
Abstract
Background
Previous studies reported that IL-38 was abnormally expressed in patients with systemic lupus erythematosus (SLE). However, the involvement of IL-38 in the pathophysiology of SLE remains unknown.
Methods
The therapeutic potential of IL-38 was tested in pristane-treated wild-type (WT) and IL-38−/− mice. Thus, SLE was induced via pristane in WT and IL-38−/− mice. Afterwards, the liver, spleen, and kidney of each mouse were obtained. The flow cytometric analysis of the immune cells, serologic expression of inflammatory cytokines and autoantibodies, renal histopathology, and inflammatory signaling were evaluated.
Results
WT mice with pristane-induced lupus exhibited hepatomegaly, splenomegaly, severe kidney damages, increased lymphoproliferation, enhanced lymphoproliferation, and upregulated inflammatory cytokines, such as IL-6, IL-13, IL-17A, MIP-3α, IL-12p70, and IFNγ, and elevated levels of autoantibodies, such as ANA IgG, anti-dsDNA IgG, and total IgG. IL-38−/− mice whose lupus progressed, had elevated cells of CD14+, CD19+, CD3+, and Th1, upregulated inflammatory cytokines and autoantibodies, and severe pathological changes in kidney. Administration of recombinant murine IL-38 to pristane-treated IL-38−/− mice improved their renal histopathology, which depended on ERK1/2, JNK1/2, p38, NF-κB p65, and STAT5 signaling pathways.
Conclusion
IL-38 regulates SLE pathogenesis. Furthermore, targeting IL-38 is critical in the treatment of SLE.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Systemic lupus erythematosus (SLE), an inflammatory autoimmune disease, is characterized by damages in tissues and organs, such as kidney and skin. Lupus nephritis (LN) is a common and life-threatening manifestation of SLE. A study showed that inflammation-mediated immune response dysfunction was significantly involved in the pathogenesis of SLE and LN [1].
Interleukin-38 (IL-38) is a novel member of the IL-1 family, which is one of the first described cytokine families. The IL-1 family has eight cytokines (IL-1α/β, IL-18, IL-33, IL-36α/β/γ, and IL-37) and three receptor antagonists (IL-1Ra, IL-36Ra, and IL-38) [2]. IL-38 was originally named as IL-1F10. It was discovered in 2001 via a high-throughput cDNA sequence and was renamed in 2010. The IL-38 gene is located at chromosome 2 between the IL-36 N and IL-1RN encoding genes (ch2q13_14.1). It has 5 exons, and it encodes 152 amino acids with a 17–18 kDa molecular weight. In addition, it lacks signal peptide. IL-38 is approximately 41% homologous with IL-1 receptor antagonists (IL-1Ra) and 43% homologous with IL-36 receptor antagonists (IL-36Ra). IL-38 potentially recruits IL-1R8 or other inhibitory co-receptors of the IL-1 family, and then bind to IL-1R6 to suppress inflammation [3]. To date, evidence from patients and animal models implies that IL-38 has a negative role in T helper 17 (Th17) cells and myeloid cells. Some studies explored the tolerogenic function of IL-38 on dendritic cell and regulatory T cell [2, 3]. Nevertheless, there were variability and inconsistency in the dose-dependent anti-inflammatory role of IL-38 and context-dependent pro-inflammatory property of IL-38, which might correlate with high heterogeneity in the materials, such as the reagents used [3]. Interestingly, in patients with rheumatoid arthritis (RA) fibroblast-like synoviocytes (FLSs), overexpression of IL-38 promoted the proliferation of synoviocytes, accelerated the migration of RA FLSs, and increased the invasion capacity of RA FLSs via autophagy [4]. Expression of IL-38 in patients with colorectal cancer was related to the progression of colorectal cancer [5]. IL-38 suppressed colorectal cancer metastasis and proliferation and facilitated apoptosis by inhibiting the activation of extracellular signal-regulated kinases (ERK) signaling [5]. In our previous studies, the plasma levels of IL-38 were increased in patients with SLE and in those with RA. In addition, the plasma level of IL-38 correlated with disease activity [6, 7]. In wild-type (WT) mice injected with pristane, there was an increase in the severe disease clinical score and a detected histopathology. However, addition of recombinant murine IL-38 to mice with pristane-induced lupus reversed the development of the disease [6]. Since several studies found that there was an aberrant expression of IL-38 in autoimmune diseases, such as SLE, it was necessary to clearly clarify how IL-38 inhibited the development of lupus. In the present study, we aimed to study IL-38 gene deficient (IL-38−/−) mice model, to investigate the involvement of IL-38 in the pathogenesis of SLE, and to discuss the therapeutic effect of IL-38 in IL-38−/− mice following lupus development.
Methods
Mice
WT female C57BL/6 mice (8 weeks) were purchased from the SPF Biotechnology (Beijing, China), while IL-38−/− mice (8 weeks) with a C57BL/6 background were purchased from Cyagen Biosciences (Suzhou, China). All mice had a supply of food and water continuously provided in a room with a controlled temperature (23 ± 1 °C) and a 12-h light/dark cycle. Their handling was in accordance with the Animal Ethics Committee of Southwest Medical University. Furthermore, the procedures conducted in this study were approved by the Animal Ethics Committee of Southwest Medical University.
Treatment protocol
Pristane (2,6,10,14-Tetramethylpentadecane)-induced lupus mice model is a widely accepted and used lupus mice model because it exhibits the clinical and laboratory characteristics of human SLE patients, such as proteinuria and glomerulonephritis [8]. Pristane is a chemical substance. Injection of 0.5 mL of pristane into WT mice, such as WT C57BL/6 mice induced a lupus-like disease, which was characterized by the following features: proteinuria, mesangial matrix, mesangial cell proliferation, deposition of immune complexes in the mesangial region, distribution of IgM, IgG and C3 in the mesangial region and mesangial capillaries, increased expression of anti-nuclear antibody (ANA) IgG (in brief ANA), and anti-double-stranded DNA (anti-dsDNA) IgG (in brief anti-dsDNA). These pathological features were similar to the changes in types III, IV, and V lupus nephritis, which were based on the classification criteria of lupus nephritis and SLE [9,10,11,12]. In this study, WT mice were classified into two groups: the control group (n = 5) and the pristane group (n = 5). On the other hand, IL-38−/− mice were classified into three groups: the control group (n = 5), the pristane group (n = 5), and the pristane + IL-38 group (n = 5). The control group of WT mice and IL-38−/− mice were both intraperitoneally injected with 500 µL of phosphate-buffered saline (PBS) at week 9 once. On the other hand, the pristane group of WT mice and the pristane and pristane + IL-38 groups of IL-38−/− mice were intraperitoneally injected with 500 µL of pristane at week 9 once. At week 21, all WT mice and IL-38−/− mice in the control group and the pristane group were intraperitoneally injected with 200 µL of PBS every other day for 14 days, whereas the IL-38−/− mice in the pristane + IL-38 group were intraperitoneally injected with recombinant murine IL-38 (AdipoGen, Hamburg, Germany) every other day for 14 days. At week 25, all mice were killed.
Liver, spleen and kidney weight
The liver, spleen, and left and right kidneys of each mouse were excised after they were killed. Extra fat and connective tissues in the organs were removed, weighed, and photographed. The average weight of the liver, spleen, and the left and right kidneys in each group represented the change among the different kinds of treatment.
Flow cytometry
Leukocytes were counted after the collection of spleen and the lysis of erythrocytes. Antibodies were used for detecting monocytes (CD14-FITC, clone: rmC5-3), dendritic cells (CD11c-FITC, clone: HL3), B cells (CD19-APC, clone: 1D3), T cells (CD3-FITC, clone: 145-2C11; CD4-FITC, clone: GK1.5; CD8-APC, clone: 53–6.7), Th1 (IFNγ-PE-CF594, clone: XMG1.2), Th2 (IL-4-APC, clone: 11B11), Th17 (IL-17A-APC-CyTM7, clone: TC11-18H10), and Treg cells (Foxp3-PE, clone: MF23). All antibodies were from the BD Biosciences (California, USA). To detect CD3, CD8, monocytes, dendritic cells, and B cells, leukocytes were stained directly and analyzed. To detect Th1, Th2, Th17 and Treg cells, leukocytes were first stained with CD4-FITC, fixed with Fixation Buffers (RD system, Minnesota, USA), permeabilized with Permeabilization/Wash Buffer (RD system, Minnesota, USA), and subsequently stained with IFNγ-PE-CF594, IL-4-APC, IL-17A-APC-CyTM7, Foxp3-PE, and the corresponding isotype control (rat IgG1, κ-PE-CF594, clone: R3-34 or rat IgG2b, κ-PE, clone: R35-38). Finally, stained cells were analyzed via flow cytometry with the FACSVerse (BD Biosciences, California, USA).
Enzyme-linked immunosorbent assay (ELISA)
The serum levels of ANA, anti-dsDNA, and total IgG were examined using ELISA kits (CUSABIO, Wuhan, China). Briefly, 100 µL standard or sample were added into a 96-well plate and incubated for 2 h at 37 °C. Afterwards, the liquid of each well was removed. Biotin-conjugate was then added, and the mixture was incubated for 1 h and washed for three times. Horseradish Peroxidase-avidin was added and incubated for 1 h and was washed for five times. TMB (3,3',5,5' tetramethyl-benzidine) substrate was added, and the mixture was incubated for 15 min. Finally, we added the Stop Solution and determined the optical density of each well. The minimum detection of ANA, anti-dsDNA, and total IgG was 1.95 pg/mL, 0.39 ng/mL, and 29 ng/mL, respectively. All samples were measured in duplicates.
Histology
Lupus nephritis and kidney damage were evaluated by morphometrical and immunofluorescence assays and ultrastructural analysis [6]. The kidneys were fixed with 10% formalin and embedded in paraffin. Sections with a thickness of 4 μm were then cut. The kidney section was then stained with hematoxylin and eosin (HE) assay. Morphometrical assessment of the capsule, cortex, medulla, glomerulus, tubules, and collecting duct was conducted. The severity of glomerular lesions (graded on a score 0–3) was also assessed [13]. On the other hand, the kidney section was stained with Ponceau, Fuchsin and Aniline blue (Masson assay), and renal fibrosis was assessed using the Image-Pro Plus 6.0 software. The Panoramic 250 Flash (3DHISTECH, Hungary) was then used to scan the images. Immunofluorescence assay for total IgG in the kidney tissue was conducted using the FITC-conjugated anti-mouse IgG (Abcam, Cambridge, UK). Fluorescence intensity was presented as a score ranging from 0 to 3 [13], and images were obtained using a Laser Confocal Microscope (Olympus, Shinjuku, Japan). Moreover, kidneys were fixed in 3% glutaraldehyde, then in 1% osmium tetroxide afterwards. They were cut into 50 nm sections and stained with uranium acetate and lead citrate, and they were then analyzed using a transmission electron microscope. Ultrastructural analysis of the kidney structure, morphology of podocyte processes, mesangial cell, mitochondria, and rough endoplasmic reticulum was then conducted.
Western blotting
Total protein was extracted from splenocytes, and the concentration was examined using a BCA protein assay kit (Beyotime, Shanghai, China). Proteins were separated via 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (for total ERK1/2, cJun N-terminal kinase 1/2 (JNK1/2), p38 and nuclear factor kappa-B p65 (NF-κB p65)) or 8% SDS-PAGE (for signal transducers and activators of transcription 5 (STAT5)). They were then transferred to a polyvinylidene difluoride membrane (Millipore, USA). The membrane was blocked with 10% skimmed milk powder in 0.5% Tween/PBS, followed by incubation with primary antibodies against ERK1/2 (Abcam, Cambridge, UK), JNK1/2 (Abcam, Cambridge, UK), p38 (Abcam, Cambridge, UK), NF-κB p65 (Abcam, Cambridge, UK), STAT5 (Abcam, Cambridge, UK) and GAPDH (Beyotime, Shanghai, China) overnight at 4 °C. After washing four times within a period of 1 h, an HRP-labeled secondary antibody was added. The mixture was then incubated for 1 h. The membrane was washed twice within 40 min. Finally, the signals on the membrane were visualized using an Enhanced Chemiluminescence detection kit (Thermo Scientific, Shanghai, China).
Inflammatory cytokine microarray
A total of 18 inflammatory cytokines in the serum were evaluated using a mouse cytokine array (RayBiotech, Georgia, USA) according to the manufacturer’s instructions. The cytokine assay included interferon γ (IFNγ), IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12p70, IL-13, IL-17A, IL-17F, IL-21, IL-22, IL-23, IL-28A, macrophage inflammatory protein-3α (MIP-3α), transforming growth factor-β1 (TGF-β1), and tumor necrosis factor α (TNFα). Briefly, the slides were blocked (blocking buffer, 100 µl/well) for 30 min in room temperature. The buffer was then discarded, and a sample and standard was added and incubated overnight at 4 °C. Afterwards, the sample and standard were discarded, and the mixture was washed five times. Biotin-Antibody Cocktail was added, and the mixture was incubated for 2 h. Afterwards, the mixture was incubated with Cy3-Streptavidin for 1 h. Finally, the streptavidin was discarded, and the remaining solution was washed five times. The signals were scanned and quantified using the GenePix 4000B Microarray Scanner (Molecular Devices, Sunnyvale, USA). Images in the microarray analysis was read by a GenePix Pro 6.0 software (Axon Instruments, Foster City, CA).
Statistical analysis
Quantitative data are expressed as mean ± standard deviation (SD) if the data are normally distributed. The Student’s t test was conducted to compare two groups. The analysis of variance (ANOVA) was conducted to compare more than two groups. The post-tests were performed to further evaluate the differences among groups more than two. Statistical analysis was conducted using the Prism 5 software (GraphPad, California, USA) and the SPSS software version 16.0 (SPSS, Chicago, USA). A p value of < 0.05 was considered significant.
Results
Increased degree of hepatomegaly and splenomegaly with IL-38 deficiency
IL-38−/− mice and WT mice were killed at week 25 after pristane treatment. Their liver, spleen, and left and right kidneys were examined. WT mice treated with pristane had hepatomegaly and splenomegaly as compared to WT mice treated with PBS (Fig. 1A, B). Similarly, IL-38−/− mice treated with pristane had hepatomegaly and splenomegaly as compared to IL-38−/− mice treated with PBS. Addition of IL-38 to pristane-treated IL-38−/− mice resulted in reduced hepatomegaly and splenomegaly (Fig. 1A, B). When compared WT mice treated with PBS, IL-38−/− mice treated with PBS had heavier livers (Fig. 1A). IL-38−/− mice treated with pristane had heavier left kidneys compared with that of WT mice treated with pristane (Fig. 1C). However, the phenotype of kidneys was not significantly affected by pristane treatment of WT and IL-38−/− mice. Contrastingly, the phenotype of the left kidneys of IL-38−/− mice treated with pristane and pristane + IL-38 was significantly affected (Fig. 1C).

Increased hepatomegaly and splenomegaly with IL-38 deficiency. A–C Weight of individual liver, spleen, and kidney (left and right) with representative photographs in wild-type and IL-38−/− mice treated with pristane, or phosphate-buffered saline (PBS) or IL-38. A total of 5 samples per group were analyzed, and symbols represent individual mice. Bars show the mean ± standard deviation (SD). **P value less than 0.005. *P value less than 0.05
Effect of IL-38 deficiency on renal involvement
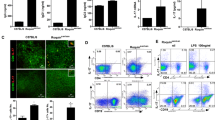
Kidney histopathology showed that pristane-treated WT mice had significantly more renal damage than PBS-treated WT mice. This was indicated by the more extensive glomerular atrophy and necrosis, mesangial proliferation, basement membrane thickening, reduced number of capillaries, nuclear swelling of endothelial cells, renal tubular degeneration, lymphocyte infiltration, and fibrous tissue hyperplasia in pristane-treated WT mice. Similarly, pristane-treated IL-38−/− mice had a significantly greater renal damage than PBS-treated IL-38−/− mice. Nevertheless, addition of IL-38 inhibited renal damage. This was indicated by the significant glomerular and tubulointerstitial damage in pristane-treated IL-38−/− mice and improved lesions in pristane-treated IL-38−/− mice when IL-38 was added (Fig. 2A–O). IgG deposition in WT and IL-38−/− mice showed that pristane-treated WT mice and IL-38−/− mice treated with pristane had significant IgG deposition (Fig. 2P).

Effect of IL-38 deficiency on renal involvement. Photomicrographic representation of renal damage. A–E Hematoxylin and eosin (HE), F–J Masson, K–O transmission electron microscope (TEM), P immunofluorescence assay of total IgG for individual kidney in wild-type and IL-38−/− mice treated with pristane, or phosphate-buffered saline (PBS) or IL-38. Original magnification × 400 for HE, Masson assay and × 1200 for TEM assay and × 400 for immunofluorescence assay. HE and Masson scores for individual kidney with symbols in right panels. HE and Masson scores A–J are the mean ± standard deviation (SD), and the fluorescence intensity scores for individual kidney with symbols in right panels (P①-⑮) are the mean ± SD. **P value less than 0.005. *P value less than 0.05
Aggravation of SLE in IL-38−/− mice is associated with the dysregulation in frequency of different immune cells
Immune cells are crucial for the maintenance of immune homeostasis. We compared the frequency of CD14+ monocytes, CD11c+ dendritic cells, CD19+ B cells, CD3+ T cells, CD8+ T cells, CD4+IFNγ+ (Th1) cells, CD4+IL-4+ (Th2) cells, CD4+IL-17A+ (Th17) cells, and CD4+FoxP3+ (regulatory T, Treg) cells between IL-38−/− mice and WT controls (Fig. 3A–E). WT mice treated with pristane had higher percentages of CD14+, CD19+, CD3+, Th1, Th2 cells as compared to those in WT mice treated with PBS (Fig. 3A–D). Pristane-treated IL-38−/− mice had higher percentages of CD14+, CD19+, CD3+, Th1 cells as compared to those in IL-38−/− mice treated with PBS. However, addition of IL-38 to pristane-treated IL-38−/− mice showed reduced percentages of these cells (Fig. 3A–D). Interestingly, IL-38−/− mice treated with PBS had higher percentages of CD14+, CD19+, Th1, and Th2 cells as compared to those in WT mice treated with PBS. IL-38−/− mice treated with pristane showed higher percentages of CD14+ and Th1 cell as compared to those in WT mice treated with pristane. Moreover, the percentage of Th17 cells was comparable in IL-38−/− mice treated with PBS or pristane compared to that in WT mice treated with PBS or pristane. Addition of IL-38 resulted in the reduction of the percentage of Th17 cells, indicating that IL-38 inhibited Th17 cell proliferation in mice with lupus (Fig. 3E). On the contrary, the percentage of Treg cells was lower in pristane-treated WT mice compared to that in WT mice treated with PBS. It was also lower in IL-38−/− mice treated with PBS. IL-38−/− mice treated with pristane showed a higher percentage of Treg cells as compared to that in IL-38−/− mice treated with PBS (Fig. 3E).

Aggravation of IL-38−/− mice from systemic lupus erythematosus is associated with dysregulated immune cells. Flow cytometry analysis for different immune cells in wild-type and IL-38−/− mice treated with pristane, or phosphate-buffered saline (PBS) or IL-38. A–E Percentages of CD14+, CD11c+, CD19+, CD3+, CD8+, T helper 1 (Th1), Th2, Th17, regulatory T (Treg) cells among splenocytes in mice in each treatment group (left panel). In A–E (right panel), symbols represent individual mice. Bars show the mean ± standard deviation (SD). **P value less than 0.005. *P value less than 0.05
Dysregulation of cytokine levels
A total of 18 inflammatory cytokines were analyzed. For IL-1β, IL-2, IL-4, IL-5, IL-10, IL-17F, IL-21, IL-23, and TNFα, some samples from the WT mice and IL-38−/− mice were not detectable. Therefore, differences in the serum levels of IFNγ, IL-6, IL-12p70, IL-13, IL-17A, IL-22, IL-28A, MIP-3α, and TGF-β1 were discussed (Fig. 4A–I). Results showed that serum levels of IL-6, IL-13, IL-17A, MIP-3α, IL-12p70, and IFNγ were significantly higher in WT mice treated with pristane compared with those in WT mice treated with PBS (Fig. 4A-C, E, F, H). Similarly, the serum levels of IL-6, IL-13, IL-17A, MIP-3α, and IL-12p70 were significantly elevated in IL-38−/− mice treated with pristane compared with those in IL-38−/− mice treated with PBS. Contrastingly, addition of IL-38 to pristane-treated IL-38−/− mice significantly reduced the levels of inflammatory cytokines (Fig. 4A-C, E, F). The serum levels of IL-28A were lower in WT mice treated with pristane compared with that in WT mice treated with PBS, which was demonstrated in IL-38−/− mice, showing that serum levels of IL-28A were lower in IL-38−/− mice treated with pristane compared with those in IL-38−/− mice treated with PBS. Addition of IL-38 to pristane-treated IL-38−/− mice increased the serum levels of IL-28A (Fig. 4D). IL-38−/− mice treated with PBS had higher levels of IL-6, IL-28A, IL-12p70, IFNγ, and TGF-β1 compared with WT mice treated with PBS (Fig. 4A, D, F, H, I). IL-38−/− mice treated with pristane had higher levels of IL-6, IL-17A, IL-12p70, IFNγ, TGF-β1 compared with WT mice treated with pristane (Fig. 4A, C, F, H, I).

Regulation of inflammatory cytokines in serum from wild-type and IL-38−/− mice. A–I Serum levels of inflammatory cytokines from wild-type and IL-38−/− mice treated with pristane, or phosphate-buffered saline (PBS) or IL-38 were examined, including interleukin-6 (IL-6), IL-13, IL-17A, IL-28A, macrophage inflammatory protein 3α (MIP-3α), IL-12p70, IL-22, interferon-γ (IFNγ) and transforming growth factor β1 (TGF-β1). Symbols represent individual mice. Bars show the mean ± standard deviation (SD). **P value less than 0.005. *P value less than 0.05
The effect of IL-38 deficiency on the production of autoantibodies in mice with lupus
The levels of total IgG, ANA, anti-dsDNA were detected in IL-38−/− and WT mice after their treatment with pristane. Compared with PBS-treated WT mice, pristane-treated WT mice had significantly higher levels of total IgG, ANA, and anti-dsDNA (Fig. 5A–C). Pristane-treated IL-38−/− mice also had higher levels of total IgG, ANA, and anti-dsDNA as compared with PBS-treated IL-38−/− mice. However, addition of IL-38 downregulated the levels of total IgG, ANA, and anti-dsDNA in pristane-treated IL-38−/− mice (Fig. 5A–C). Interestingly, IL-38−/− mice treated with PBS showed higher levels of ANA as compared with that in WT mice treated with PBS (Fig. 5A).

Promotion of serologic manifestations of lupus in IL-38−/− mice. A–C Wild-type and IL-38−/− mice were treated with pristane, phosphate-buffered saline (PBS) or IL-38, and serum levels of anti-nuclear antibody (ANA), double-stranded DNA (dsDNA), total IgG were examined as described in “Methods”. **P value less than 0.005. *P value less than 0.05
Reduced inflammatory signaling pathway in IL-38-deficient mice
Several signaling pathways were analyzed in WT and IL-38−/− mice (Fig. 6A). In WT mice treated with PBS, the expression of total JNK1/2, ERK1/2, p38, STAT5 and NF-κB p65 was significantly lower compared with those in WT mice treated with pristane (Fig. 6B–F). Similarly, the expression of total JNK1/2 and ERK1/2 was significantly lower in IL-38−/− mice treated with PBS than in IL-38−/− mice treated with pristane. However, the addition of IL-38 to pristane-treated IL-38−/− mice significantly reduced the expression of total JNK1/2 and ERK1/2 (Fig. 6B–C). There were no significant differences in the expression of total p38, NF-κβ p65, and STAT5 between IL-38−/− mice treated with PBS and those treated with pristane. On the other hand, the addition of IL-38 to pristane-treated IL-38−/− mice significantly reduced the expression of total p38, NF-κB p65, and STAT5 (Fig. 6D–F). However, WT mice treated with PBS showed a lower expression of total JNK1/2, ERK1/2, p38, STAT5, and NF-κB p65 compared with IL-38−/− mice treated with PBS (Fig. 6B–F). WT mice treated with pristane also had lower expression levels of total JNK1/2, ERK1/2, and p38 compared with IL-38−/− mice treated with pristane (Fig. 6B–D).

IL-38 deficiency upregulates inflammatory signaling. Protein extracts were obtained from splenocytes. Total expression of extracellular signal-regulated kinases 1/2 (ERK1/2), c-Jun N-terminal kinase 1/2 (JNK1/2), p38, nuclear factor kappa-B (NF-κB) p65 and signal transducer and activator of transcription 5 (STAT5) in splenocytes from wild-type and IL-38−/− mice treated with pristane, phosphate-buffered saline (PBS) or IL-38, was examined by western blotting (WB). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as invariant control. Representative blot from 1 mouse per treatment group was shown. Data are the mean ± standard deviation (SD) of 5 mice per group. **P value less than 0.005. *P value less than 0.05
Discussion
In previous studies, dysregulation of IL-38 expression was reportedly present in inflammatory autoimmune diseases. In multiple sclerosis (MS) patients, serum levels of IL-38 were higher in newly diagnosed patients than in treated patients [14]. The levels of IL-38 were elevated in the intestines of patients with inflammatory bowel diseases (IBD) and mice with dextran sulfate sodium (DSS)-induced colitis [15]. On the contrary, patients with primary Sjogren’s syndrome (pSS) had lower expression levels of IL-38 compared with healthy controls [16]. Interestingly, a study analyzed the serum levels of IL-38 and showed that IL-38 abundance was higher in samples from SLE patients than in those from healthy controls [17]. SLE patients with active disease had a much higher IL-38 level than those with inactive disease. Furthermore, IL-38 detection was related to an increased risk of the development of renal lupus and central nervous system lupus [17]. Therefore, it is suggested that IL-38 may play a critical role in the pathogenesis of the aforementioned diseases. In this study, we used WT and IL-38−/− mice, both of which were treated with PBS and pristane. (1) WT mice treated with PBS were compared with WT mice treated with pristane. This aimed to see whether there were pathogenic changes in mice with pristane-induced lupus. (2) IL-38−/− mice were divided into three groups: those treated with PBS, those treated with pristane, and those treated with pristane and recombinant IL-38. This aimed to determine whether pristane could induce a lupus-like disease in IL-38−/− mice. Furthermore, this aimed to discuss the potential of recombinant IL-38 as a therapeutic agent for lupus. (3) We also compared WT mice and IL-38−/− mice treated with PBS and WT mice and IL-38−/− mice treated with pristane. This aimed to determine whether IL-38 deficiency promoted inflammation and autoimmunity development in PBS-treated groups and aggravated the lupus-like disease in pristane-treated groups. In our previous study [6], we only used WT mice to induce lupus through treatment with pristane and discussed the role of recombinant IL-38 in inhibiting lupus progression. The design of the current study is novel compared with the previous study. (1) In the current study, IL-38 gene knockout mice were directly used to confirm the role of IL-38 in SLE development. (2) Recombinant IL-38 was used to discuss the therapeutic effect of IL-38 in IL-38−/− lupus mice. (3) Either IL-38−/− mice treated with PBS or IL-38−/− mice treated with pristane (lupus mice model) were used to compare the histological and serological changes in WT mice treated with PBS or WT mice treated with pristane, respectively, because the current study aimed to discuss whether IL-38 gene deficiency was able to induce dysregulation of immunity, inflammation, and autoimmunity.
Both WT mice and IL-38−/− mice treated with pristane had significant hepatomegaly and splenomegaly, suggesting that IL-38 deficiency promoted hepatomegaly and splenomegaly during lupus development. Interestingly, the addition of IL-38 in IL-38−/− mice treated with pristane inhibited hepatomegaly and splenomegaly. Therefore, IL-38 has a potential to inhibit hepatomegaly and splenomegaly in mice with lupus. Similarly, both WT mice and IL-38−/− mice treated with pristane had severe histological scores and significant kidney damages. In contrast, the administration of IL-38 to IL-38−/− mice treated with pristane inhibited the role of IL-38 deficiency in mice with lupus. This corresponded to the improvement in the pathological changes associated with lupus. This was confirmed in WT mice and IL-38−/− mice treated with pristane whose IgG deposition in kidneys was evaluated. We found that both of the mice had significant IgG deposition in their kidneys compared to those treated with PBS. Moreover, addition of IL-38 to IL-38−/− mice treated with pristane resulted in the downregulation of IgG, suggesting that IL-38 suppressed autoantibody deposition in the kidney. In a study discussing the effect of IL-38 in MRL/lpr lupus mice, treatment of MRL/lpr mice with IL-38 resulted in the reduction of their glomerulonephritis score compared with the PBS-treated control group. Moreover, the mesangial thickening and proliferation in IL-38 treated mice were ameliorated [18]. However, IgG renal deposition was comparable between IL-38-treated mice and the control group [18]. This difference may be attributed to several reasons. First, the MRL/lpr mice were spontaneous lupus-prone mice with damages in their kidneys. We used WT and IL-38−/− mice to induce lupus by pristane, where there were no kidney lesions in the mice before pristane treatment. Second, the MRL/lpr mice were 10–20 week-old when they were recruited into this the study. On the other hand, the age of the WT and IL-38−/− mice was 8 weeks. Third, the MRL/lpr mice were culled one day after they were injected with recombinant murine IL-38 for 7 days, then. The kidney changes were then analyzed. In our study, WT and IL-38−/− mice were treated with pristane when they were 9 weeks old and were culled when they were 25 weeks old. Moreover, a group of pristane-treated IL-38−/− mice administrated IL-38 for 14 days at 21 weeks old and were culled at 25 weeks old. In our study, IL-38−/− mice treated with PBS had larger and heavier livers compared with those of WT mice treated with PBS. In addition, IL-38−/− mice treated with pristane had larger and heavier left kidneys compared with those of WT mice treated with pristane. The findings suggested that IL-38 deficiency might promote liver and kidney swelling. It must be noted that kidney may be more severely inflamed in IL-38−/− mice. However, the clear mechanism needs to be clarified in the future.
Innate and adaptive immune responses are required for maintaining homeostasis. In this study, we analyzed the frequencies of CD14+, CD11c+, CD19+, CD3+, CD8+, CD4+INFγ+ (Th1), CD4+IL-4+ (Th2), CD4+IL-17A+ (Th17), and CD4+FoxP3+ (Treg) cell in WT and IL-38−/− mice. WT mice treated with pristane had higher frequencies of CD14+, CD19+, CD3+, Th1, and Th2 cells. On the contrary, WT mice treated with pristane had lower frequencies of Treg cells compared with WT mice treated with PBS. Interestingly, frequencies of CD11c+, CD8+, Th17 cells were comparable between WT mice treated with pristane and those treated with PBS. The aforementioned findings were partly confirmed in IL-38−/− mice whose frequencies of CD14+, CD19+, CD3+, and Th1 cells were examined. We found increased percentages of CD14+, CD19+, CD3+, and Th1 cells in IL-38−/− mice treated with pristane compared with those in IL-38−/− mice treated with PBS. It is notable that the frequencies of these cells were significantly reduced after the addition of IL-38 in pristane-treated IL-38−/− mice. Therefore, IL-38 inhibited the proliferation of CD14, CD19, CD3, and Th1 cells in lupus development. The frequencies of CD11c+ and Th2 cells were not significantly different among IL-38−/− mice treated with PBS, pristane, and pristane + IL-38, suggesting that IL-38 deficiency did not affect the proliferation of cells in lupus development. Similarly, there was a comparable percentage of Th17 cells between IL-38−/− mice treated with PBS and pristane. However, the addition of IL-38 in pristane-treated IL-38−/− mice significantly reduced the frequency of Th17 cells, demonstrating that IL-38 inhibited Th17 cell proliferation. In IL-38-treated MRL/lpr mice, the proportion of Th17 cells was reduced as compared to that in MRL/lpr mice treated with PBS.18 However, the Th1, CD4+, CD8+, and CD19+ cells were comparable between MRL/lpr mice treated with IL-38 and those treated with PBS [18]. In our study, the frequency of Treg cells was higher in IL-38−/− mice treated with pristane compared with IL-38−/− mice treated with PBS, which was similar to that in IL-38−/− mice treated with pristane + IL-38, suggesting that IL-38 inhibited Treg cell proliferation during lupus development. A study discussed the expression of IL-38 and Treg cells in childhood asthma, showing that serum levels of IL-38 were higher in asthmatic patients, while the percentage of CD4+FoxP3+ Treg cells was decreased in asthmatic patients [19]. Elevated IL-38 expression was negatively related to the percentage of Treg cells in asthmatic patients [19]. Treatment of septic mice induced by cecal ligation and puncture (CLP) with IL-38 significantly promoted the immunosuppressive activity of Treg cell [20]. These different findings may correlate with the negative feedback mechanism for regulating the role of IL-38 in lupus development. There was significant immune stringent status in lupus mice, exhibiting dysregulated autoantibodies, inflammatory cytokine production, and inflammatory immune cell proliferations as discussed above. The excessive immune stringent status in lupus may aggravate the feedback mechanism, which then upregulates IL-38 expression, leading to a higher percentage of Treg cells in pristane-treated IL-38−/− mice as compared to that in WT mice treated with pristane and IL-38−/− mice treated with PBS. However, it is necessary to elucidate the feedback mechanism responsible for regulating IL-38 in lupus. In our study, we found that IL-38−/− mice treated with PBS had much higher percentages of CD14+, CD19+, Th1, and Th2 cells and lower percentages of CD11c+ and CD8+ cells compared with those in WT mice treated with PBS. The findings were demonstrated in IL-38−/− mice treated with pristane, where there were much higher percentages of CD14+ and Th1 cells and lower percentages of CD11c+ and CD8+ cells compared to those in WT mice treated with pristane. The results implied that IL-38 inhibited CD14+, CD19+, Th1, and Th2 cell proliferation and promoted CD11c+ and CD8+ cell proliferation under physiological conditions. Furthermore, IL-38 inhibited CD14+ and Th1 cell proliferation and promoted CD11c+ cell proliferation under pathological conditions, such as lupus.
The production of inflammatory cytokines and autoantibodies are hallmarks of lupus. To discuss the role of IL-38 in regulating the inflammatory response, we evaluated the serum levels the of total IgG, anti-dsDNA, ANA, and different inflammatory cytokines in WT and IL-38−/− mice. We found that all autoantibodies were higher in WT mice treated with pristane than in WT mice treated with PBS. The findings were confirmed in IL-38−/− mice treated with pristane, which had increased serum levels of total IgG, anti-dsDNA, and ANA in pristane-treated mice compared with those in mice treated with PBS. Interestingly, the addition of IL-38 significantly downregulated the elevation of autoantibodies in pristane-treated IL-38−/− mice. In MRL/lpr mice treated with IL-38, both levels of total IgG and anti-dsDNA were similar with PBS-treated MRL/lpr mice [19]. To better discuss the potential inflammatory response regulated by IL-38, 18 inflammatory cytokines were evaluated in WT and IL-38−/− mice. We found a higher expression of IL-6, IL-13, IL-17A, MIP-3α, and IL-12p70, and a lower expression of IL-28A in IL-38−/− mice treated with pristane, which were significantly changed after the addition of IL-38. The levels of IL-6, IL-13, IL-17A, and IL-12p70 were dysregulated in lupus and might play a significant role in the pathogenesis of lupus [21, 22]. Collectively, the findings demonstrated that IL-38 was necessary to inhibit pro-inflammatory serological changes in lupus development mediated by inflammatory cytokines and autoantibodies.
Mitogen-activated protein kinases (MAPKs), NF-κB, and STAT5 are necessary for innate and adaptive immune response, which contribute to SLE pathogenesis [23, 24]. In mice with allergic asthma, IL-38 treatment resulted in the downregulation of the expression of p38, ERK1/2, and NF-κB pathways [25]. In addition, IL-38 alleviated the poly(I:C) induced lung inflammation by inhibiting the p38, ERK1/2, and NF-κB signaling pathways [26]. Nucleus pulposus cells from the intervertebral disc degeneration (IVDD) patients were stimulated with IL-38, which resulted in the inhibition of the expression of the NF-κB p65 protein [27]. Thus, IL-38 may suppress the inflammatory signaling pathways. In this study, WT mice treated with PBS had downregulated total ERK1/2, JNK1/2, p38, NF-κB p65, and STAT5 compared with WT mice treated with pristane, demonstrating that lupus mice had excessive inflammation, which was characterized by increased expression of inflammatory signaling pathways. This was confirmed in IL-38−/− mice, whereby IL-38−/− mice treated with pristane had a higher expression of total JNK1/2, ERK1/2, p38, NF-κB p65, and STAT5 compared with those in IL-38−/− mice treated with PBS, although there were no significant differences in the expression of total p38, NF-κB p65, and STAT5 between the two groups of mice. Interestingly, administration of IL-38 to pristane-treated IL-38−/− mice significantly downregulated the expression of total JNK1/2, ERK1/2, p38, NF-κB p65, and STAT5. Therefore, IL-38 potentially inhibits the inflammatory signaling pathways in SLE development. MAPKs, NF-κB, and STAT5 signaling pathways regulate different immune cells, which then contribute to SLE development. For instance, p38 interacted with IL-17, promoting plasma cell survival and enhancing autoantibody production in mice with lupus [28]. Inhibition of STAT5 in mice with lupus was accompanied by reduced Th1 and Th17 cells, ameliorated proteinuria and renal lesion severity, decreased serum levels of anti-dsDNA antibody, and reduced spleen size [24, 29]. Together, IL-38 inhibits the above signaling pathways, which then downregulate the differentiation and proliferation of immune cells, thereby inhibiting lupus development.
This study has several limitations. Administration of murine recombinant IL-38 in IL-38−/− mice treated with pristane may raise some questions. Although the dose injected into the mice model and the time when the mice were killed were similar to those in a previous study [6], there were still questions. For example, the most effective dose was unknown. The best time to kill the mice was unknown. Therefore, in the future, the dose and time should be discussed and evaluated. This may provide useful evidence for the treatment of lupus with IL-38. The present study showed that IL-38 inhibited lupus development. However, it did not clearly determine the negative feedback mechanism in which IL-38 was involved in. Nevertheless, we discussed some possible reasons above. In the previous study [6], SLE patients with arthritis, pericarditis, hematuria, proteinuria, pyuria, and anti-dsDNA had higher plasma levels of IL-38 compared with those in SLE patients without the aforementioned features, respectively. In this study, IL-38−/− mice treated with pristane showed severe kidney damage, such as glomerulonephritis and higher levels of anti-dsDNA antibody, and yet IL-38−/− mice treated with pristane improved when more IL-38 was administered. These findings suggest that there is a negative feedback mechanism in which IL-38 is involved and the potential mechanisms in particular. First, we found higher serum levels of autoantibodies (total IgG, ANA, anti-dsDNA) in IL-38−/− mice treated with pristane, suggesting that much autoantibodies may accumulate at the joints of the mice, which then recruit inflammatory cells, such as neutrophils and macrophages and secrete more pro-inflammatory cytokines at the joints or distribute the secreted pro-inflammatory cytokines in the circulation in IL-38−/− mice treated with pristane. Therefore, the deficiency of IL-38 in lupus may promote the development of arthritis, while addition of IL-38 may inhibit arthritis. Second, SLE patients with proteinuria had higher plasma levels of IL-38 when compared with that in SLE patients without proteinuria, and both WT mice and IL-38−/− mice treated with pristane showed kidney damage, such as glomerulonephritis. Addition of IL-38 to both of the mice improved glomerulonephritis. This may be attributed to various reasons. SLE patients with proteinuria/lupus nephritis or lupus mice had immune complex deposition in their kidneys. This was demonstrated in this study and in other previous studies as well [6, 30]. The deficiency of IL-38 in lupus may promote immune complex deposition at the kidneys. Immune complexes, such as much autoantibodies ANA, and anti-dsDNA deposition, led to nephritis. Addition of IL-38 inhibited the deposition of autoantibodies, improving kidney damage. These hypotheses should be clarified in future studies.
In conclusion, we demonstrated that inhibition of IL-38 is related to the aggravation of lupus-like disease, while addition of IL-38 improved lupus-like disease. Thus, targeting IL-38 may be a novel therapeutic approach for SLE patients.
Data availability
Datasets are available from the corresponding author on reasonable request.
Abbreviations
- SLE:
-
Systemic lupus erythematosus
- WT:
-
Wild-type
- IL-38:
-
Interleukin-38
- LN:
-
Lupus nephritis
- IL-1Ra:
-
IL-1 receptor antagonist
- IL-36Ra:
-
IL-36 receptor antagonist
- RA:
-
Rheumatoid arthritis
References
Xu WD, Huang AF. Role of interleukin-38 in chronic inflammatory diseases: a comprehensive review. Front Immunol. 2018;9:1462.
Esmaeilzadeh A, Bahmaie N, Nouri E, Hajkazemi MJ, Zareh RM. Immunobiological properties and clinical applications of interleukin-38 for immune-mediated disorders: a systematic review study. Int J Mol Sci. 2021;22(22):12552.
Han Y, Huard A, Mora J, da Silva P, Brüne B, Weigert A. IL-36 family cytokines in protective versus destructive inflammation. Cell Signal. 2020;75: 109773.
Hao Z, Liu Y. IL-38 and IL-36 target autophagy for regulating synoviocyte proliferation, migration, and invasion in rheumatoid arthritis. Dis Markers. 2021;2021:7933453.
Huang L, Zhang H, Zhao D, Hu H, Lu Z. Interleukin-38 suppresses cell migration and proliferation and promotes apoptosis of colorectal cancer cell through negatively regulating extracellular signal-regulated kinases signaling. J Interferon Cytokine Res. 2021;41(10):375–84.
Xu WD, Su LC, Liu XY, et al. IL-38: a novel cytokine in systemic lupus erythematosus pathogenesis. J Cell Mol Med. 2020;24(21):12379–89.
Xu WD, Su LC, He CS, Huang AF. Plasma interleukin-38 in patients with rheumatoid arthritis. Int Immunopharmacol. 2018;65:1–7.
Freitas EC, de Oliveira MS, Monticielo OA. Pristane-induced lupus: considerations on this experimental model. Clin Rheumatol. 2017;36(11):2403–14.
Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725.
Markowitz GS, D’Agati VD. The ISN/RPS 2003 classification of lupus nephritis: an assessment at 3 years. Kidney Int. 2007;71(6):491–5.
Satoh M, Kumar A, Kanwar YS, Reeves WH. Anti-nuclear antibody production and immune-complex glomerulonephritis in BALB/c mice treated with pristane. Proc Natl Acad Sci USA. 1995;92(24):10934–8.
Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009;30(9):455–64.
Fu R, Xia Y, Li M, et al. Pim-1 as a therapeutic target in lupus nephritis. Arthritis Rheumatol. 2019;71(8):1308–18.
Zarrabi M, Nazarinia M, Rahimi Jaberi A, Gholijani N, Amirghofran Z. Elevated IL-38 serum levels in newly diagnosed multiple sclerosis and systemic sclerosis patients. Med Princ Pract. 2021;30(2):146–53.
Xie C, Yan W, Quan R, et al. Interleukin-38 is elevated in inflammatory bowel diseases and suppresses intestinal inflammation. Cytokine. 2020;127: 154963.
Luo D, Chen Y, Zhou N, Li T, Wang H. Blockade of Th17 response by IL-38 in primary Sjogren’s syndrome. Mol Immunol. 2020;127:107–11.
Rudloff I, Godsell J, Nold-Petry CA, et al. Brief report: interleukin-38 exerts anti-inflammatory functions and is associated with disease activity in systemic lupus erythematosus. Arthritis Rheumatol. 2015;67(12):3219–25.
Chu M, Tam LS, Zhu J, et al. In vivo anti-inflammatory activities of novel cytokine IL-38 in Murphy Roths Large (MRL)/lpr mice. Immunobiology. 2017;222(3):483–93.
Chu M, Chu IM, Yung EC, et al. Aberrant expression of novel cytokine IL-38 and regulatory T lymphocytes in childhood asthma. Molecules. 2016;21(7):933.
Ge Y, Huang M, Wu Y, Dong N, Yao YM. Interleukin-38 protects against sepsis by augmenting immunosuppressive activity of CD4(+) CD25(+) regulatory T cells. J Cell Mol Med. 2020;24(2):2027–39.
Sigdel KR, Duan L, Wang Y, et al. Serum cytokines Th1, Th2, and Th17 expression profiling in active lupus nephritis-IV: from a Southern Chinese Han population. Mediators Inflamm. 2016;2016:4927530.
Mao YM, Zhao CN, Leng J, et al. Interleukin-13: a promising therapeutic target for autoimmune disease. Cytokine Growth Factor Rev. 2019;45:9–23.
Xiao ZX, Hu X, Zhang X, et al. High salt diet accelerates the progression of murine lupus through dendritic cells via the p38 MAPK and STAT1 signaling pathways. Signal Transduct Target Ther. 2020;5(1):34.
Goropevšek A, Holcar M, Pahor A, Avčin T. STAT signaling as a marker of SLE disease severity and implications for clinical therapy. Autoimmun Rev. 2019;18(2):144–54.
Sun X, Hou T, Cheung E, et al. Anti-inflammatory mechanisms of the novel cytokine interleukin-38 in allergic asthma. Cell Mol Immunol. 2020;17(6):631–46.
Gao X, Chan PKS, Lui GCY, et al. Interleukin-38 ameliorates poly(I:C) induced lung inflammation: therapeutic implications in respiratory viral infections. Cell Death Dis. 2021;12(1):53.
Yu H, Liu Y, Xie W, Xie Q, Liu Q, Cheng L. IL-38 alleviates the inflammatory response and the degeneration of nucleus pulposus cells via inhibition of the NF-κB signaling pathway in vitro. Int Immunopharmacol. 2020;85: 106592.
Ma K, Du W, Xiao F, et al. IL-17 sustains the plasma cell response via p38-mediated Bcl-xL RNA stability in lupus pathogenesis. Cell Mol Immunol. 2021;18(7):1739–50.
Hou LF, He SJ, Li X, et al. Oral administration of artemisinin analog SM934 ameliorates lupus syndromes in MRL/lpr mice by inhibiting Th1 and Th17 cell responses. Arthritis Rheum. 2011;63(8):2445–55.
Chang A, Clark MR, Ko K. Cellular aspects of the pathogenesis of lupus nephritis. Curr Opin Rheumatol. 2021;33(2):197–204.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81701606), and Sichuan Provincial Natural Science Foundation (2022NSFSC0697, 2022NSFSC0694).
Author information
Authors and Affiliations
Contributions
Study conception and design: WX, LS, and AH. Acquisition of data: LF, YL, and XL. Analysis and interpretation of data: QH, QW, and JZ. Drafting the article: WX, LS, and AH. Final approval of the version of the article to be published: all the authors, and all the authors agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval and consent to participate
This study was approved Animal Ethics Committee of Southwest Medical University.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xu, WD., Su, LC., Fu, L. et al. IL-38, a potential therapeutic agent for lupus, inhibits lupus progression. Inflamm. Res. 71, 963–975 (2022). https://doi.org/10.1007/s00011-022-01581-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-022-01581-3