Effects of anti-IL-33Ab treatment on the renal function in MRL/lpr mice. a Urine protein score was assessed every 2 weeks in MRL/lpr mice treated with anti-IL-33Ab or vehicle. Urinary protein excretion was tested every 2 weeks on freshly obtained 24-h urine using a semi-quantitative test. Proteinuria was evaluated as 0–4 according to the manufacturer (Bayer Clinitek). b Serum creatinine was determined at the end of the study. n = 12 mice per group. Data are presented as means ± SEM. Data were tested for homogeneity of variances and then compared at each time interval by one-way analysis of variance (b) or Kruskal–Wallis test (a). # P < 0.01 compared with control-treated group.
Anti-IL-33Ab Reduced the Circulating Levels of Anti-dsDNA, Immune Complex, and C3
Production of anti-dsDNA antibodies and circulating immune complex is associated with lupus-like renal disease in MRL/lpr mice. There was a remarkable rise in serum anti-dsDNA antibody levels in the control group at week 20, whereas anti-IL-33Ab-treated group had obviously reduced anti-dsDNA antibody levels at week 20. We also assessed serum immune complex levels. Similar to the change seen in serum anti-dsDNA antibody levels, anti-IL-33Ab-treatment significantly reduced the levels of circulating immune complex and C3 in their serum compared to the control group at week 20 (Fig. 2a–c). We detected the total IgG to confirm that anti-dsDNA suppression was not related to a more global effect on B cells (Fig. 2d, p > 0.05).

Anti-IL-33Ab reduced the serum levels of anti-dsDNA autoantibody (a) and circulating immune complex (b) and increased the C3 level (c) in MRL/lpr mice. The total IgG did not change significantly (d). At week 20, sera were collected from mice and subjected to ELISA. n = 12 mice per group. Data are presented as means ± SEM. Data were compared by one-way analysis of variance. # P < 0.01 compared with control-treated group.
Anti-IL-33Ab Reduced Renal Pathology and Deposition of IgG and C3
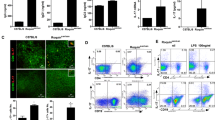
H&E-stained kidney sections were assessed by histological scoring for overall glomerular inflammation and vasculitis. MRL/lpr mice in the control group exhibited classical features of lupus renal disease with diffuse glomerulonephritis and vasculitis. Anti-IL-33Ab-treated mice, however, exhibited a significant improvement in glomerular inflammation and vasculitis in the kidneys of MRL/lpr mice (Fig. 3a, b).

Anti-IL-33Ab reduced renal pathology and deposition of IgG and C3. a Renal pathology (H&E, ×100). b Semiquantitative analysis of histological damage. c Renal deposition of IgG and C3 (×200). d Semiquantitative analysis of fluorescence intensity. n = 12 mice per group. Data are presented as means ± SEM. Data were compared by Kruskal–Wallis test. # P < 0.01 compared with control-treated group.
Moreover, the glomerular IgG and C3 deposition was significantly reduced in anti-IL-33Ab-treated mice compared with controls (Fig. 3c, d). These results are consistent with the effect of anti-IL-33Ab on anti-dsDNA antibody levels.
Anti-IL-33Ab Reduced Proinflammatory Cytokines
Proinflammatory cytokines play indispensable roles in the development of SLE. First,we tested the levels of IL-1β, IL-33, IL-6, and IL-17 in young MRL/lpr mice aged 6 weeks (before disease onset) and older mice with renal damage (aged 22 weeks), and we found that renal IL-33, IL-1β, IL-6, and IL-17 were upregulated in this model with disease progression (Fig. 4a). To ascertain whether IL-33 blockade inhibits these cytokines, anti-IL-33Ab-treated and untreated lupus-prone mice were bled at the end of the study. The levels of IL-1β, IL-6, and IL-17 in the serum and kidneys were measured by ELISA. The anti-IL-33Ab-treated mice showed lower serum and renal levels of IL-1β, IL-6, and IL-17 than the untreated mice (Fig. 4b, c). These results suggest that IL-33 blockade may have a therapeutic effect on SLE by inhibiting the production of inflammatory cytokines.

Anti-IL-33Ab reduced production of proinflammatory cytokines. a Proinflammatory cytokines in the young MRL/lpr mice aged 6 weeks (before disease onset) and older mice with renal damage (aged 22 weeks). We found renal IL-33, IL-1β, IL-6, and IL-17 were upregulated in this model with disease progression. b Serum levels of IL-1β, IL-6, and IL-17 after anti-IL-33Ab or control treatment. c Renal levels of cytokines after anti-IL-33Ab or or control treatment. n = 12 mice per group. Data are means ± SEM. Data were compared by one-way analysis of variance test. # P < 0.01 compared with control-treated group.
Anti-IL-33Ab Regulated Tregs MDSCs and Th17 Cells and Reduced the Mortality
We also evaluated the T cell subset in the spleens of MRL/lpr mice. We found that anti-IL-33Ab significantly increased the percentage of Tregs and MDSCs and reduced the percentage of Th17 cells as compared with the control group (Fig. 5a, b).

Anti-IL-33Ab treatment increased Tregs and MDSCs, reduced the Th17 cells and improved mouse survival. a Representative flow cytometrical data of Tregs, MDSCs and Th17 cells in the spleens in the mice aged 20 weeks (n = 10 mice per group). b Flow cytometrical analysi (n = 10 mice per group). c Survival was significantly prolonged in mice treated with anti-IL-33Ab compared with control (n = 14 mice per group). At 38 weeks, survival was 80 % in the anti-IL-33Ab group compared with 20 % in control group (P = 0.011). Data are presented as means ± SEM. Data were compared by Kruskal–Wallis test. # P < 0.01 compared with control-treated group.
In addition, we assess the effects of anti-IL-33Ab treatment on the mouse survival in MRL/lpr mice (n = 14 per group). By 38 weeks of age, there was 80 % mortality of control treated mice, while 80 % of anti-IL-33Ab-treated mice survived (Fig. 5c, p = 0.011). This suggests that anti-IL-33Ab treatment significantly prolonged the survival of MRL/lpr mice.
Increased Serum IL-33 Level Was Correlated with SLEDAI in SLE Patients
Furthermore, we also investigate the clinical serum samples in SLE patients. We found that serum IL-33 level was markedly increased in the SLE patients compared with healthy volunteers (Fig. 6a). Correlation analysis showed that serum IL-33 was positively correlated with SLEDAI in SLE patients (Fig. 6b, r = 0.563, p = 0.0013), suggesting IL-33 as an indicator for disease activity in SLE.

Increased serum IL-33 level was correlated with SLEDAI in 28 SLE patients. a Serum IL-33 level was markedly increased in the 28 SLE patients compared with 20 healthy volunteers. b Correlation analysis showed that serum IL-33 was positively correlated with SLEDAI in 28 SLE patients (r = 0.563, p = 0.0013). Data are the mean ± SEM for 28 patients and 20 healthy controls. Data were compared by ANOVA test. # P < 0.01 compared with control-treated group.
DISCUSSION
SLE is a chronic autoimmune inflammatory disease that affects various organs, especially the kidney. Therapeutic advances over the last few decades have led to significant improvements in prognosis; however, multiple aspects of the management of SLE patients are still far from optimal [19]. In this study, we present evidence that IL-33 played a critical role in the pathological process of the SLE and LN and verified that administration of anti-IL-33Ab in vivo could significantly ameliorate the severity of SLE disease, as demonstrated by reduced levels of anti-dsDNA antibodies, circulating immune complex, and C3, reduced renal immune complex deposition, lessened proteinuria, and reduced score of glomerulonephritis. This therapeutic effect was closely associated with expansion of Tregs and MDSCs, accompanied by significantly reduced Th17 cells and inflammatory cytokines in the serum and kidneys of anti-IL-33Ab-treated MRL/lpr mice. These data suggest the new IL-1 family member IL-33 is a potential therapeutic target in SLE and LN.
Increasing evidence has shown that IL-33 and its receptor ST2 contribute to the pathogenesis of autoimmune diseases. It was reported that IL-33 increased significantly in patients with SLE compared to that in controls, and they suggested that IL-33 might play a role in the acute phase of SLE [20]. In another study, serum sST2 was significantly higher in patients with active SLE compared with that in inactive patients and correlated significantly with SLEDAI and anti-dsDNA levels [21]. Studies have also indicated a similar relationship between the IL-33/ST2 system and rheumatoid arthritis (RA), wherein the levels of serum and synovial fluid IL-33 are significantly higher in patients with RA than those in controls [22]. Data showed that serum levels of IL-33 and sST2 are correlated with IL-1β and IL-6 [23]. Meanwhhile, silencing IL-33 significantly reduces tumor necrosis factor-α-induced synthesis of IL-33, IL-6, IL-8, and monocyte chemotactic protein-1 in RA at the mRNA and protein levels [24]. However, the functional role of IL-33 in SLE remains to be explored. In our present study, we observed higher levels of IL-33 in patients with SLE compared with those in the control. In addition, serum IL-33 levels were correlated with disease activity (SLEDAI), which was consistent with previous results. More importantly, we found that anti-IL-33Ab treatment provided therapeutic and survival benefit for lupus-prone mice.
Tregs and MDSCs, with immunosuppressive properties, have been verified in the animal models in suppressing autoimmune responses [9, 10]. MDSCs significantly prevented autoimmune diabetes onset, and the protective effects of MDSCs might be mediated by inducing anergy in autoreactive T cells and the development of Tregs [25]. Studies have also indicated that transfer of Tregs inhibited experimental anti-glomerular basement membrane glomerulonephritis in mice and prolonged drug-induced disease remission in (NZBxNZW) F1 lupus mice [26, 27]. In contrast, emerging data show a body of evidence that IL-17 and Th17 cells contribute to the pathogenesis of SLE [28]. Data have shown that type I IFN and IL-17 act in concert to sustain and amplify autoimmune and inflammatory responses involved in the pathogenesis of systemic autoimmune diseases [29]. Further studies have found that the immune imbalance between Th17 and Tregs is involved in the development of SLE and LN [30–32]. Of note, IL-33 plays critical roles in T cell differentiation. Recent data demonstrate IL-33 is closely related to a Th1-to-Th2/Treg switch [33]. Studies also provide clear evidence that IL-33 plays a role in switching a predominantly pathogenic Th17/Th1 response to Th2 activity [34]. As shown in this study, our data showed that anti-IL-33Ab treatment increased Tregs and MDSCs and reduced the Th17 cells, suggesting that the protective role of IL-33 blockade was, at least partly, mediated via expansion of MDSCs and Tregs.
In addition, the therapeutic effects of IL-33 blockade might also be related to a systemic blunting of autoimmunity and proinflammatory responses, as reflected by reduced serum levels of autoantibodies, immune complex, C3, and proinflammatory cytokines in kidney extracts and serum as well as renal immune complex deposition. In agreement with our study, elevated serum IL-33 is associated with autoantibody production in patients with RA [35].
In summary, this study demonstrated that IL-33 blockade improves lupus nephritis and mouse survival mainly by suppressing abnormal autoimmunity and inflammation of SLE. Our analysis proves the therapeutic efficacy of IL-33 blockade in the treatment of SLE in MRL/lpr mice. Taken with the current data, IL-33 blockade might have a potential for treating SLE patients.
References
Kurowska-Stolarska, M., A. Hueber, B. Stolarski, and I.B. McInnes. 2011. Interleukin-33: a novel mediator with a role in distinct disease pathologies. Journal of Internal Medicine 269: 29–35.
Lloyd, C.M. 2010. IL-33 family members and asthma–bridging innate and adaptive immune responses. Current Opinion in Immunology 22: 800–806.
Mirchandani, A.S., R.J. Salmond, and F.Y. Liew. 2012. Interleukin-33 and the function of innate lymphoid cells. Trends in Immunology 33: 389–396.
McLaren, J.E., D.R. Michael, R.C. Salter, T.G. Ashlin, C.J. Calder, A.M. Miller, et al. 2010. IL-33 reduces macrophage foam cell formation. The Journal of Immunology 185: 1222–1229.
Miller, A.M., D. Xu, D.L. Asquith, L. Denby, Y. Li, N. Sattar, et al. 2008. IL-33 reduces the development of atherosclerosis. The Journal of Experimental Medicine 205: 339–346.
Moffatt, M.F., I.G. Gut, F. Demenais, D.P. Strachan, E. Bouzigon, S. Heath, et al. 2010. A large-scale, consortium-based genomewide association study of asthma. The New England Journal of Medicine 363: 1211–1221.
Xu, D., H.R. Jiang, P. Kewin, Y. Li, R. Mu, A.R. Fraser, et al. 2008. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proceedings of the National Academy of Sciences of the United States of America 105: 10913–10918.
Talabot-Ayer, D., T. McKee, P. Gindre, S. Bas, D.L. Baeten, C. Gabay, et al. 2012. Distinct serum and synovial fluid interleukin (IL)-33 levels in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Joint, Bone, Spine 79: 32–37.
Humrich, J.Y., H. Morbach, R. Undeutsch, P. Enghard, S. Rosenberger, O. Weigert, et al. 2010. Homeostatic imbalance of regulatory and effector T cells due to IL-2 deprivation amplifies murine lupus. Proceedings of the National Academy of Sciences of the United States of America 107: 204–209.
Trigunaite A, Khan A, Der E, Song A, Varikuti S, Jørgensen TN. Gr1(high) CD11b(+) cells suppress B cell differentiation and lupus-like disease in lupus-prone male mice. Arthritis Rheum. 2013. doi:10.1002/art.38048.
Gabrilovich, D.I., and S. Nagaraj. 2009. Myeloid-derived suppressor cells as regulators of the immune system. Nature Reviews Immunology 9: 162–174.
Peranzoni, E., S. Zilio, I. Marigo, L. Dolcetti, P. Zanovello, S. Mandruzzato, et al. 2010. Myeloid-derived suppressor cell heterogeneity and subset definition. Current Opinion in Immunology 22: 238–244.
Ioannou, M., T. Alissafi, I. Lazaridis, G. Deraos, J. Matsoukas, A. Gravanis, et al. 2012. Crucial role of granulocytic myeloid-derived suppressor cells in the regulation of central nervous system autoimmune disease. The Journal of Immunology 188: 1136–1146.
Fujii, W., E. Ashihara, H. Hirai, H. Nagahara, N. Kajitani, K. Fujioka, et al. 2013. Myeloid-derived suppressor cells play crucial roles in the regulation of mouse collagen-induced arthritis. The Journal of Immunology 191: 1073–1081.
Tsokos, G.C. 2011. Systemic lupus erythematosus. The New England Journal of Medicine 365: 2110–2121.
Kim, Y.H., T.Y. Yang, C.S. Park, S.H. Ahn, B.K. Son, J.H. Kim, et al. 2012. Anti-IL-33 antibody has a therapeutic effect in a murine model of allergic rhinitis. Allergy 67: 183–190.
Sekine, H., K.L. Graham, S. Zhao, M.K. Elliott, P. Ruiz, P.J. Utz, et al. 2006. Role of MHC-linked genes in autoantigen selection and renal disease in a murine model of systemic lupus erythematosus. The Journal of Immunology 177: 7423–7434.
Wellmann, U., M. Letz, A. Schneider, K. Amann, and T.H. Winkler. 2001. An Ig μ-heavy chain transgene inhibits systemic lupus erythematosus immunopathology in autoimmune (NZB × NZW)F1 mice. International Immunology 13: 1461–1469.
Lateef, A., and M. Petri. 2012. Unmet medical needs in systemic lupus erythematosus. Arthritis Research and Therapy 14(Suppl 4): S4.
Yang, Z., Y. Liang, W. Xi, C. Li, and R. Zhong. 2011. Association of increased serum IL-33 levels with clinical and laboratory characteristics of systemic lupus erythematosus in Chinese population. Clinical and Experimental Medicine 11: 75–80.
Mok, M.Y., F.P. Huang, W.K. Ip, Y. Lo, F.Y. Wong, E.Y. Chan, et al. 2010. Serum levels of IL-33 and soluble ST2 and their association with disease activity in systemic lupus erythematosus. Rheumatology (Oxford) 49: 520–527.
Oboki, K., T. Ohno, N. Kajiwara, K. Arae, H. Morita, A. Ishii, et al. 2010. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proceedings of the National Academy of Sciences of the United States of America 107: 18581–18586.
Hong, Y.S., S.J. Moon, Y.B. Joo, C.H. Jeon, M.L. Cho, J.H. Ju, et al. 2011. Measurement of interleukin-33 (IL-33) and IL-33 receptors (sST2 and ST2L) in patients with rheumatoid arthritis. Journal of Korean Medical Science 26: 1132–1139.
Kunisch, E., S. Chakilam, M. Gandesiri, and R.W. Kinne. 2012. IL-33 regulates TNF-α dependent effects in synovial fibroblasts. International Journal of Molecular Medicine 29: 530–540.
Yin, B., G. Ma, C.Y. Yen, Z. Zhou, G.X. Wang, C.M. Divino, et al. 2010. Myeloid-derived suppressor cells prevent type 1 diabetes in murine models. The Journal of Immunology 185: 5828–5834.
Wolf, D., K. Hochegger, A.M. Wolf, H.F. Rumpold, G. Gastl, H. Tilg, et al. 2005. CD4 + CD25+ regulatory T cells inhibit experimental anti-glomerular basement membrane glomerulonephritis in mice. Journal of the American Society of Nephrology 16: 1360–1370.
Weigert, O., C. von Spee, R. Undeutsch, L. Kloke, J.Y. Humrich, and G. Riemekasten. 2013. CD4 + Foxp3+ regulatory T cells prolong drug-induced disease remission in (NZBxNZW) F1 lupus mice. Arthritis Research and Therapy 15: R35.
Shin, M.S., N. Lee, and I. Kang. 2011. Effector T-cell subsets in systemic lupus erythematosus: update focusing on Th17 cells. Current Opinion in Rheumatology 23: 444–448.
Ambrosi, A., A. Espinosa, and M. Wahren-Herlenius. 2012. IL-17: a new actor in IFN-driven systemic autoimmune diseases. European Journal of Immunology 42: 2274–2284.
Ma, J., J. Yu, X. Tao, L. Cai, J. Wang, and S.G. Zheng. 2010. The imbalance between regulatory and IL-17-secreting CD4+ T cells in lupus patients. Clinical Rheumatology 29: 1251–1258.
Mengya, Z., M. Hanyou, L. Dong, L. Xiaohong, and Z. Lihua. 2013. Th17/Treg imbalance induced by increased incidence of atherosclerosis in patients with systemic lupus erythematosus (SLE). Clinical Rheumatology 32: 1045–1052.
Zhao, J., H. Wang, C. Dai, H. Wang, H. Zhang, Y. Huang, et al. 2013. P2X7 blockade attenuates lupus nephritis by inhibiting NLRP3/ASC/caspase-1 activation. Arthritis and Rheumatism 65: 3176–3185.
Duan, L., J. Chen, H. Zhang, H. Yang, P. Zhu, A. Xiong, et al. 2012. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3+ regulatory T-cell responses in mice. Molecular Medicine 18: 753–761.
Jiang, H.R., M. Milovanović, D. Allan, W. Niedbala, A.G. Besnard, S.Y. Fukada, et al. 2012. IL-33 attenuates EAE by suppressing IL-17 and IFN-γ production and inducing alternatively activated macrophages. European Journal of Immunology 42: 1804–1814.
Mu, R., H.Q. Huang, Y.H. Li, C. Li, H. Ye, and Z.G. Li. 2010. Elevated serum interleukin 33 is associated with autoantibody production in patients with rheumatoid arthritis. The Journal of Rheumatology 37: 2006–2013.
Conflict of Interest
All authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, P., Lin, W. & Zheng, X. IL-33 Neutralization Suppresses Lupus Disease in Lupus-Prone Mice. Inflammation 37, 824–832 (2014). https://doi.org/10.1007/s10753-013-9802-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-013-9802-0